
Industrially Compatible Transfusable iPSC-Derived RBCs: Progress, Challenges and Prospective Solutions



 and
and
Abstract
:1. Introduction
1.1. Overwhelming Worldwide Demand for Blood
1.2. Storage-Induced Lesions in Donor Blood
1.3. Lack of Potential Blood Substitutes
2. Cell Sources for In Vitro Erythropoiesis
2.1. Hematopoietic Stem and Progenitor Cells (HSPCs)
2.2. Immortalized Erythroblasts
2.3. Human Embryonic Stem Cells and Human Induced Pluripotent Stem Cells

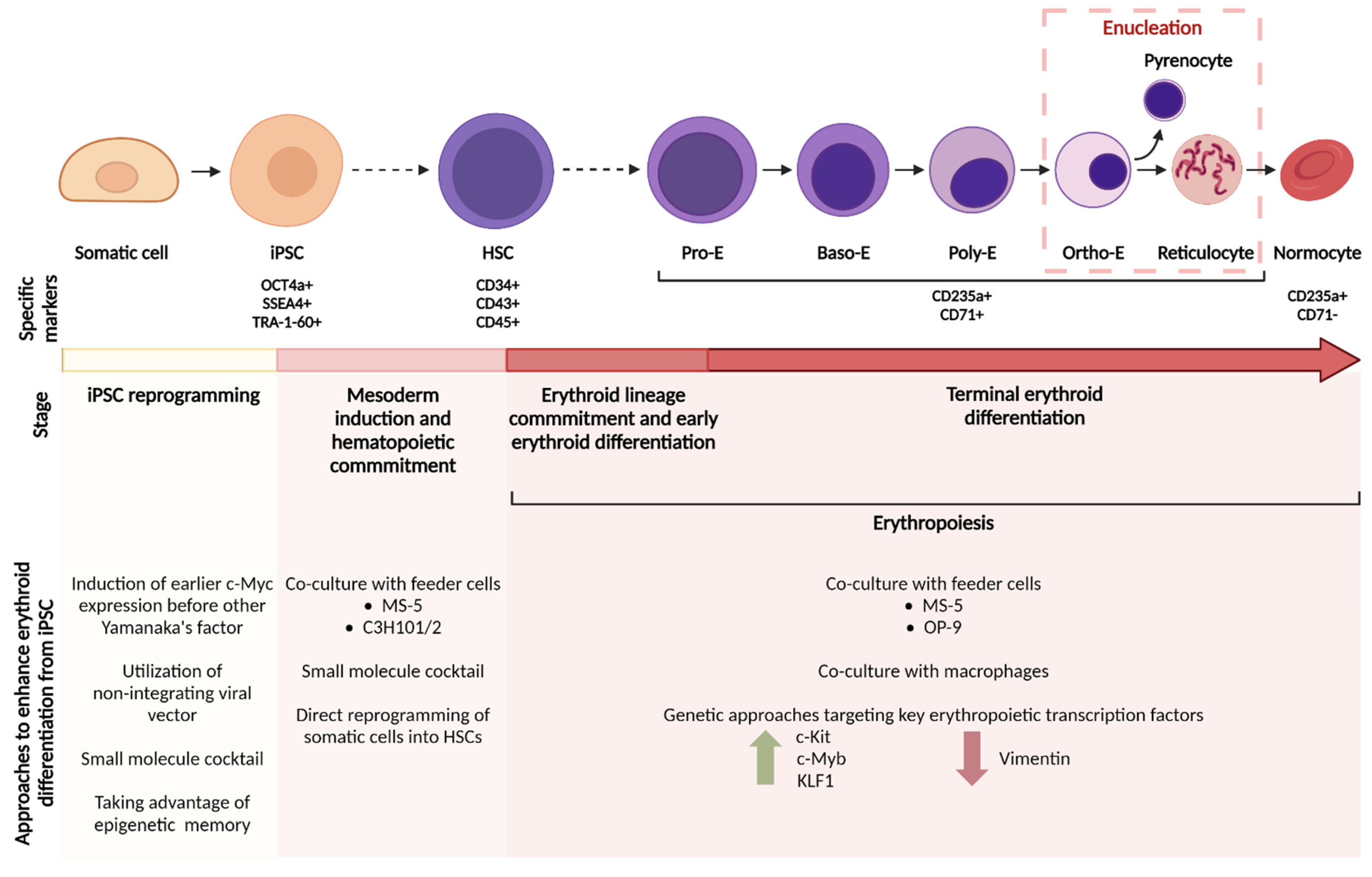
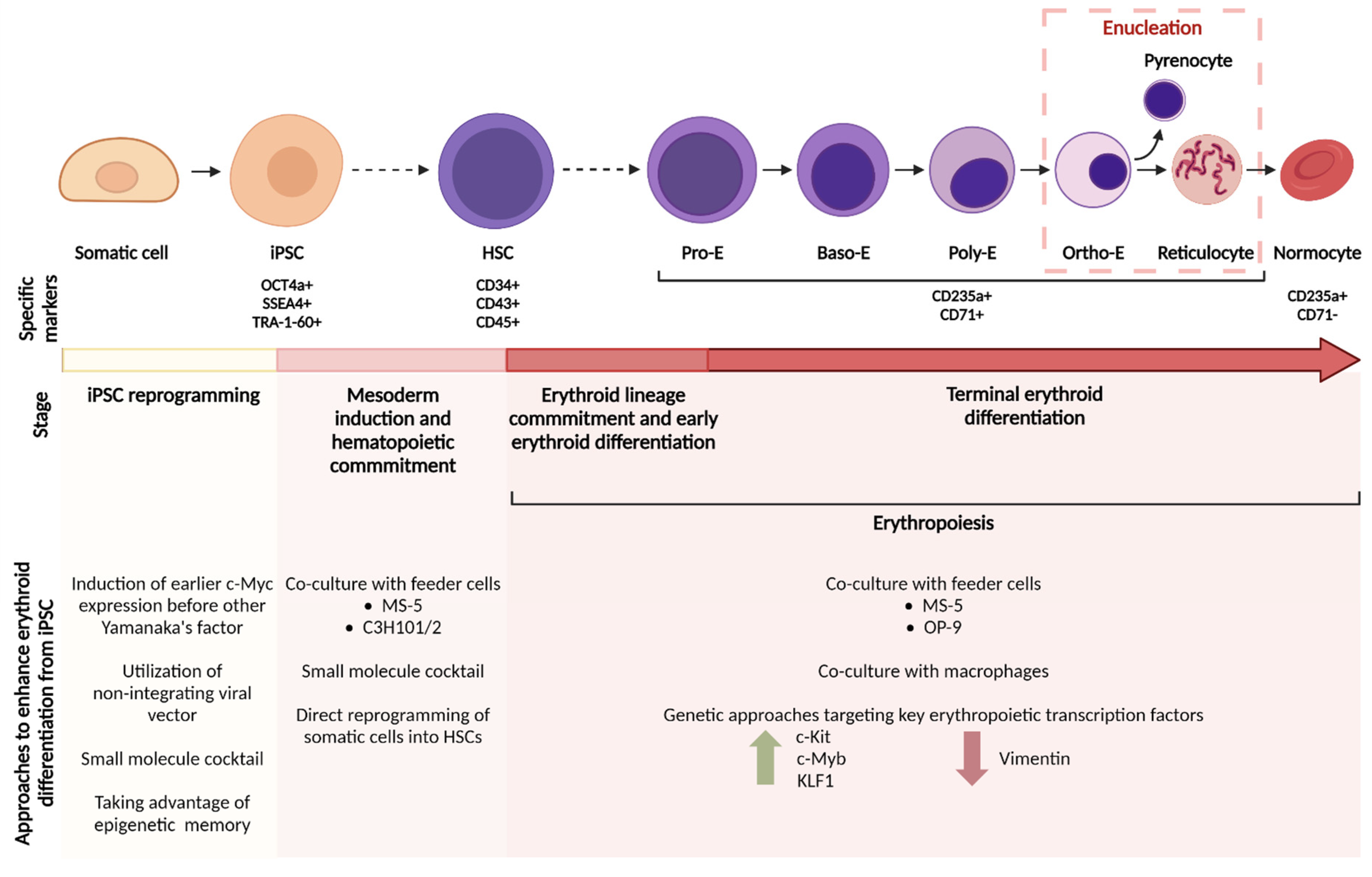{kind=link}
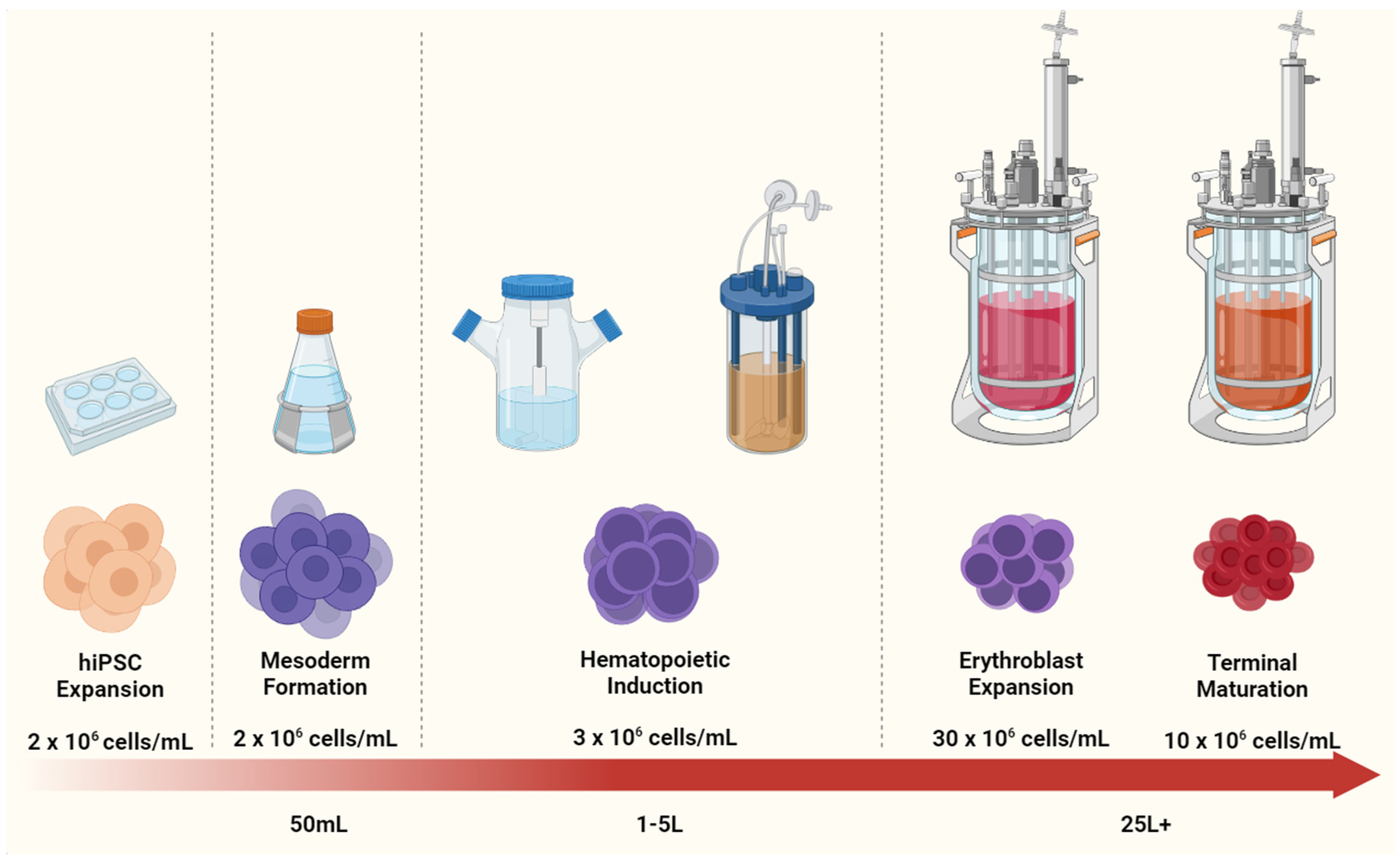
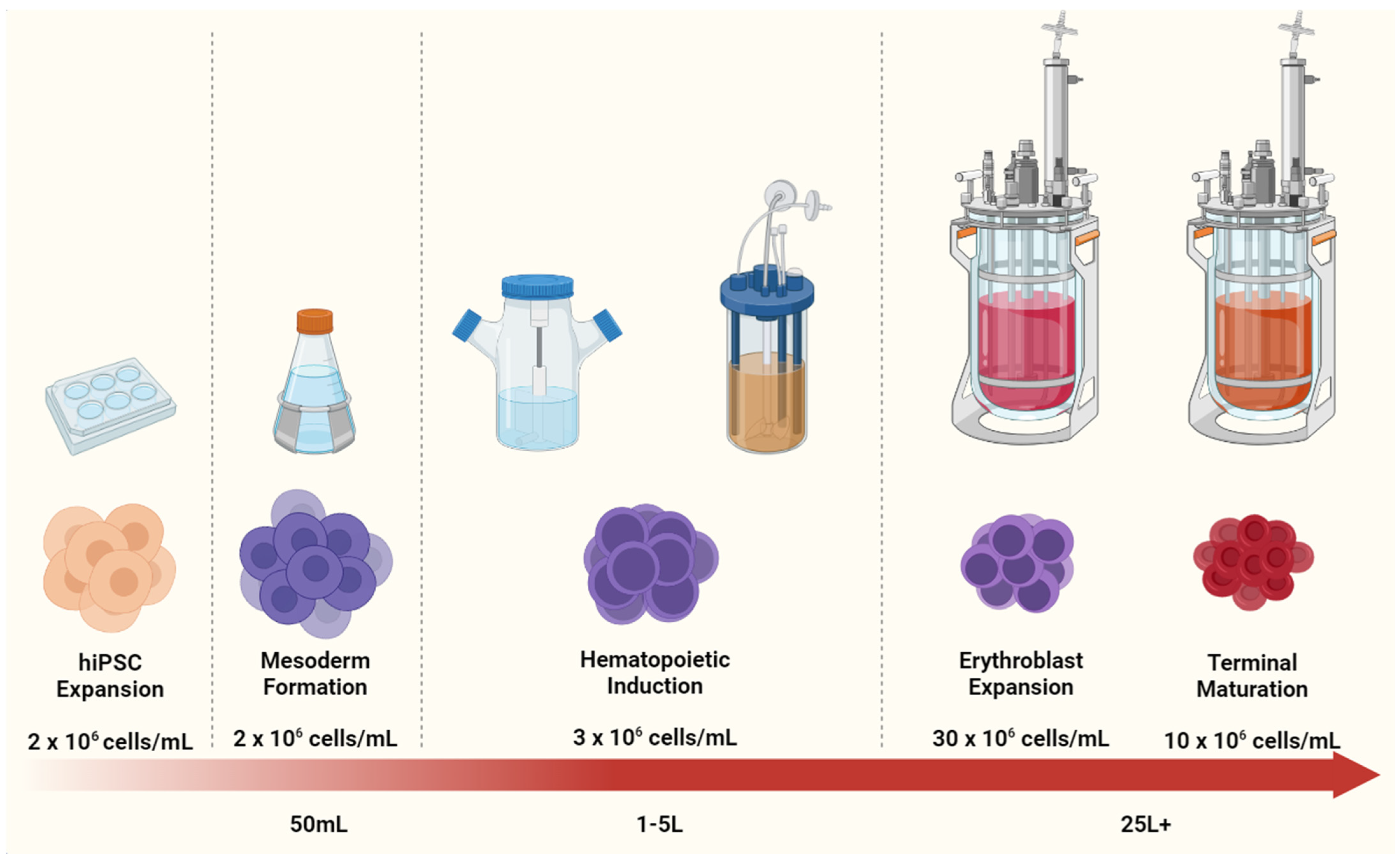{kind=link}
| Reference | Cell Source and Cell Lines Used | Culture Platform (Vessel Format) | Feeder Cell for Enucleation | Novel Components for Erythroid Expansion or Enucleation | Culture Period | Fold Increase for Erythroid Diff’n | Percentage of Enucleated Cells (In Vitro) |
|---|---|---|---|---|---|---|---|
| Kaufman et al., 2001 [58] | hESCs (H1,H1.1 and H9.2) | Monolayer | S17, C166 | - | 18 | Not reported | Not reported |
| Vodyanik et al., 2005 [61] | hESCs (H1 and H9) | Monolayer | OP9, S17, mMS-5 | - | 9 | Not reported | Not reported |
| Olivier et al., 2006 [64] | hESCs (H1) | Suspension | FH-B-hTERT, mMS-5 | - | 39 | 5000 fold | No enucleation |
| Lu et al., 2008 [60] | hESCs (MA01) | Suspension | hMSCs, OP9 | - | 42 | >104-fold | 10–30% feeder-free, 30–65% with OP9 |
| Ma et al., 2008 [63] | hESCs (H1) | Monolayer | mFLSCs | - | 18 | 100-fold | 11% |
| Klimchenko et al., 2009 [59] | hESCs (H1 and H9) | Suspension | OP9 | - | 14 | Not reported | Not reported |
| Lapillonne et al., 2010 [66] | hESCs (H1), hiPSCs (IMR90 and FD136) | Suspension | - | 5–10% Human Plasma | 26 | ~3500-fold, ~225- to 440-fold | 52–66% 4–10% |
| Dias et al., 2011 [62] | hESCs (H1), Transgene/free hiPSCs (SK46-M4-10,Foreskin-1, 19-9-7T and 4-3-7T) | Monolayer | OP9, mMS-5 | - | 70–90 | ~4000-fold | 2–10% |
| Kobari et al., 2012 [68] | hiPSCs (PB04 cell line from SCD) | Suspension | - | - | 52 | - | 20–26% |
| Rouzbeh et al., 2015 [67] | hESCs (H1 and H9) | Suspension | - | - | 34 | 75-fold | ~50% |
| Olivier et al., 2016 [69] | hiPSCs (33D6) | T75 Flask | - | CHIR99021, Activin A, IBMX, SR1 and Pluripotin | 31 | 2 × 105-fold | 10% |
| Wang et al., 2016 [70] | hiPSCs (BC1, TNC1 and E2) | 1L S.Flask | - | - | 29 | 240- to 370-fold | 2–15% |
| Sivalingam et al., 2018 [71] | hiPSCs (IMR90, BR2, BR7, D5, D9, D11, D12 and X13) | Suspension | hMSCs | CHIR99021 | 42 | >104-fold | 28–40.6 % |
| Bernecker et al., 2019 [73] | hiPSCs (CD34-iPSCs and PEB-iPSCs) | Monolayer | HCFC | - | 56 | 100- to 1000-fold | 40–60%) |
| Lopez-Yrigoyen et al., 2019 [74] | hiPSCs (SFCi55 and SFCi55-iKLF1.2) | Suspension | KLF1-activated iPSC-DMs | ANGPTL7, IL33 and SERPINB2 | 28 | - | 73% (CB HSC-derived erythroblasts) 6% (iPSC-derived erythroblasts) |
| Olivier et al., 2019 [75] | hiPSCs (NY22, OM1, OM2, OM3. and OM4) | Suspension | - | RED + and FeIII-EDTA | 39 | ~1000-fold | 20–76.4% |
| Sivalingam et al., 2021 [72] | hiPSCs (IMR90, BM1, CB6, FR202, BR7, D9, D12 and X13) | 500 mL S.Flask | OP9 | SR1 and Pluripotin | 35 | ~1000-fold | 6% w/o OP9, 18.1–59.3% with OP9 |
3. Approaches to Enhance Erythroid Differentiation from hiPSCs
3.1. Reprogramming to hiPSCs
3.1.1. First Generation: Integrating Methods
3.1.2. Second Generation: Non-Integrating Methods
3.1.3. The Use of Small Molecules
3.1.4. Concerns Regarding Cell Quality
3.2. Generation of Hematopoietic Cells In Vitro
3.2.1. Directed Differentiation with Growth Factors and Small Molecules
3.2.2. Recapitulation of Primitive vs. Definitive Hematopoiesis
3.2.3. Transcription Factor-Mediated Conversion to HSCs
3.3. Erythropoiesis
3.3.1. Co-Culture with Feeder Cells—Mimicking the Bone Marrow Microenvironment
3.3.2. Co-Culture with Macrophages—Mimicking the Erythroblastic Island
3.3.3. Genetic Approaches to Expand Erythroid Cells
4. Generating Clinically Suitable iPSC-RBCs for Transfusion
4.1. GMP-Compliant RBC Products (Feeder and Serum-Free, Xenogeneic-Free)
4.2. Scaling Up iPSC-RBC Generation
4.2.1. In Vitro RBC Cost Evaluation
4.2.2. Bioprocess Intensification
4.3. Expression of Fetal vs. Adult Hemoglobin
5. Novel Applications for hiPSCs-Derived, Engineered RBCs
5.1. Genetic Amenability and Application of hiPSC-Derived RBCs for Basic Research
5.2. Universal hiPSCs-Derived RBC-EVs for Medical Treatments (Drug Delivery Vehicles and oncomiR Gene Editing)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Here’s How Much Snow the “Bomb Cyclone” Dropped on the East Coast. Available online: https://time.com/5089443/snow-totals-bomb-cyclone-east-coast/ (accessed on 25 June 2021).
- Lee, E.; Sivalingam, J.; Lim, Z.R.; Chia, G.; Shi, L.G.; Roberts, M.; Loh, Y.-H.; Reuveny, S.; Oh, S.K.-W. Review: In vitro generation of red blood cells for transfusion medicine: Progress, prospects and challenges. Biotechnol. Adv. 2018, 36, 2118–2128. [Google Scholar] [CrossRef]
- Red Cross Issues Urgent Call for Blood Donations Heading into Holiday Season. Available online: https://www.redcross.org/about-us/news-and-events/press-release/2018/red-cross-issues-urgent-call-for-blood-donations-heading-into-ho.html (accessed on 25 June 2021).
- Roberts, N.; James, S.; Delaney, M.; Fitzmaurice, C. The global need and availability of blood products: A modelling study. Lancet Haematol. 2019, 6, e606–e615. [Google Scholar] [CrossRef]
- Stocks of Blood Type AB−, AB+ and A+ at Critical Levels. Available online: https://www.straitstimes.com/singapore/health/national-blood-stocks-down-by-a-third-as-donations-fall-amid-pandemic (accessed on 25 June 2021).
- American Red Cross Faces Severe Blood Shortage as Coronavirus Outbreak Threatens Availability of Nation’s Supply. Available online: https://www.redcross.org/about-us/news-and-events/press-release/2020/american-red-cross-faces-severe-blood-shortage-as-coronavirus-outbreak-threatens-availability-of-nations-supply.html (accessed on 25 June 2021).
