
Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Results
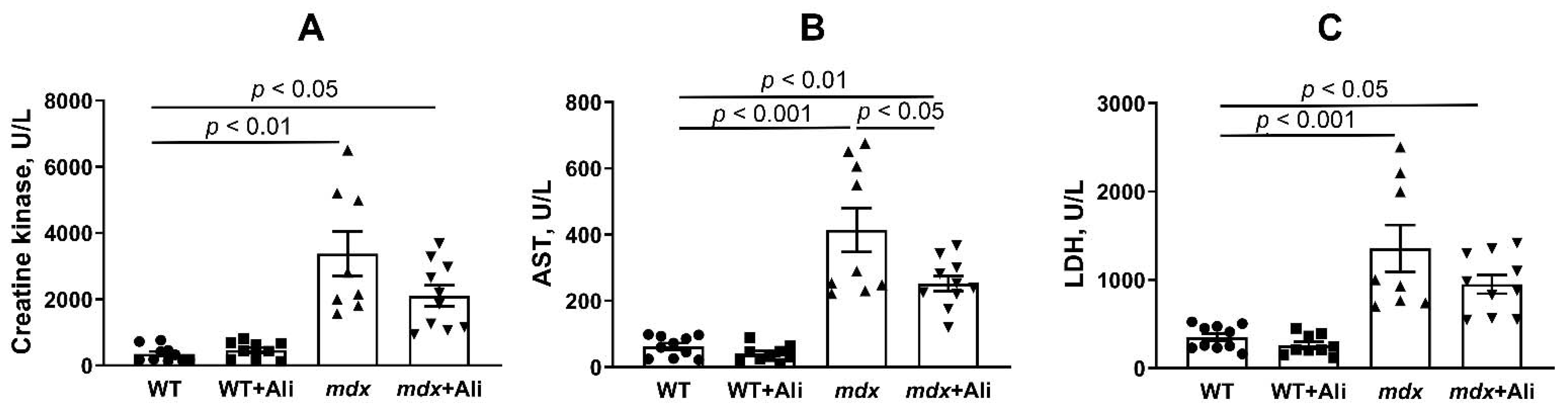
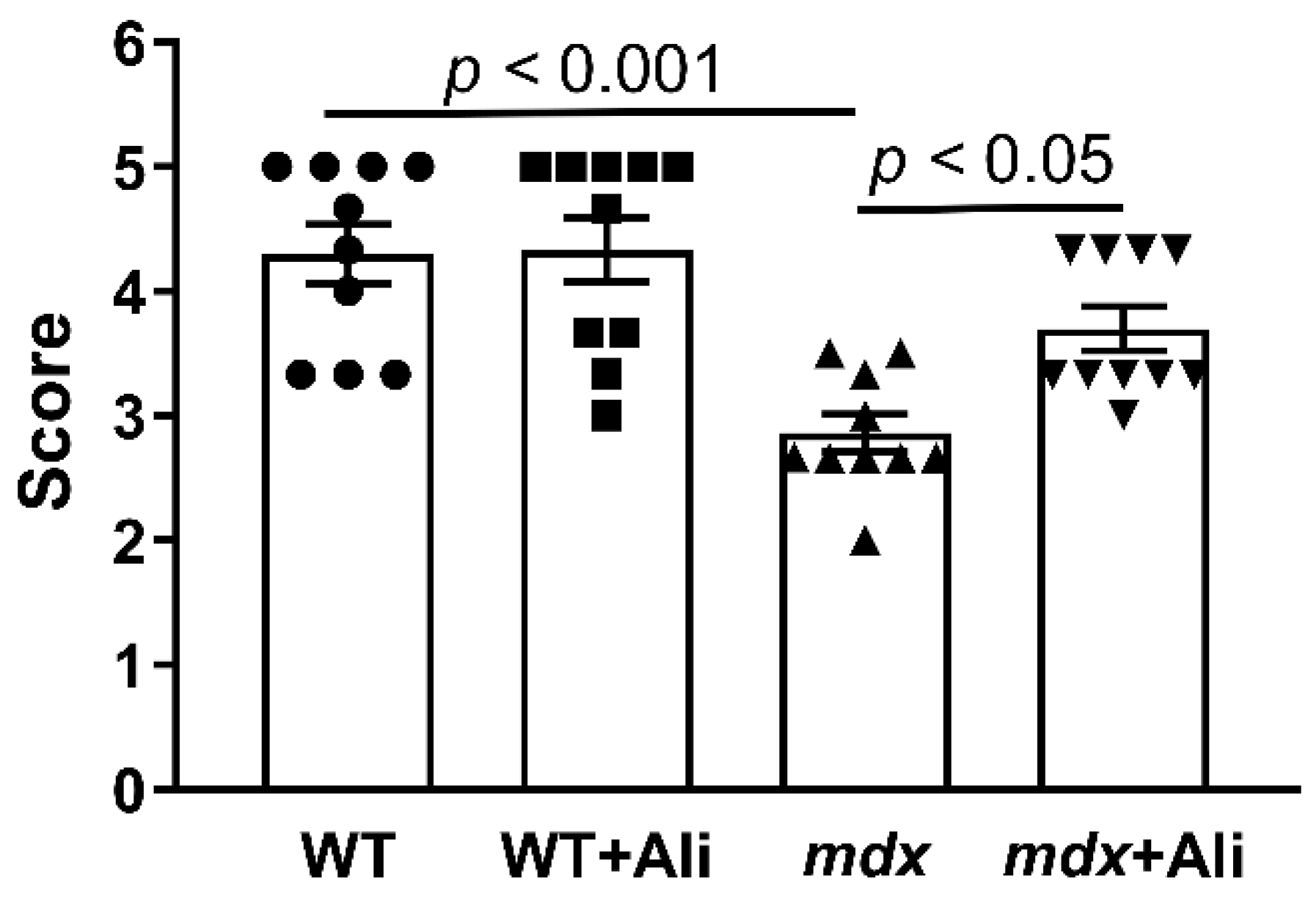
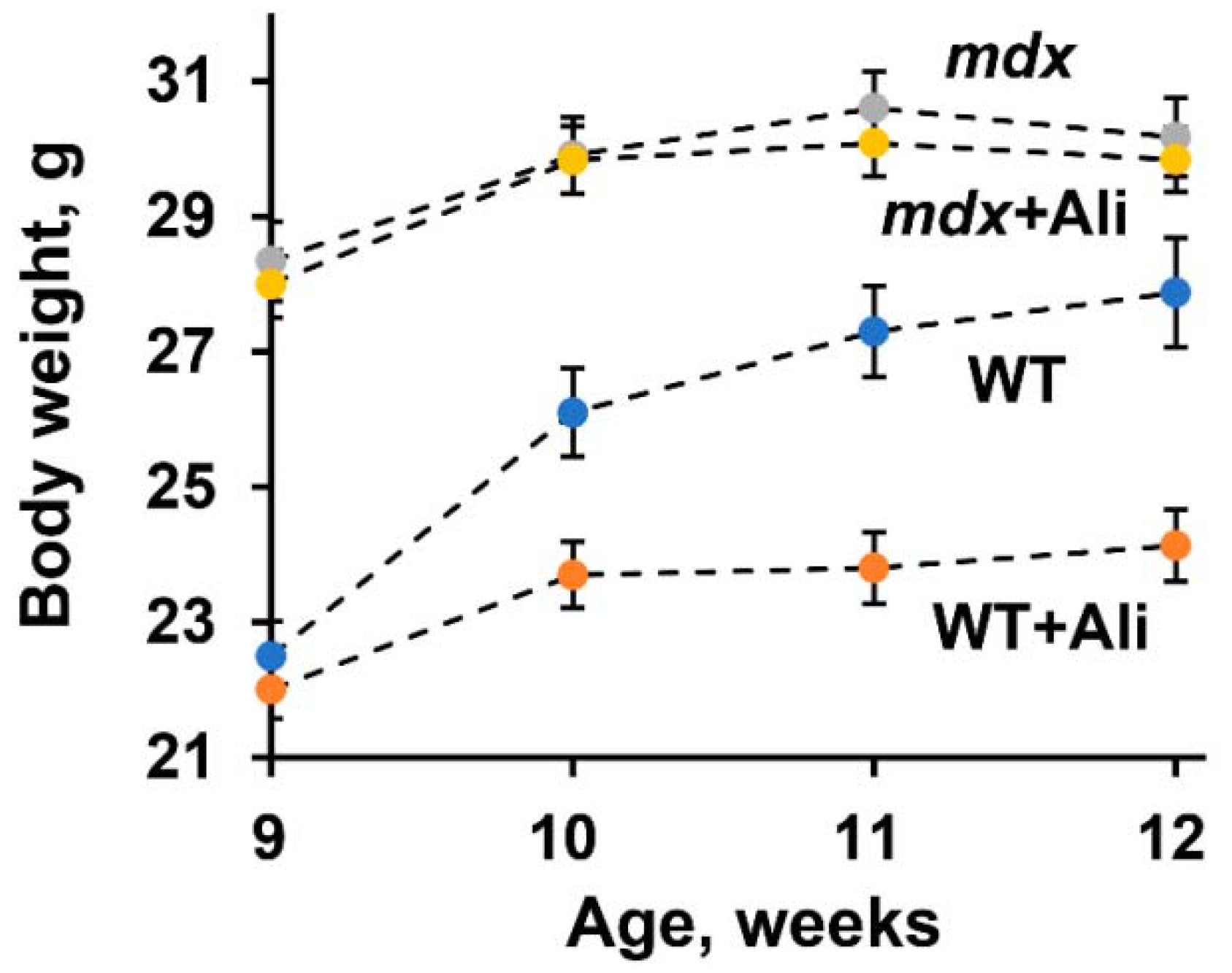
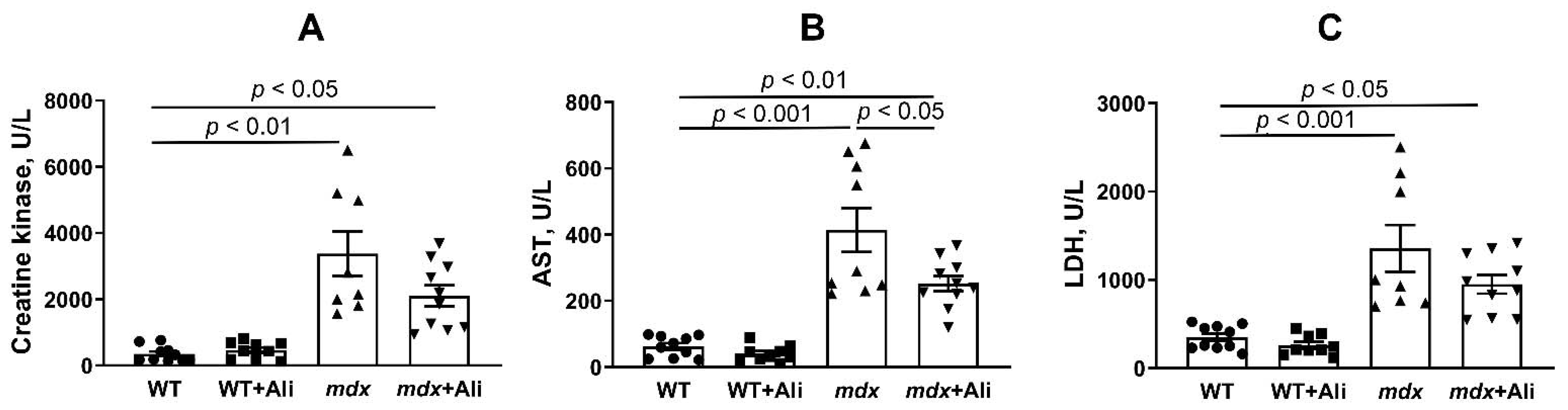