- Hess, J.R. An update on solutions for red cell storage. Vox Sang. 2006, 91, 13–19. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Liumbruno, G.M.; Grazzini, G.; Zolla, L. Red blood cell storage: The story so far. Blood Transfus. 2010, 8, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Raphael, J.L.; Council, P.P. The role of policy in red blood cell storage and transfusion in children. Pediatr. Res. 2017, 82, 894–896. [Google Scholar] [CrossRef]
- Orlov, D.; Karkouti, K. The pathophysiology and consequences of red blood cell storage. Anaesthesia 2014, 70, 29-e12. [Google Scholar] [CrossRef] [PubMed]
- Högman, C.F.; Meryman, H.T. Storage parameters affecting red blood cell survival and function after transfusion. Transfus. Med. Rev. 1999, 13, 275–296. [Google Scholar] [CrossRef]
- Kalhan, T.G.; Bateman, D.A.; Bowker, R.M.; Hod, E.A.; Kashyap, S. Effect of red blood cell storage time on markers of hemolysis and inflammation in transfused very low birth weight infants. Pediatr. Res. 2017, 82, 964–969. [Google Scholar] [CrossRef]
- Scott, A.V.; Nagababu, E.; Johnson, D.J.; Kebaish, K.M.; Lipsitz, J.A.; Dwyer, I.M.; Zuckerberg, G.S.; Barodka, V.M.; Berkowitz, D.E.; Frank, S.M. 2,3-diphosphoglycerate concentrations in autologous salvaged versus stored red blood cells and in surgical patients after transfusion. Anesth. Analg. 2016, 122, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancarelli, A.; Birrer, K.L.; Alban, R.F.; Hobbs, B.P.; Liu-DeRyke, X. Hypocalcemia in trauma patients receiving massive transfusion. J. Surg. Res. 2015, 202, 182–187. [Google Scholar] [CrossRef]
- Cancelas, J.A.; Dumont, L.J.; Maes, L.A.; Rugg, N.; Herschel, L.; Whitley, P.H.; Szczepiokowski, Z.M.; Siegel, A.H.; Hess, J.R.; Zia, M. Additive solution-7 reduces the red blood cell cold storage lesion. Transfusion 2014, 55, 491–498. [Google Scholar] [CrossRef]
- Barshtein, G.; Gural, A.; Manny, N.; Zelig, O.; Yedgar, S.; Arbell, D. Storage-induced damage to red blood cell mechanical properties can be only partially reversed by rejuvenation. Transfus. Med. Hemotherapy 2014, 41, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrales, P.; Intaglietta, M. Blood substitutes. ASAIO J. 2013, 59, 337–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modery-Pawlowski, C.L.; Tian, L.L.; Pan, V.; Gupta, A.S. Synthetic approaches to RBC mimicry and oxygen carrier systems. Biomacromolecules 2013, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Singh, K.; Friedman, M.T. Artificial blood: The history and current perspectives of blood substitutes. Discoveries 2020, 8, e104. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.C.; Gollan, F. Survival of mammals breathing organic liquids equilibrated with oxygen at atmospheric pressure. Science 1966, 152, 1755–1756. [Google Scholar] [CrossRef]
- Spence, R.K.; Norcross, E.D.; Costabile, J.; McCoy, S.; Cernaianu, A.C.; Alexander, J.B.; Pello, M.J.; Atabek, U.; Camishion, R.C. Perfluorocarbons as blood substitutes: The early years: Experience with Fluosol DA-20% in the 1980s. Artif. Cells Blood Substit. Biotechnol. 1994, 22, 955–963. [Google Scholar] [CrossRef]
- Castro, C.I.; Briceno, J.C. Perfluorocarbon-based oxygen carriers: Review of products and trials. Artif. Organs 2010, 34, 622–634. [Google Scholar] [CrossRef]
- Fabian, T.C. Perfluorocarbons. J. Trauma Inj. Infect. Crit. Care 2011, 70, S42–S44. [Google Scholar] [CrossRef]
- Riess, J.G. Oxygen carriers (“blood substitutes”)—Raison d’etre, chemistry, and some physiology—Blut ist ein ganz besondrer Saft. Chem. Rev. 2001, 101, 2797–2920. [Google Scholar] [CrossRef]
- Latson, G.W. Perftoran (Vidaphor)—Introduction to western Medicine. Shock 2019, 52, 65–69. [Google Scholar] [CrossRef]
- Gupta, A.S. Hemoglobin-based oxygen carriers: Current state-of-the-art and novel molecules. Shock 2019, 52, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Browdie, D.; Smith, H. Stroma-free hemoglobin: Simplified preparation and in vivo and in vitro effects on coagulation in rabbits. Am. J. Surg. 1975, 129, 365–368. [Google Scholar] [CrossRef]
- Jahr, J.S.; Walker, V.; Manoochehri, K. Blood substitutes as pharmacotherapies in clinical practice. Curr. Opin. Anaesthesiol. 2007, 20, 325–330. [Google Scholar] [CrossRef]
- Mer, M.; Hodgson, E.; Wallis, L.; Jacobson, B.; Levien, L.; Snyman, J.; Sussman, M.J.; James, M.; Van Gelder, A.; Allgaier, R.; et al. Hemoglobin glutamer-250 (bovine) in South Africa: Consensus usage guidelines from clinician experts who have treated patients. Transfusion 2016, 56, 2631–2636. [Google Scholar] [CrossRef] [PubMed]
- Farcas, A.D.; Al Toma, V.; Roman, I.; Sevastre, B.; Scurtu, F.; Silaghi-Dumitrescu, R. Glutaraldehyde-polymerized hemoglobin: In search of improved performance as oxygen carrier in hemorrhage models. Bioinorg. Chem. Appl. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Krafft, M.P.; Riess, J.G. Therapeutic oxygen delivery by perfluorocarbon-based colloids. Adv. Colloid Interface Sci. 2021, 294, 102407. [Google Scholar] [CrossRef]
- Sakai, H.; Kobayashi, N.; Kure, T.; Okuda, C. Translational research of hemoglobin vesicles as a transfusion alternative. Curr. Med. Chem. 2021, 28, 1. [Google Scholar] [CrossRef]
- Wong, N.S.W.; Chang, T.M.S. Polyhemoglobin-fibrinogen: A novel oxygen carrier with platelet-like properties in a hemodiluted setting. Artif. Cells Blood Substit. Biotechnol. 2007, 35, 481–489. [Google Scholar] [CrossRef]
- Abu Jawdeh, B.G.; Woodle, E.S.; Leino, A.D.; Brailey, P.; Tremblay, S.; Dorst, T.; Abdallah, M.H.; Govil, A.; Byczkowski, D.; Misra, H.; et al. A phase Ib, open-label, single arm study to assess the safety, pharmacokinetics, and impact on humoral sensitization of SANGUINATE infusion in patients with end-stage renal disease. Clin. Transplant. 2017, 32, e13155. [Google Scholar] [CrossRef]
- Parco, S.; Vascotto, F.; Visconti, P. Public banking of umbilical cord blood or storage in a private bank: Testing social and ethical policy in northeastern Italy. Dovepress 2013, 4, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, C.C.; Cazzola, M.; de Fabritiis, P.; De Vincentiis, A.; Gianni, A.M.; Lanza, F.; Lauria, F.; Lemoli, R.M.; Tarella, C.; Zanon, P. CD34-positive cells: Biology and clinical relevance. Haematologica 1995, 80, 367–387. [Google Scholar] [PubMed]
- Fujimi, A.; Matsunaga, T.; Kobune, M.; Kawano, Y.; Nagaya, T.; Tanaka, I.; Iyama, S.; Hayashi, T.; Sato, T.; Miyanishi, K.; et al. Ex vivo large-scale generation of human red blood cells from cord blood CD34+ cells by co-culturing with macrophages. Int. J. Hematol. 2008, 87, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Giarratana, M.-C.; Kobari, L.; Lapillonne, H.; Chalmers, D.J.; Kiger, L.; Cynober, T.; Marden, M.C.; Wajcman, H.; Douay, L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat. Biotechnol. 2005, 23, 69–74. [Google Scholar] [CrossRef]
- Giarratana, M.-C.; Rouard, H.; Dumont, A.; Kiger, L.; Safeukui, I.; Le Pennec, P.-Y.; François, S.; Trugnan, G.; Peyrard, T.; Marie, T.; et al. Proof of principle for transfusion of in vitro-generated red blood cells. Blood 2011, 118, 5071–5079. [Google Scholar] [CrossRef] [Green Version]
- Leberbauer, C.; Boulmé, F.; Unfried, G.; Huber, J.; Beug, H.; Müllner, E.W. Different steroids co-regulate long-term expansion versus terminal differentiation in primary human erythroid progenitors. Blood 2005, 105, 85–94. [Google Scholar] [CrossRef]
- Timmins, N.E.; Athanasas, S.; Günther, M.; Buntine, P.; Nielsen, L.K. Ultra-high-yield manufacture of red blood cells from hematopoietic stem cells. Tissue Eng. Part C Methods 2011, 17, 1131–1137. [Google Scholar] [CrossRef]
- Neildez-Nguyen, T.M.A.; Wajcman, H.; Marden, M.C.; Bensidhoum, M.; Moncollin, V.; Giarratana, M.-C.; Kobari, L.; Thierry, D.; Douay, L. Human erythroid cells produced ex vivo at large scale differentiate into red blood cells in vivo. Nat. Biotechnol. 2002, 20, 467–472. [Google Scholar] [CrossRef]
- Baek, E.J.; Kim, H.-S.; Kim, S.; Jin, H.; Choi, T.-Y.; Kim, H.O. In vitro clinical-grade generation of red blood cells from human umbilical cord blood CD34+ cells. Transfusion 2008, 48, 2235–2245. [Google Scholar] [CrossRef]
- Miharada, K.; Hiroyama, T.; Sudo, K.; Nagasawa, T.; Nakamura, Y. Efficient enucleation of erythroblasts differentiated in vitro from hematopoietic stem and progenitor cells. Nat. Biotechnol. 2006, 24, 1255–1256. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, C.; Wang, L.; Shen, B.; Guan, X.; Tian, J.; Ren, Z.; Ding, X.; Ma, Y.; Dai, W.; et al. Large-scale ex vivo generation of human red blood cells from cord blood CD34+ cells. Stem Cells Transl. Med. 2017, 6, 1698–1709. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Baek, E.J.; Kim, H.-S.; Kim, J.-H.; Kim, N.J.; Kim, H.O. Stroma-free mass production of clinical-grade red blood cells (RBCs) by using poloxamer 188 as an RBC survival enhancer. Transfusion 2009, 49, 2285–2295. [Google Scholar] [CrossRef]
- Simamura, E.; Arikawa, T.; Ikeda, T.; Shimada, H.; Shoji, H.; Masuta, H.; Nakajima, Y.; Otani, H.; Yonekura, H.; Hatta, T. Melanocortins contribute to sequential differentiation and enucleation of human erythroblasts via melanocortin receptors 1, 2 and 5. PLoS ONE 2015, 10, e0123232. [Google Scholar] [CrossRef]
- Griffiths, R.E.; Kupzig, S.; Cogan, N.; Mankelow, T.J.; Betin, V.M.S.; Trakarnsanga, K.; Massey, E.J.; Lane, J.D.; Parsons, S.F.; Anstee, D.J. Maturing reticulocytes internalize plasma membrane in glycophorin A-containing vesicles that fuse with autophagosomes before exocytosis. Blood 2012, 119, 6296–6306. [Google Scholar] [CrossRef] [Green Version]
- Kupzig, S.; Parsons, S.F.; Curnow, E.; Anstee, D.J.; Blair, A. Superior survival of ex vivo cultured human reticulocytes following transfusion into mice. Haematologica 2016, 102, 476–483. [Google Scholar] [CrossRef] [Green Version]
- Heshusius, S.; Heideveld, E.; Burger, P.; Thiel-Valkhof, M.; Sellink, E.; Varga, E.; Ovchynnikova, E.; Visser, A.; Martens, J.H.A.; von Lindern, M.; et al. Large-scale in vitro production of red blood cells from human peripheral blood mononuclear cells. Blood Adv. 2019, 3, 3337–3350. [Google Scholar] [CrossRef] [Green Version]
- Hirose, S.-I.; Takayama, N.; Nakamura, S.; Nagasawa, K.; Ochi, K.; Hirata, S.; Yamazaki, S.; Yamaguchi, T.; Otsu, M.; Sano, S.; et al. Immortalization of erythroblasts by c-MYC and BCL-XL enables large-scale erythrocyte production from human pluripotent stem cells. Stem Cell Rep. 2013, 1, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 2013, 8, e59890. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Shah, S.; Wang, J.; Ye, Z.; Dowey, S.N.; Tsang, K.M.; Mendelsohn, L.G.; Kato, G.J.; Kickler, T.S.; Cheng, L. Extensive ex vivo expansion of functional human erythroid precursors established from umbilical cord blood cells by defined factors. Mol. Ther. 2014, 22, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trakarnsanga, K.; Griffiths, R.E.; Wilson, M.C.; Blair, A.; Satchwell, T.J.; Meinders, M.; Cogan, N.; Kupzig, S.; Kurita, R.; Nakamura, Y.; et al. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017, 8, 14750. [Google Scholar] [CrossRef] [Green Version]