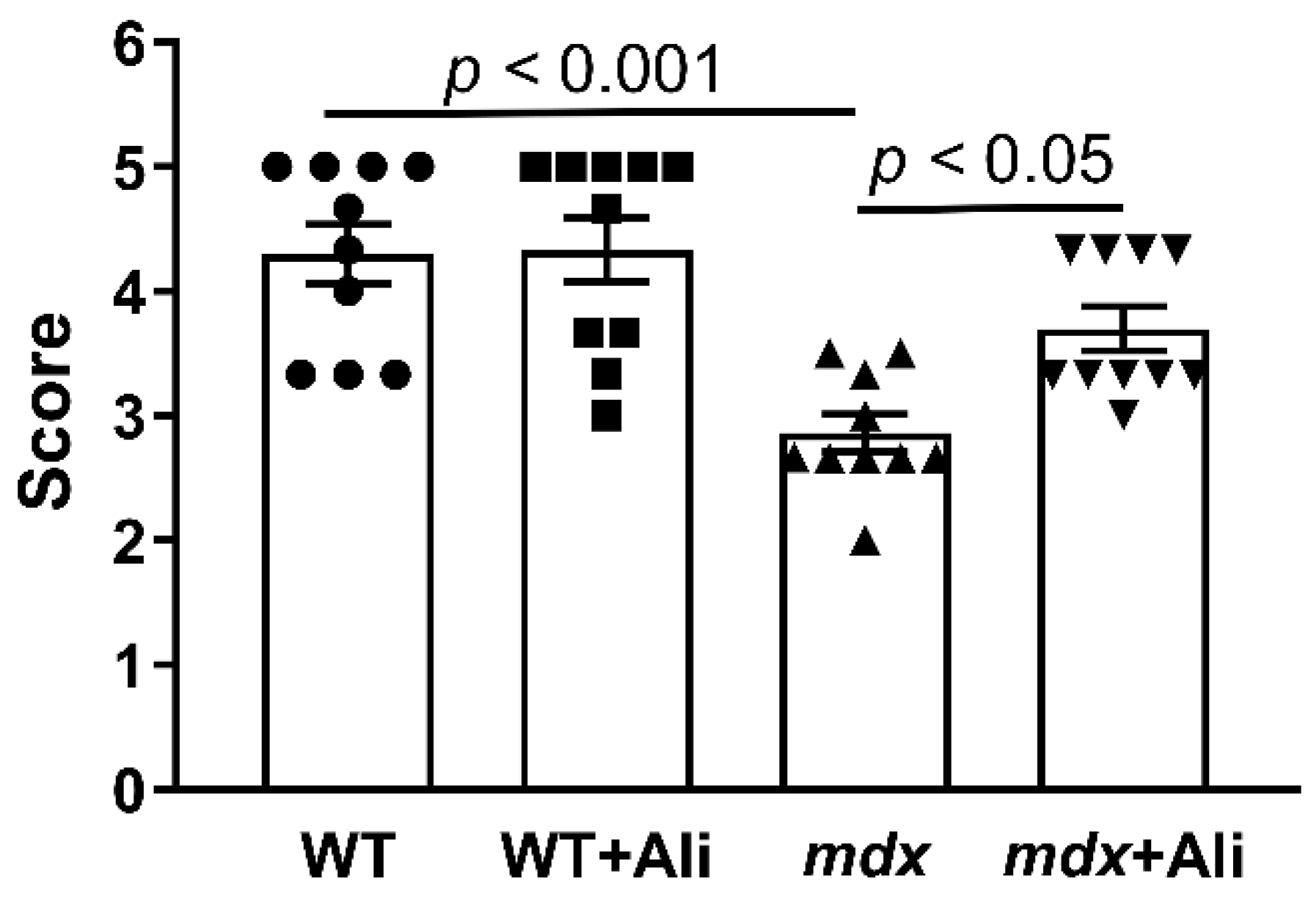
2.1. Alisporivir Reduces the Intensity of the Inflammatory Process and Improves the Muscle Function of Dystrophin-Deficient Mice