- Trakarnsanga, K.; Tipgomut, C.; Metheetrairut, C.; Wattanapanitch, M.; Khuhapinant, A.; Poldee, S.; Kurita, R.; Nakamura, Y.; Srisawat, C.; Frayne, J. Generation of an immortalised erythroid cell line from haematopoietic stem cells of a haemoglobin E/β-thalassemia patient. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Kurita, R.; Funato, K.; Abe, T.; Watanabe, Y.; Shiba, M.; Tadokoro, K.; Nakamura, Y.; Nagai, T.; Satake, M. Establishment and characterization of immortalized erythroid progenitor cell lines derived from a common cell source. Exp. Hematol. 2019, 69, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, D.; Hanson, E.T.; Lewis, R.L.; Auerbach, R.; Thomson, J.A. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 10716–10721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimchenko, O.; Mori, M.; DiStefano, A.; Langlois, T.; Larbret, F.; Lecluse, Y.; Feraud, O.; Vainchenker, W.; Norol, F.; Debili, N. A common bipotent progenitor generates the erythroid and megakaryocyte lineages in embryonic stem cell–derived primitive hematopoiesis. Blood 2009, 114, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.-J.; Feng, Q.; Park, J.S.; Vida, L.; Lee, B.-S.; Strausbauch, M.; Wettstein, P.J.; Honig, G.R.; Lanza, R. Biologic properties and enucleation of red blood cells from human embryonic stem cells. Blood 2008, 112, 4475–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodyanik, M.A.; Bork, J.A.; Thomson, J.A.; Slukvin, I.I. Human embryonic stem cell-derived CD34+ cells: Efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005, 105, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.; Gumenyuk, M.; Kang, H.; Vodyanik, M.; Yu, J.; Thomson, J.A.; Slukvin, I.I. Generation of red blood cells from human induced pluripotent stem cells. Stem Cells Dev. 2011, 20, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Ebihara, Y.; Umeda, K.; Sakai, H.; Hanada, S.; Zhang, H.; Zaike, Y.; Tsuchida, E.; Nakahata, T.; Nakauchi, H.; et al. Generation of functional erythrocytes from human embryonic stem cell-derived definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 13087–13092. [Google Scholar] [CrossRef] [Green Version]
- Olivier, E.; Qiu, C.; Velho, M.; Hirsch, R.E.; Bouhassira, E.E. Large-scale production of embryonic red blood cells from human embryonic stem cells. Exp. Hematol. 2006, 34, 1635–1642. [Google Scholar] [CrossRef]
- Yamanaka, S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif. 2007, 41, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, H.; Kobari, L.; Mazurier, C.; Tropel, P.; Giarratana, M.-C.; Zanella-Cleon, I.; Kiger, L.; Wattenhofer-Donzé, M.; Puccio, H.; Hebert, N.; et al. Red blood cell generation from human induced pluripotent stem cells: Perspectives for transfusion medicine. Haematologica 2010, 95, 1651–1659. [Google Scholar] [CrossRef] [Green Version]
- Rouzbeh, S.; Kobari, L.; Cambot, M.; Mazurier, C.; Hebert, N.; Faussat, A.-M.; Durand, C.; Douay, L.; Lapillonne, H. Molecular signature of erythroblast enucleation in human embryonic stem cells. Stem Cells 2015, 33, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Kobari, L.; Yates, F.; Oudrhiri, N.; Francina, A.; Kiger, L.; Mazurier, C.; Rouzbeh, S.; El-Nemer, W.; Hebert, N.; Giarratana, M.-C.; et al. Human induced pluripotent stem cells can reach complete terminal maturation: In vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica 2012, 97, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Olivier, E.; Marenah, L.; McCahill, A.; Condie, A.; Cowan, S.; Mountford, J.C. High-efficiency serum-free feeder-free erythroid differentiation of human pluripotent stem cells using small molecules. Stem Cells Transl. Med. 2016, 5, 1394–1405. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Y.; He, C.; Ye, Z.; Gerecht, S.; Cheng, L. Scalable production of human erythrocytes from induced pluripotent stem cells. bioRxiv 2016, 050021. [Google Scholar] [CrossRef]
- Sivalingam, J.; Chen, H.Y.; Yang, B.-X.; Lim, Z.R.; Lam, A.T.L.; Woo, T.L.; Chen, A.K.-L.; Reuveny, S.; Loh, Y.-H.; Oh, S.K.-W. Improved erythroid differentiation of multiple human pluripotent stem cell lines in microcarrier culture by modulation of Wnt/β-Catenin signaling. Haematologica 2018, 103, e279–e283. [Google Scholar] [CrossRef] [Green Version]
- Sivalingam, J.; SuE, Y.; Lim, Z.R.; Lam, A.T.; Lee, A.P.; Lim, H.L.; Chen, H.Y.; Tan, H.K.; Warrier, T.; Hang, J.W.; et al. A scalable suspension platform for generating high-density cultures of universal red blood cells from human induced pluripotent stem cells. Stem Cell Rep. 2020, 16, 182–197. [Google Scholar] [CrossRef]
- Bernecker, C.; Ackermann, M.; Lachmann, N.; Rohrhofer, L.; Zaehres, H.; Araúzo-Bravo, M.J.; Akker, E.V.D.; Schlenke, P.; Dorn, I. Enhanced ex vivo generation of erythroid cells from human induced pluripotent stem cells in a simplified cell culture system with low cytokine support. Stem Cells Dev. 2019, 28, 1540–1551. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Yrigoyen, M.; Yang, C.-T.; Fidanza, A.; Cassetta, L.; Taylor, A.H.; McCahill, A.; Sellink, E.; Von Lindern, M.; Akker, E.V.D.; Mountford, J.C.; et al. Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nat. Commun. 2019, 10, 881. [Google Scholar] [CrossRef] [Green Version]
- Olivier, E.; Zhang, S.; Yan, Z.; Suzuka, S.; Roberts, K.; Wang, K.; Bouhassira, E.E. PSC-RED and MNC-RED: Albumin-free and low-transferrin robust erythroid differentiation protocols to produce human enucleated red blood cells. Exp. Hematol. 2019, 75, 31–52.e15. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef] [Green Version]
- Markoulaki, S.; Hanna, J.H.; Beard, C.; Carey, B.W.; Cheng, A.; Lengner, C.; Dausman, J.A.; Fu, D.; Gao, Q.; Wu, S.; et al. Transgenic mice with defined combinations of drug-inducible reprogramming factors. Nat. Biotechnol. 2009, 27, 169–171. [Google Scholar] [CrossRef]
- Shao, L.; Wu, W.-S. Gene-delivery systems for iPS cell generation. Expert Opin. Biol. Ther. 2009, 10, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Hanna, J.H.; Zhang, X.; Ku, M.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced pluripotent stem cells: Reprogramming platforms and applications in cell replacement therapy. BioRes. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Szymczak, A.L.; Workman, C.J.; Wang, Y.; Vignali, K.M.; Dilioglou, S.; Vanin, E.F.; Vignali, D.A.A. Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide–based retroviral vector. Nat. Biotechnol. 2004, 22, 589–594. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Nemes, C.; Varga, E.; Freund, C.; Kosmidis, G.; Gkatzis, K.; de Jong, D.; Szuhai, K.; Dinnyés, A.; Mummery, C.L. Generation of induced pluripotent stem cells from human foetal fibroblasts using the Sleeping Beauty transposon gene delivery system. Differentiation 2013, 86, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Grabundzija, I.; Wang, J.; Sebe, A.; Erdei, Z.; Kajdi, R.; Devaraj, A.; Steinemann, D.; Szuhai, K.; Stein, U.; Cantz, T.; et al. Sleeping Beauty transposon-based system for cellular reprogramming and targeted gene insertion in induced pluripotent stem cells. Nucleic Acids Res. 2012, 41, 1829–1847. [Google Scholar] [CrossRef]
- Woltjen, K.; Michael, I.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2014, 33, 58–63. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in pluripotent stem cells: History, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2019, 16, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-I.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Su, R.-J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient generation of integration-free iPS cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, T.; Guan, J.; Zhang, X.; Fu, Y.; Ye, J.; Zhu, J.; Meng, G.; Ge, J.; Yang, S.; et al. A XEN-like state bridges somatic cells to pluripotency during chemical reprogramming. Cell 2015, 163, 1678–1691. [Google Scholar] [CrossRef] [Green Version]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Frampton, G.; Soldner, F.; Hockemeyer, D.; Mitalipova, M.; Jaenisch, R.; Young, R.A. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010, 7, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Hongguang, H.; Loh, Y.-H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Mitra, K.; Koya, M.; Velho, M.; Desprat, R.; Lenz, J.; Bouhassira, E.E. Production of embryonic and fetal-like red blood cells from human induced pluripotent stem cells. PLoS ONE 2011, 6, e25761. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; An, B.; Kim, J.-H.; Han, H.-W.; Heo, H.-R.; Ha, K.-S.; Han, E.-T.; Park, W.S.; Hong, S.-H. BMP4 and perivascular cells promote hematopoietic differentiation of human pluripotent stem cells in a differentiation stage-specific manner. Exp. Mol. Med. 2020, 52, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ivanovs, A.; Rybtsov, S.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G.; Medvinsky, A. Human haematopoietic stem cell development: From the embryo to the dish. Development 2017, 144, 2323–2337. [Google Scholar] [CrossRef] [Green Version]
- Sivalingam, J.; Lam, T.L.; Chen, H.Y.; Yang, B.X.; Chen, A.K.-L.; Reuveny, S.; Loh, Y.-H.; Oh, S.K.-W. Superior red blood cell generation from human pluripotent stem cells through a novel microcarrier-based embryoid body platform. Tissue Eng. Part C Methods 2016, 22, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Tsuneyoshi, N.; Nakatsuji, N.; Suemori, H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/β-catenin, Activin/Nodal and BMP signaling. Development 2008, 135, 2969–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, S.; Neijts, R.; Simmini, S.; van Rooijen, C.; Tan, S.C.; Kester, L.; van Oudenaarden, A.; Creyghton, M.P.; Deschamps, J. Cdx and T brachyury co-activate growth signaling in the embryonic axial progenitor niche. Cell Rep. 2016, 17, 3165–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, P.P.L.; Loebel, D.A.F. Gene function in mouse embryogenesis: Get set for gastrulation. Nat. Rev. Genet. 2007, 8, 368–381. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, L.J.; Gupta, S.; Figueroa, M.E.; Mullins, M.C.; Evans, T. Smad1 and Smad5 differentially regulate embryonic hematopoiesis. Blood 2007, 110, 3881–3890. [Google Scholar] [CrossRef] [Green Version]
- Nostro, M.C.; Cheng, X.; Keller, G.M.; Gadue, P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell 2008, 2, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Lyu, C.; Zhu, Y.; Feng, Z.; Zhang, S.; Hoyle, D.L.; Ji, G.; Brodsky, R.A.; Cheng, T.; Wang, Z.Z. Defining early hematopoietic-fated primitive streak specification of human pluripotent stem cells by the orchestrated balance of Wnt, activin, and BMP signaling. J. Cell. Physiol. 2019, 234, 16136–16147. [Google Scholar] [CrossRef] [PubMed]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.; Chi, N.C.; Santoso, B.; Teng, S.; Stainier, D.; Traver, D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 2010, 464, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, J.L.; Butler, J.M.; Chan, Y.-Y.; Chandrasekaran, D.; Poulos, M.G.; Ginsberg, M.; Nolan, D.J.; Elemento, O.; Wood, B.L.; Adair, J.; et al. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J. Clin. Investig. 2015, 125, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, E.S.; Azzola, L.; Bruveris, F.F.; Calvanese, V.; Phipson, B.; Vlahos, K.; Hirst, C.; Jokubaitis, V.J.; Yu, Q.C.; Maksimovic, J.; et al. Differentiation of human embryonic stem cells to HOXA+ hemogenic vasculature that resembles the aorta-gonad-mesonephros. Nat. Biotechnol. 2016, 34, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef]
- Haro-Mora, J.J.; Uchida, N.; Demirci, S.; Wang, Q.; Zou, J.; Tisdale, J.F. Biallelic correction of sickle cell disease-derived induced pluripotent stem cells (iPSCs) confirmed at the protein level through serum-free iPS-sac/erythroid differentiation. Stem Cells Transl. Med. 2020, 9, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, T.; Sudo, K.; Hiroyama, T.; Miharada, K.; Ninomiya, H.; Chiba, S.; Nagasawa, T.; Nakamura, Y. Human hematopoietic stem cells can survive in vitro for several months. Adv. Hematol. 2009, 2009, 936761. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhu, Y.; Lyu, C.; Feng, Z.; Lyu, S.; Zhao, Y.; Hoyle, D.L.; Ji, G.; Miao, W.; Zhang, X.; et al. Sequential cellular niches control the generation of enucleated erythrocytes from human pluripotent stem cells. Haematologica 2019, 105, e48–e51. [Google Scholar] [CrossRef]