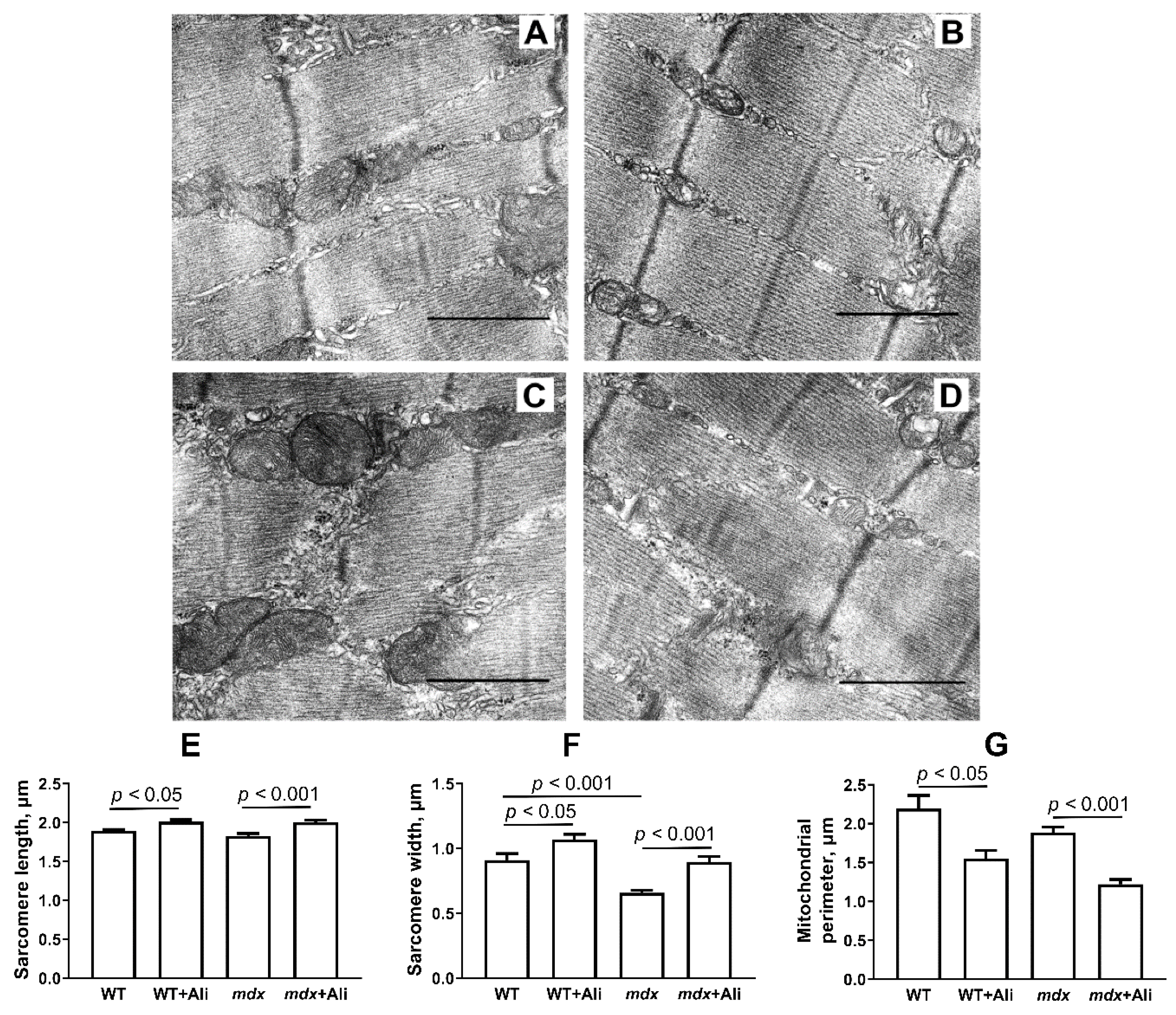
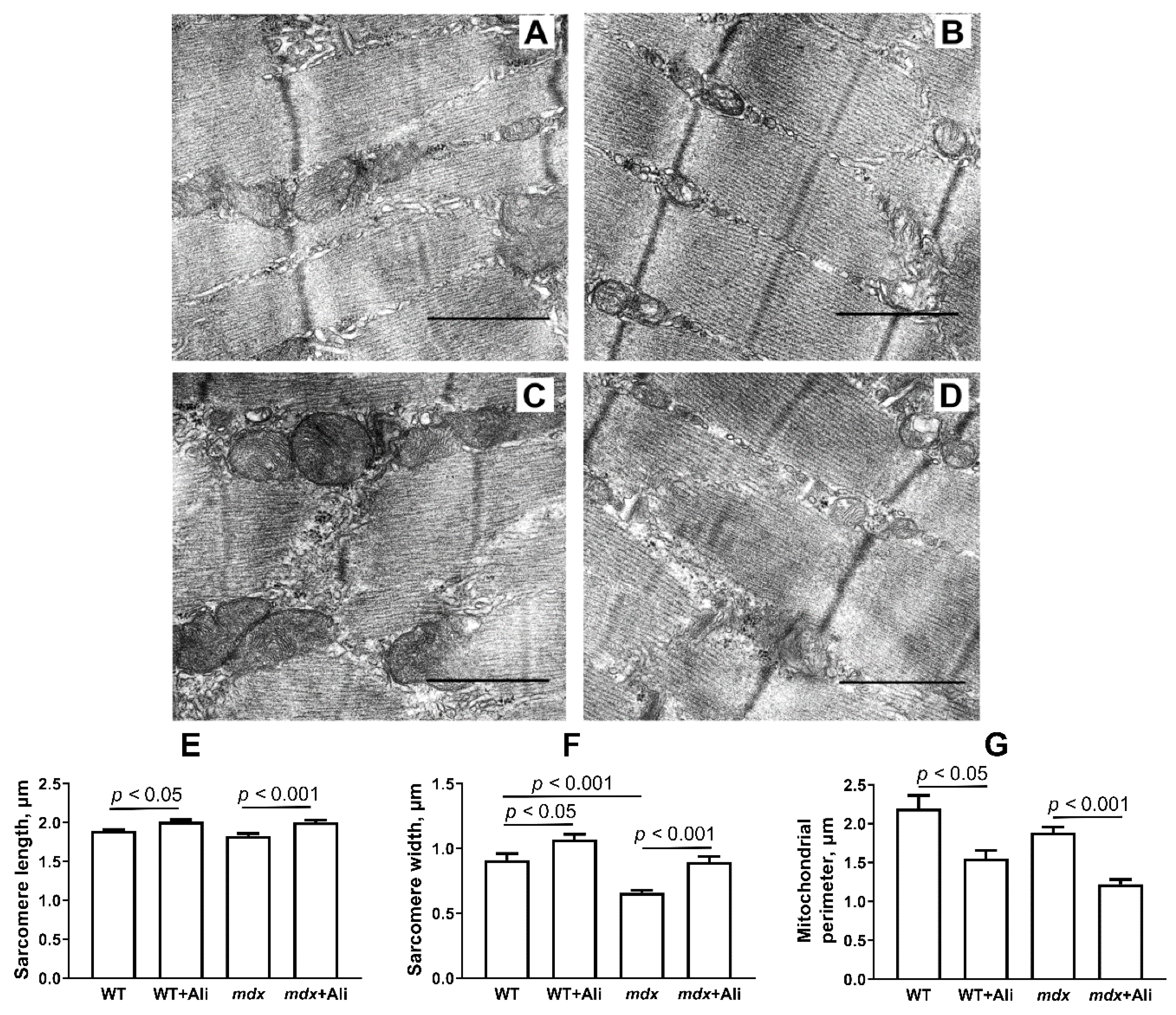
2.2. The Effect of Alisporivir on the Ultrastructure of Skeletal Muscle Mitochondria
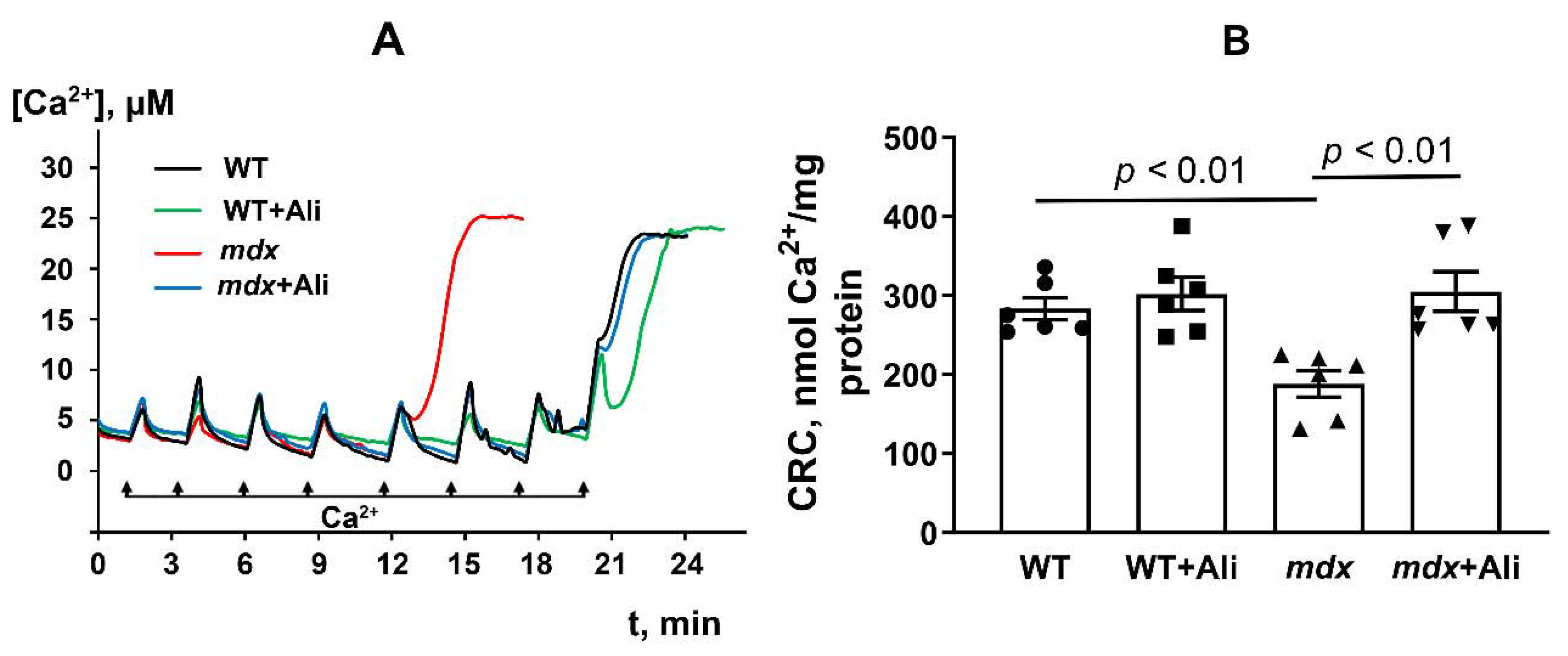