- Fares, I.; Chagraoui, J.; Gareau, Y.; Gingras, S.; Ruel, R.; Mayotte, N.; Csaszar, E.; Knapp, D.J.; Miller, P.; Ngom, M.; et al. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 2014, 345, 1509–1512. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xia, C.; Wang, T.; Liu, L.; Zhao, Q.; Yang, D.; Hu, F.; Zhang, M.; Huang, K.; Geng, Y.; et al. Pyrimidoindole derivative UM171 enhances derivation of hematopoietic progenitor cells from human pluripotent stem cells. Stem Cell Res. 2017, 21, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Togarrati, P.P.; Choi, K.-D.; Suknuntha, K. StemRegenin 1 selectively promotes expansion of multipotent hematopoietic progenitors derived from human embryonic stem cells. J. Stem Cells Regen. Med. 2017, 13, 75–79. [Google Scholar] [PubMed]
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef] [Green Version]
- Slukvin, I.I. Generating human hematopoietic stem cells in vitro-exploring endothelial to hematopoietic transition as a portal for stemness acquisition. FEBS Lett. 2016, 590, 4126–4143. [Google Scholar] [CrossRef] [Green Version]
- Hutt, D. Engraftment, graft failure, and rejection. In The European Blood and Marrow Transplantation Textbook for Nurses: Under the Auspices of EBMT; Kenyon, M., Babic, A., Eds.; Springer: Cham, Switzerland, 2018; pp. 259–270. [Google Scholar]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, R.; Jha, D.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-H.; Nelson, A.M.; Cao, H.; Wang, L.; Nakamoto, B.; Ware, C.B.; Papayannopoulou, T. Definitive-like erythroid cells derived from human embryonic stem cells coexpress high levels of embryonic and fetal globins with little or no adult globin. Blood 2006, 108, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zuniga-Pflucker, J.C.; Keller, G. T Lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturgeon, C.M.; Ditadi, A.; Awong, G.; Kennedy, M.; Keller, G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Hematopoietic stem cells from pluripotent stem cells: Clinical potential, challenges, and future perspectives. Stem Cells Transl. Med. 2020, 9, 1549–1557. [Google Scholar] [CrossRef]
- Sandler, V.M.; Lis, R.; Liu, Y.; Kedem, A.; James, D.; Elemento, O.; Butler, J.M.; Scandura, J.; Rafii, S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature 2014, 511, 312–318. [Google Scholar] [CrossRef]
- Capellera-Garcia, S.; Pulecio, J.; Dhulipala, K.; Siva, K.; Rayon-Estrada, V.; Singbrant, S.; Sommarin, M.; Walkley, C.; Soneji, S.; Karlsson, G.; et al. Defining the minimal factors required for erythropoiesis through direct lineage conversion. Cell Rep. 2016, 15, 2550–2562. [Google Scholar] [CrossRef] [Green Version]
- Julie, J.; Hale, J.; Bhagia, P.; Xue, F.; Chen, L.; Jaffray, J.; Yan, H.; Lane, J.; Gallagher, P.G.; Mohandas, N.; et al. Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood 2014, 124, 3636–3645. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, J.; Ginzburg, Y.; Li, H.; Xue, F.; De Franceschi, L.; Chasis, J.A.; Mohandas, N.; An, X. Quantitative analysis of murine terminal erythroid differentiation in vivo: Novel method to study normal and disordered erythropoiesis. Blood 2013, 121, e43–e49. [Google Scholar] [CrossRef] [Green Version]
- Hirschi, K.K. Hemogenic endothelium during development and beyond. Blood 2012, 119, 4823–4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Crocker, P.; Westaby, S.; Key, N.; Mason, D.Y.; Gordon, S.; Weatherall, D.J. Isolation and immunocytochemical characterization of human bone marrow stromal macrophages in hemopoietic clusters. J. Exp. Med. 1988, 168, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- De Back, D.Z.; Kostova, E.; Van Kraaij, M.; Berg, T.K.V.D.; Van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J. Cell. Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef]
- Yoshida, H.; Kawane, K.; Koike, M.; Mori, Y.; Uchiyama, Y.; Nagata, S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 2005, 437, 754–758. [Google Scholar] [CrossRef]
- Belay, E.; Hayes, B.J.; Blau, C.A.; Torok-Storb, B. Human Cord blood and bone marrow CD34+ cells generate macrophages that support erythroid islands. PLoS ONE 2017, 12, e0171096. [Google Scholar] [CrossRef] [PubMed]
- Heideveld, E.; Masiello, F.; Marra, M.; Esteghamat, F.; Nurcan, Y.; Von Lindern, M.; Migliaccio, A.R.F.; Akker, E.V.D. CD14+ cells from peripheral blood positively regulate hematopoietic stem and progenitor cell survival resulting in increased erythroid yield. Haematologica 2015, 100, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Heideveld, E.; Hampton-O’Neil, L.A.; Cross, S.J.; van Alphen, F.P.J.; Biggelaar, M.V.D.; Toye, A.M.; Akker, E.V.D. Glucocorticoids induce differentiation of monocytes towards macrophages that share functional and phenotypical aspects with erythroblastic island macrophages. Haematologica 2017, 103, 395–405. [Google Scholar] [CrossRef]
- Ramos, P.; Casu, C.; Gardenghi, S.; Breda, L.; Crielaard, B.; Guy, E.; Marongiu, M.F.; Gupta, R.; Levine, R.L.; Abdel-Wahab, O.; et al. Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat. Med. 2013, 19, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; James, W.; Moore, M.D. Human induced pluripotent stem cell-derived macrophages share ontogeny with MYB -independent tissue-resident macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, K.E.; Kingsley, P.D.; Koniski, A.D.; Porter, R.L.; Bushnell, T.P.; Palis, J. Enucleation of primitive erythroid cells generates a transient population of “pyrenocytes” in the mammalian fetus. Blood 2008, 111, 2409–2417. [Google Scholar] [CrossRef] [Green Version]
- Van Handel, B.; Prashad, S.L.; Hassanzadeh-Kiabi, N.; Huang, A.; Magnusson, M.; Atanassova, B.; Chen, A.; Hamalainen, E.I.; Mikkola, H.K.A. The first trimester human placenta is a site for terminal maturation of primitive erythroid cells. Blood 2010, 116, 3321–3330. [Google Scholar] [CrossRef] [Green Version]
- Merryweather-Clarke, A.T.; Tipping, A.J.; Lamikanra, A.A.; Fa, R.; Abu-Jamous, B.; Tsang, H.P.; Carpenter, L.; Robson, K.J.H.; Nandi, A.K.; Roberts, D.J. Distinct gene expression program dynamics during erythropoiesis from human induced pluripotent stem cells compared with adult and cord blood progenitors. BMC Genom. 2016, 17, 817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vegiopoulos, A.; Garcia, P.; Emambokus, N.; Frampton, J. Coordination of erythropoiesis by the transcription factor c-Myb. Blood 2006, 107, 4703–4710. [Google Scholar] [CrossRef]
- Bianchi, E.; Zini, R.; Salati, S.; Tenedini, E.; Norfo, R.; Tagliafico, E.; Manfredini, R.; Ferrari, S. c-myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 2010, 116, e99–e110. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, E.; Bulgarelli, J.; Ruberti, S.; Rontauroli, S.; Sacchi, G.; Norfo, R.; Pennucci, V.; Zini, R.; Salati, S.; Prudente, Z.; et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486-3p-mediated downregulation of MAF. Cell Death Differ. 2015, 22, 1906–1921. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-T.; Ma, R.; Axton, R.A.; Jackson, M.; Taylor, A.H.; Fidanza, A.; Marenah, L.; Frayne, J.; Mountford, J.C.; Forrester, L.M. Activation of KLF1 enhances the differentiation and maturation of red blood cells from human pluripotent stem cells. Stem Cells 2017, 35, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Trakarnsanga, K.; Wilson, M.C.; Griffiths, R.E.; Toye, A.; Carpenter, L.; Heesom, K.J.; Parsons, S.F.; Anstee, D.J.; Frayne, J. Qualitative and quantitative comparison of the proteome of erythroid cells differentiated from human iPSCs and adult erythroid cells by multiplex TMT labelling and nanoLC-MS/MS. PLoS ONE 2014, 9, e100874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thom, C.S.; Traxler, E.A.; Khandros, E.; Nickas, J.M.; Zhou, O.; Lazarus, J.; Silva, A.P.; Prabhu, D.; Yao, Y.; Aribeana, C.; et al. Trim58 degrades dynein and regulates terminal erythropoiesis. Dev. Cell 2014, 30, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, D.G.; Pushkaran, S.; Johnson, J.F.; Cancelas, J.A.; Manganaris, S.; Harris, C.E.; Williams, D.A.; Zheng, Y.; Kalfa, T.A. Signaling and cytoskeletal requirements in erythroblast enucleation. Blood 2012, 119, 6118–6127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.P.; Zhang, S.F.; Du, Q.; Sun, H.; Xin, J.; Liu, S.Q.; Ma, J. The role of cytoskeletal elements in the two-phase denucleation process of mammalian erythroblasts in vitro observed by laser confocal scanning microscope. Cell. Mol. Boil. 1997, 43, 851–860. [Google Scholar]
- Sangiorgi, F.; Woods, C.; Lazarides, E. Vimentin downregulation is an inherent feature of murine erythropoiesis and occurs independently of lineage. Development 1990, 110, 85–96. [Google Scholar] [CrossRef]
- Ngai, J.; Capetanaki, Y.G.; Lazarides, E. Differentiation of murine erythroleukemia cells results in the rapid repression of vimentin gene expression. J. Cell Biol. 1984, 99, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, B.L.; Lazarides, E. Structural associations of synemin and vimentin filaments in avian erythrocytes revealed by immunoelectron microscopy. Cell 1982, 30, 263–275. [Google Scholar] [CrossRef]
- Trakarnsanga, K.; Ferguson, D.; Daniels, D.; Griffiths, R.E.; Wilson, M.C.; Mordue, K.E.; Gartner, A.; Andrienko, T.N.; Calvert, A.; Condie, A.; et al. Vimentin expression is retained in erythroid cells differentiated from human iPSC and ESC and indicates dysregulation in these cells early in differentiation. Stem Cell Res. Ther. 2019, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Tursky, M.L.; Loi, T.H.; Artuz, C.M.; Alateeq, S.; Wolvetang, E.J.; Tao, H.; Ma, D.D.; Molloy, T.J. Direct comparison of four hematopoietic differentiation methods from human induced pluripotent stem cells. Stem Cell Rep. 2020, 15, 735–748. [Google Scholar] [CrossRef]
- LaRochelle, A. Generation of red blood cells in vitro: Monitoring the process for improved efficiency. Cytotherapy 2013, 15, 1043–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toner, R.W.; Pizzi, L.; Leas, B.; Ballas, S.K.; Quigley, A.; Goldfarb, N.I. Costs to hospitals of acquiring and processing blood in the US. Appl. Health Econ. Health Policy 2011, 9, 29–37. [Google Scholar] [CrossRef]
- Zeuner, A.; Martelli, F.; Vaglio, S.; Federici, G.; Whitsett, C.; Migliaccio, A.R. Concise review: Stem cell-derived erythrocytes as upcoming players in blood transfusion. Stem Cells 2012, 30, 1587–1596. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Saini, A.; Tsuji, K.; Sharma, P.B.; Chandra, R. Manufacturing blood ex vivo: A futuristic approach to deal with the supply and safety concerns. Front. Cell Dev. Biol. 2014, 2, 26. [Google Scholar] [CrossRef]
- Bayley, R.; Ahmed, F.; Glen, K.; McCall, M.; Stacey, A.; Thomas, R. The productivity limit of manufacturing blood cell therapy in scalable stirred bioreactors. J. Tissue Eng. Regen. Med. 2017, 12, e368–e378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, C.K.; Ueda, Y.; Kadari, A.; Günther, K.; Ergün, S.; Heron, A.; Schnitzler, A.C.; Rook, M.; Edenhofer, F. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single-use bioreactors. J. Tissue Eng. Regen. Med. 2017, 12, e1076–e1087. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, B.; Aguiar, T.; Arnault, A.; Costa, R.; Gomes-Alves, P.; Aspegren, A.; Serra, M.; Alves, P. Expansion of 3D human induced pluripotent stem cell aggregates in bioreactors: Bioprocess intensification and scaling-up approaches. J. Biotechnol. 2017, 246, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.L.; Li, J.; Chen, A.K.-L.; Reuveny, S.; Oh, S.K.-W.; Birch, W.R. Cationic surface charge combined with either vitronectin or laminin dictates the evolution of human embryonic stem cells/microcarrier aggregates and cell growth in agitated cultures. Stem Cells Dev. 2014, 23, 1688–1703. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.R.; Tomney, A.; Woon, M.T.; Uth, N.; Shafighi, F.; Ngabo, I.; Vallabhaneni, H.; Levinson, Y.; Abraham, E.; Ben-Nun, I.F. End-to-end platform for human pluripotent stem cell manufacturing. Int. J. Mol. Sci. 2019, 21, 89. [Google Scholar] [CrossRef] [Green Version]