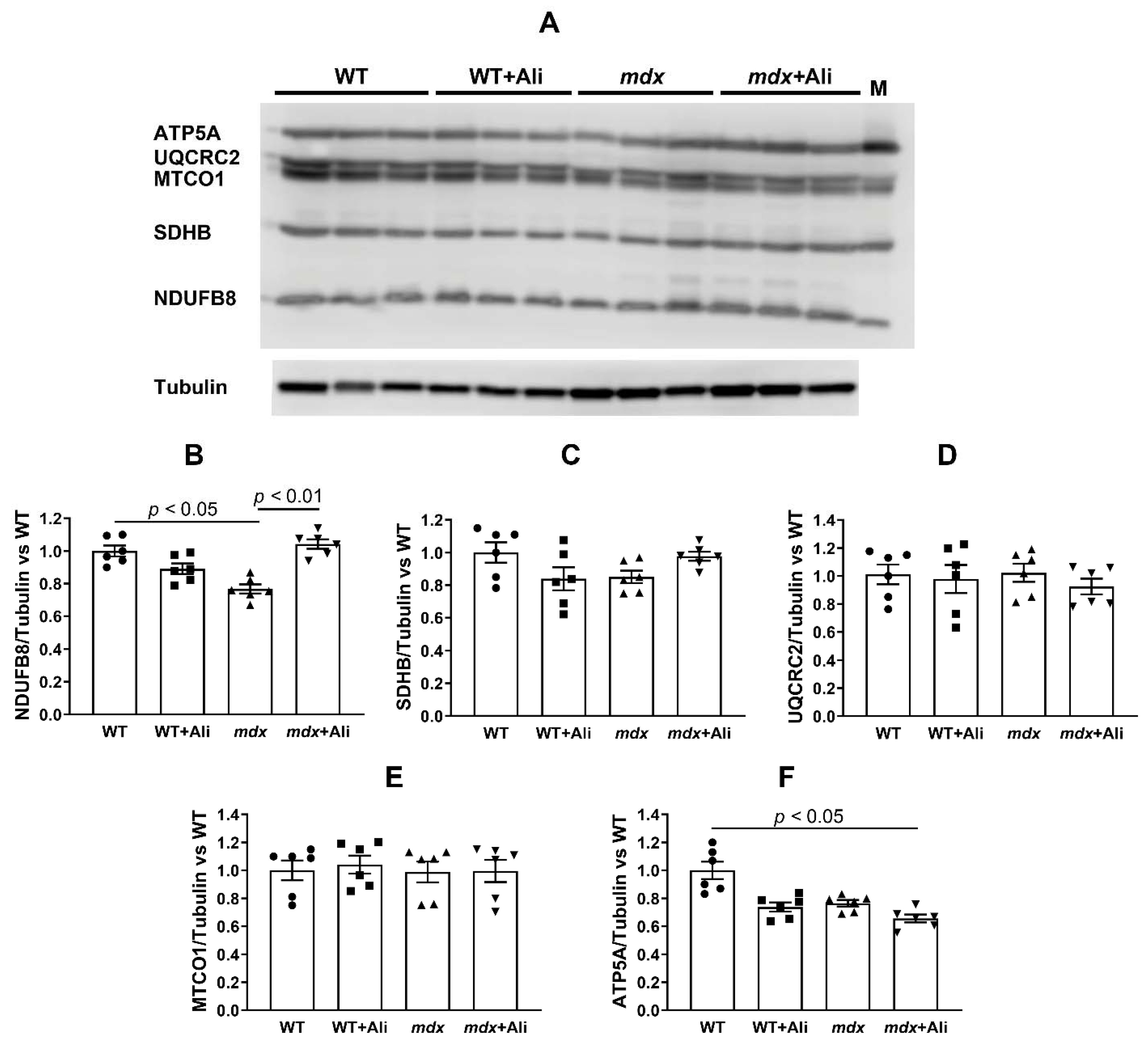
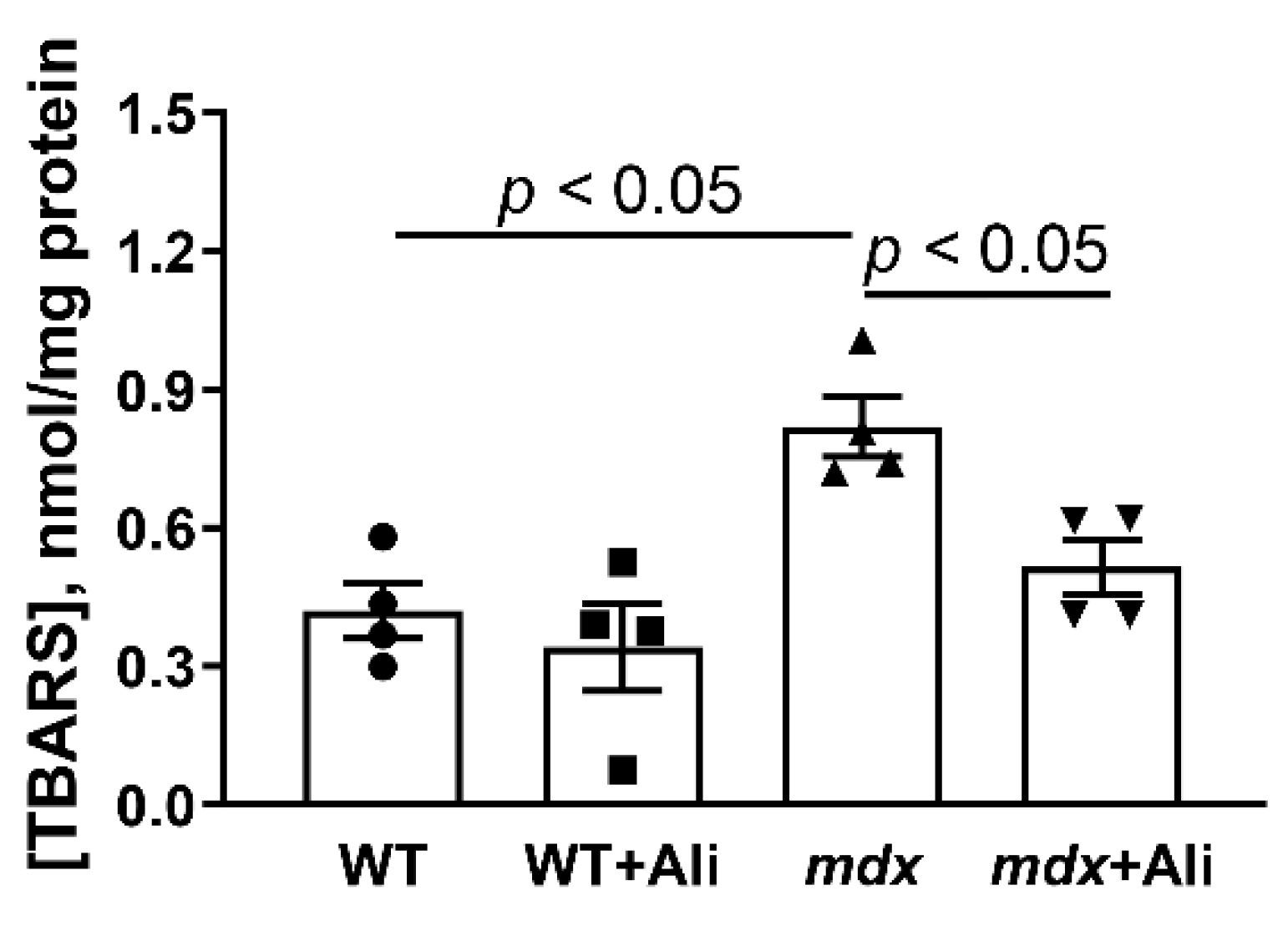
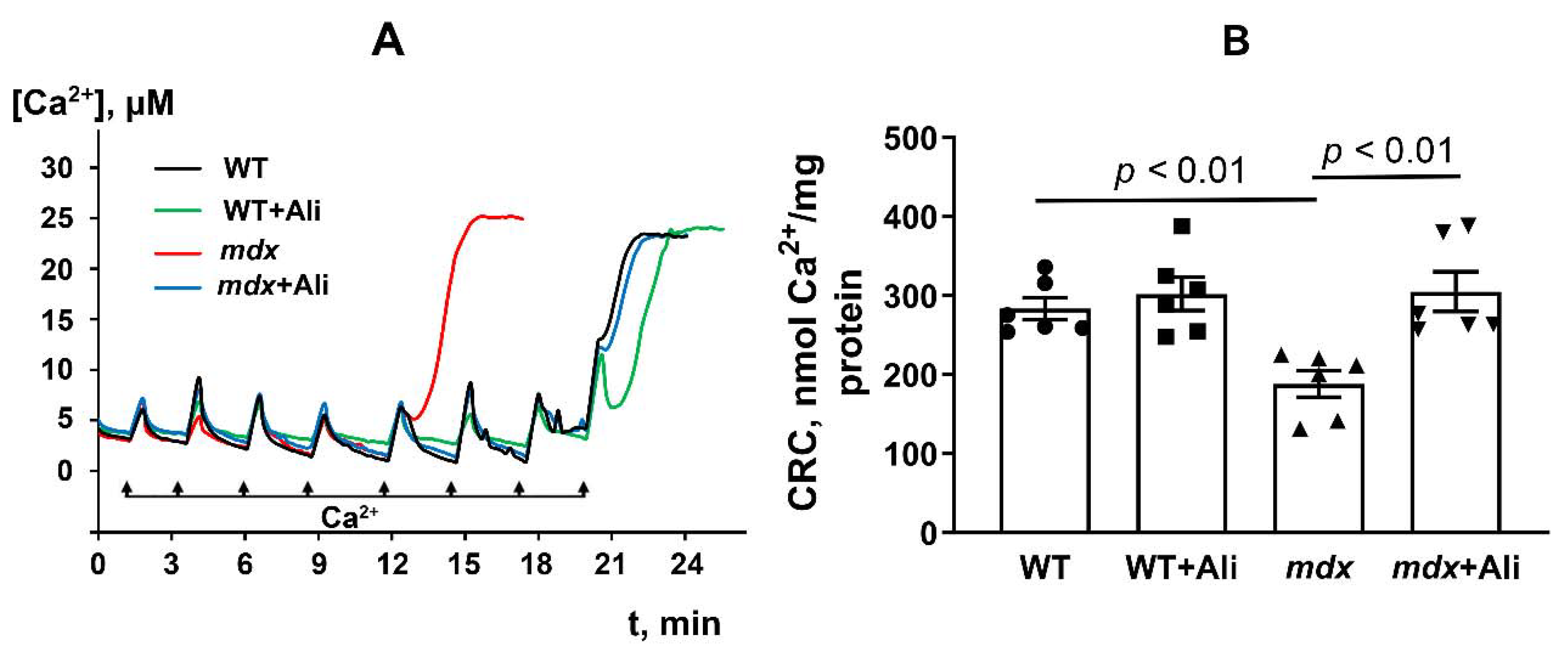
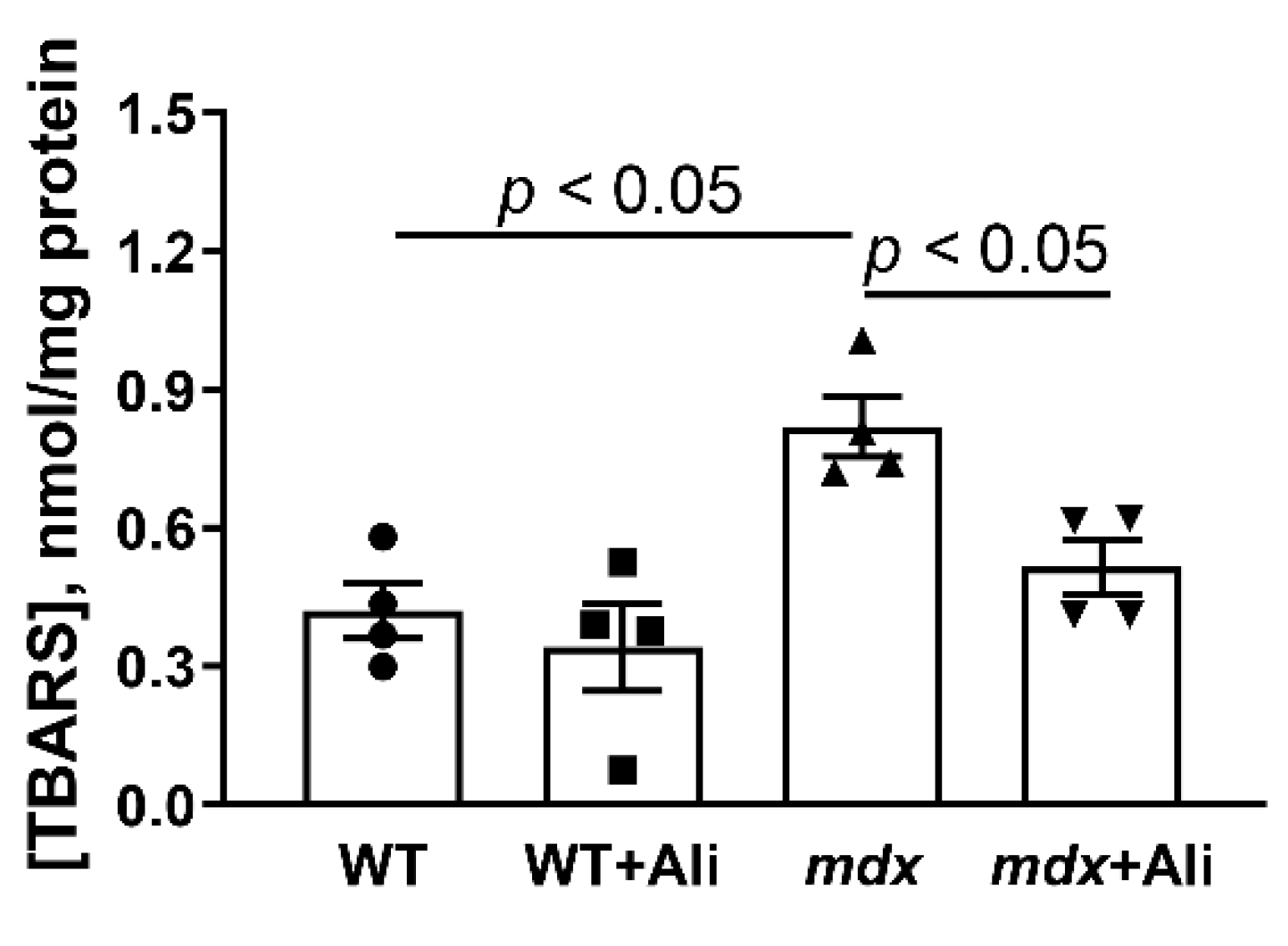
2.3. Alisporivir Desensitizes the PTP to Ca2+ and Improves the Functioning of Skeletal Muscle Mitochondria in mdx Mice
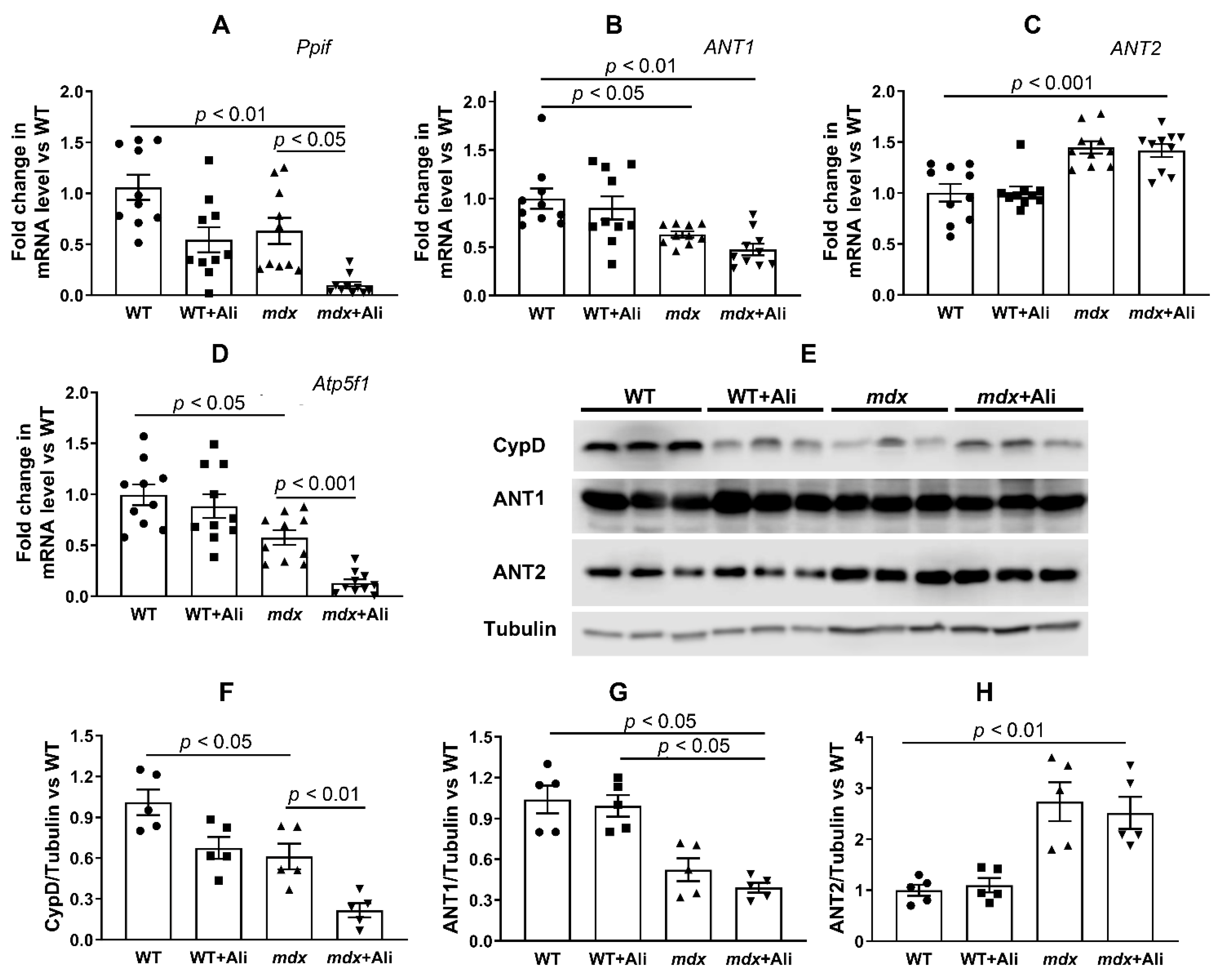
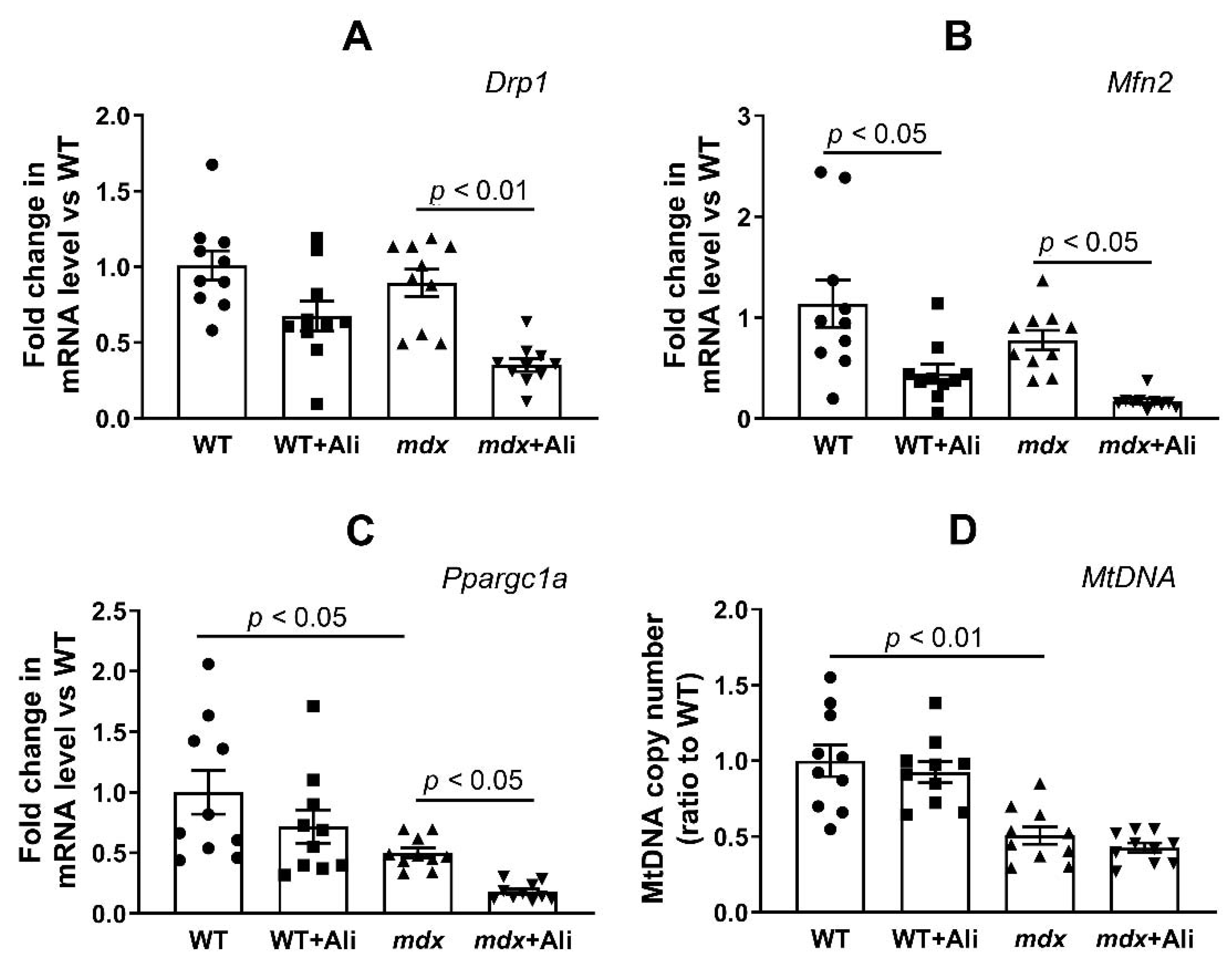
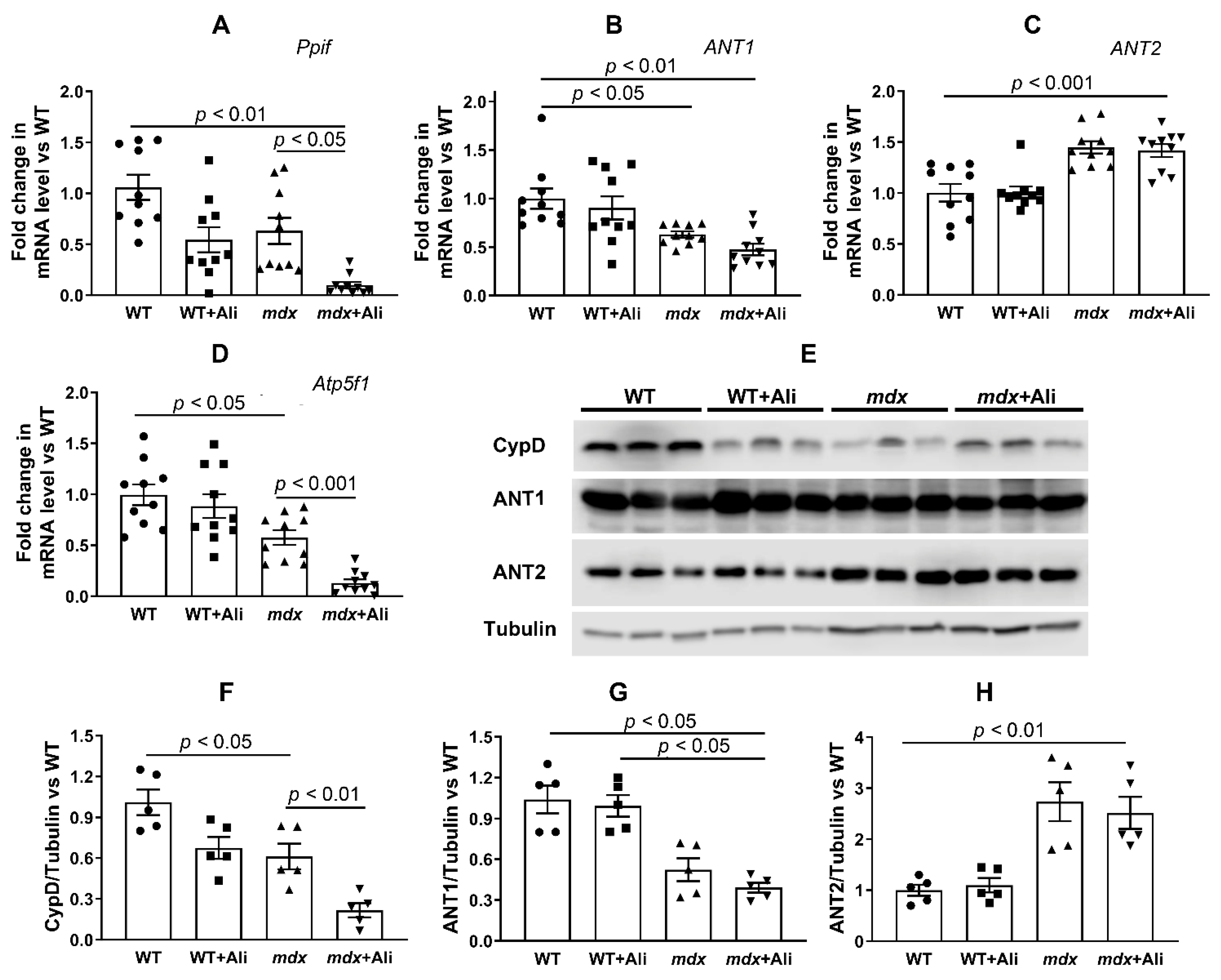
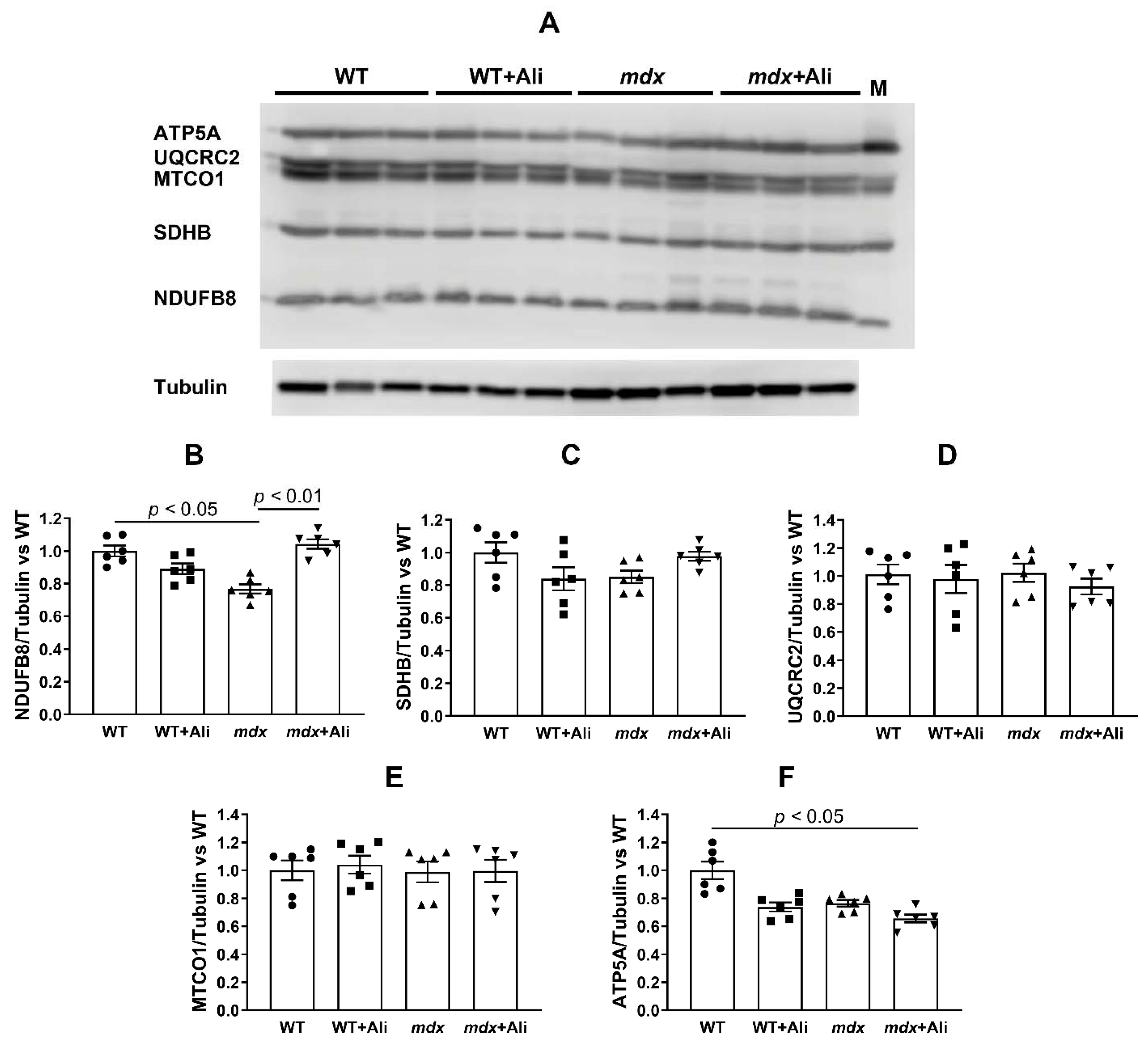