- Borys, B.S.; Dang, T.; So, T.; Rohani, L.; Revay, T.; Walsh, T.; Thompson, M.; Argiropoulos, B.; Rancourt, D.E.; Jung, S.; et al. Overcoming bioprocess bottlenecks in the large-scale expansion of high-quality hiPSC aggregates in vertical-wheel stirred suspension bioreactors. Stem Cell Res. Ther. 2021, 12, 55. [Google Scholar] [CrossRef]
- Meng, G.; Liu, S.; Poon, A.; Rancourt, D.E. Optimizing human induced pluripotent stem cell expansion in stirred-suspension culture. Stem Cells Dev. 2017, 26, 1804–1817. [Google Scholar] [CrossRef]
- Allenby, M.C.; Panoskaltsis, N.; Tahlawi, A.; Dos Santos, S.B.; Mantalaris, A. Dynamic human erythropoiesis in a three-dimensional perfusion bone marrow biomimicry. Biomaterials 2018, 188, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Guzniczak, E.; Otto, O.; Whyte, G.; Chandra, T.; Robertson, N.; Willoughby, N.; Jimenez, M.; Bridle, H. Purifying stem cell-derived red blood cells: A high-throughput label-free downstream processing strategy based on microfluidic spiral inertial separation and membrane filtration. Biotechnol. Bioeng. 2020, 117, 2032–2045. [Google Scholar] [CrossRef]
- Zeming, K.K.; Sato, Y.; Yin, L.; Huang, N.-J.; Wong, L.H.; Loo, H.L.; Lim, Y.B.; Lim, C.T.; Chen, J.; Preiser, P.R.; et al. Microfluidic label-free bioprocessing of human reticulocytes from erythroid culture. Lab Chip 2020, 20, 3445–3460. [Google Scholar] [CrossRef] [PubMed]
- Siatecka, M.; Bieker, J.J. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 2011, 118, 2044–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; Van Der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 2010, 42, 801–805. [Google Scholar] [CrossRef]
- Shah, S.; Huang, X.; Cheng, L. Concise review: Stem cell-based approaches to red blood cell production for transfusion. Stem Cells Transl. Med. 2013, 3, 346–355. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeon, S.-H.; Kim, H.-K.; Suh, E.J.; Choi, S.J.; Kim, S. Human induced pluripotent stem cell line banking for the production of rare blood type erythrocytes. J. Transl. Med. 2020, 18, 236. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koh, H.; Zhen, X.; Lee, D.-S.; Ha, H.-Y.; Lee, J.-H. Establishment of iPSC (KRIBBi001-A) from CD34+ group O D-negative bone marrow blood. Stem Cell Res. 2021, 51, 102199. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J. RBCs as targets of infection. Hematology 2014, 2014, 404–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowman, A.F.; Tonkin, C.J.; Tham, W.-H.; Duraisingh, M.T. The molecular basis of erythrocyte invasion by malaria parasites. Cell Host Microbe 2017, 22, 232–245. [Google Scholar] [CrossRef]
- Salinas, N.; Tolia, N.H. Red cell receptors as access points for malaria infection. Curr. Opin. Hematol. 2016, 23, 215–223. [Google Scholar] [CrossRef]
- Hang, J.-W.; Tukijan, F.; Lee, E.-Q.; Abdeen, S.; Aniweh, Y.; Malleret, B. Zoonotic malaria: Non-Laverania Plasmodium biology and invasion mechanisms. Pathogens 2021, 10, 889. [Google Scholar] [CrossRef]
- Lobo, C.-A. Babesia divergens and Plasmodium falciparum use common receptors, glycophorins A and B, to invade the human red blood cell. Infect. Immun. 2005, 73, 649–651. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.; Dehio, C. Intruders below the radar: Molecular pathogenesis of Bartonella spp. Clin. Microbiol. Rev. 2012, 25, 42–78. [Google Scholar] [CrossRef] [Green Version]
- Horta, M.F.; Andrade, L.O.; Martins-Duarte, S.; Castro-Gomes, T. Cell invasion by intracellular parasites—The many roads to infection. J. Cell Sci. 2020, 133, jcs232488. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Dzieciatkowska, M.; Issaian, A.; Nemkov, T.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Buehler, P.W.; Zimring, J.C.; et al. Evidence of structural protein damage and membrane lipid remodeling in red blood cells from COVID-19 patients. J. Proteome Res. 2020, 19, 4455–4469. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.P.; Tigges, J.C.; Toxavidis, V.; Ghiran, I. Red blood cells: A source of extracellular vesicles. Extracell. Vesicles 2017, 1660, 15–22. [Google Scholar] [CrossRef]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, T.L.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Al Azad, M.A.R.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Z.R.; Vassilev, S.; Leong, Y.W.; Hang, J.W.; Rénia, L.; Malleret, B.; Oh, S.K.-W. Industrially Compatible Transfusable iPSC-Derived RBCs: Progress, Challenges and Prospective Solutions. Int. J. Mol. Sci. 2021, 22, 9808. https://doi.org/10.3390/ijms22189808
Lim ZR, Vassilev S, Leong YW, Hang JW, Rénia L, Malleret B, Oh SK-W. Industrially Compatible Transfusable iPSC-Derived RBCs: Progress, Challenges and Prospective Solutions. International Journal of Molecular Sciences. 2021; 22(18):9808. https://doi.org/10.3390/ijms22189808
Chicago/Turabian StyleLim, Zhong Ri, Svetlan Vassilev, Yew Wai Leong, Jing Wen Hang, Laurent Rénia, Benoit Malleret, and Steve Kah-Weng Oh. 2021. "Industrially Compatible Transfusable iPSC-Derived RBCs: Progress, Challenges and Prospective Solutions" International Journal of Molecular Sciences 22, no. 18: 9808. https://doi.org/10.3390/ijms22189808
APA StyleLim, Z. R., Vassilev, S., Leong, Y. W., Hang, J. W., Rénia, L., Malleret, B., & Oh, S. K.-W. (2021). Industrially Compatible Transfusable iPSC-Derived RBCs: Progress, Challenges and Prospective Solutions. International Journal of Molecular Sciences, 22(18), 9808. https://doi.org/10.3390/ijms22189808