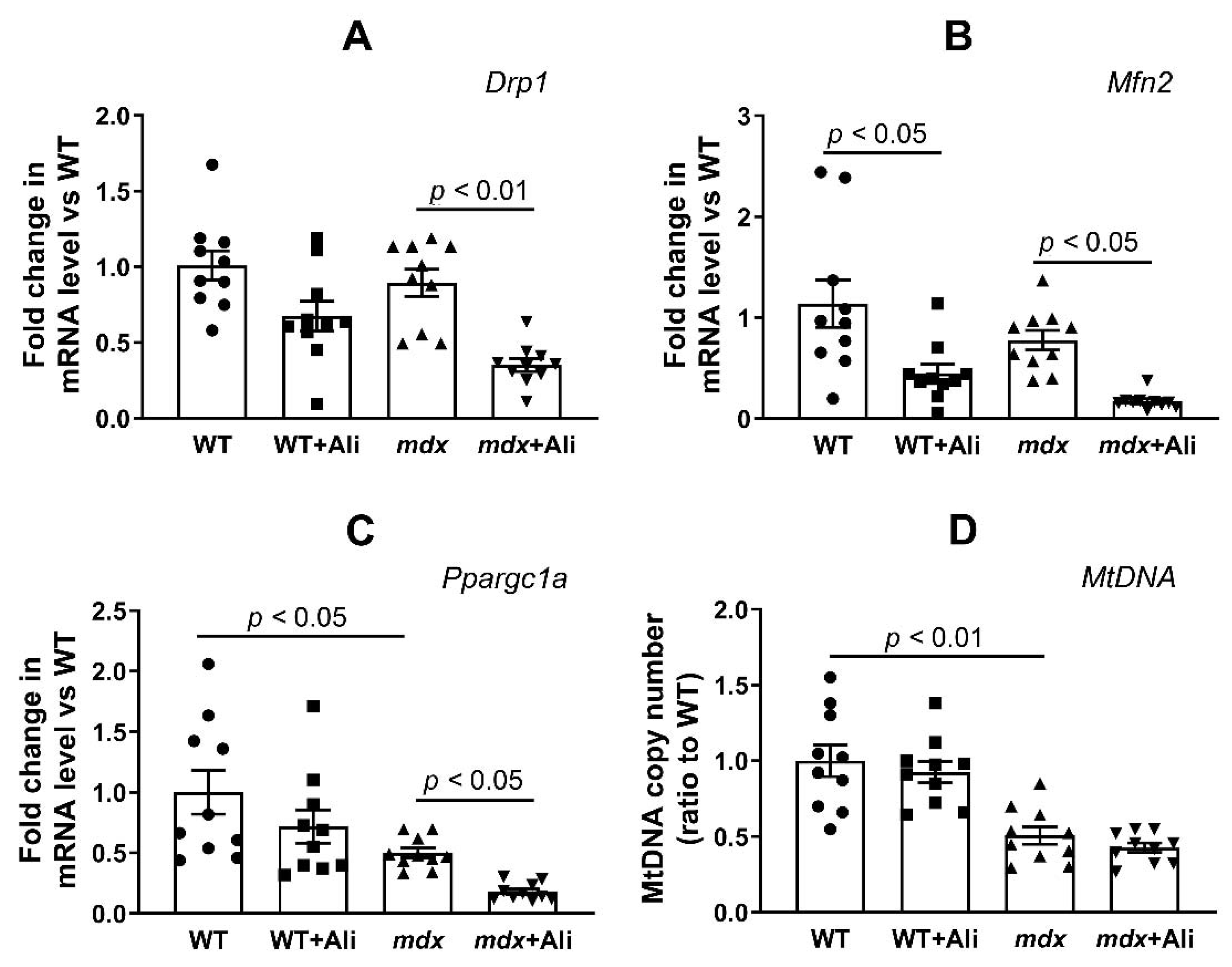
2.4. Alisporivir Suppresses Mitochondrial Dynamics and Biogenesis in Skeletal Muscle
3. Discussion
4. Materials and Methods
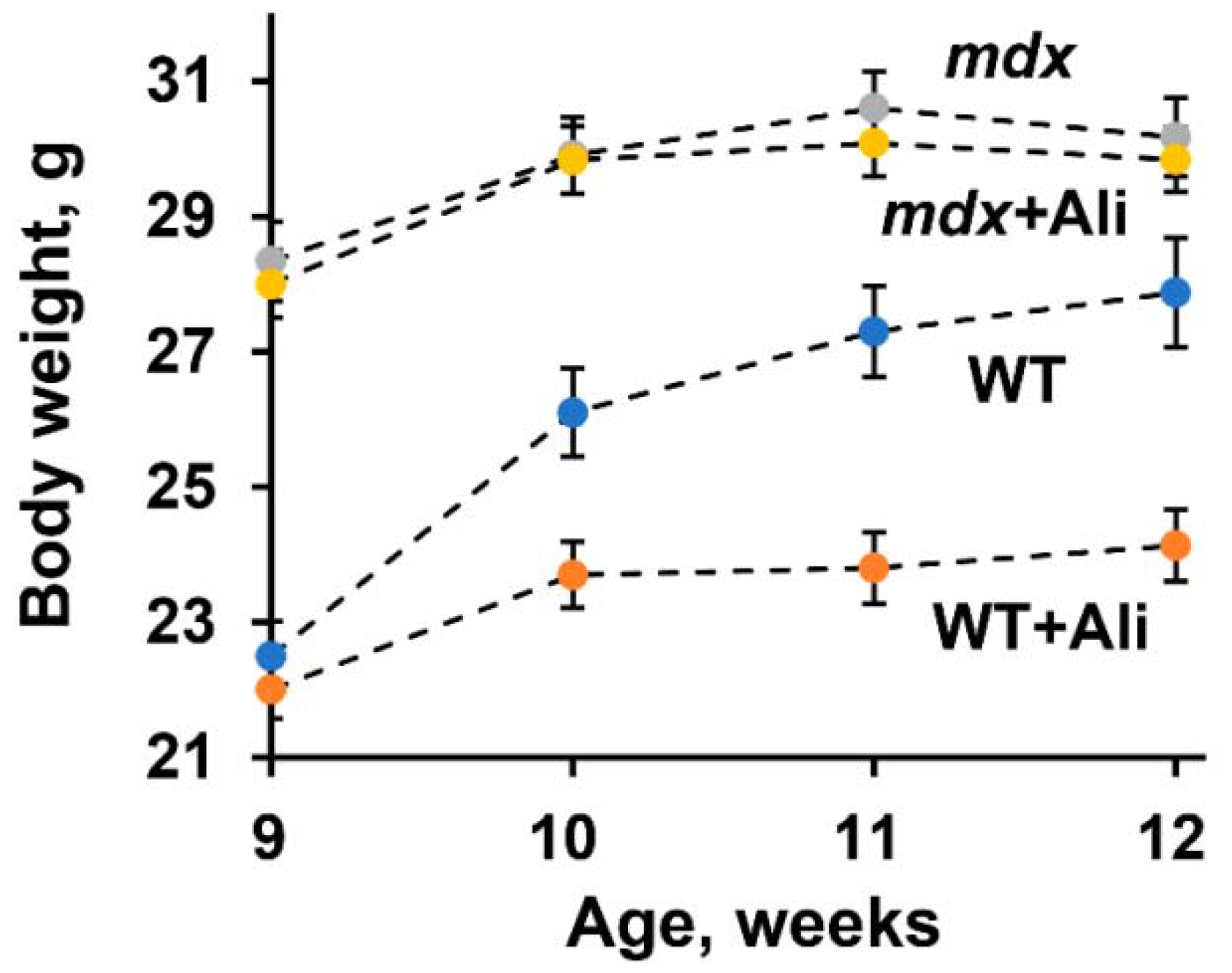
4.1. Animals
4.2. Wire-Hanging Test
4.3. Electron Microscopy
4.4. Mitochondria Isolation and Determination of Respiration and Oxidative Phosphorylation
4.5. Determination of Ca2+ Retention by Mitochondria, MPT Pore Opening Assay
4.6. Lipid Peroxidation
4.7. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
4.8. Analysis of mtDNA/nDNA Ratio
4.9. Electrophoresis and Immunoblotting of Mitochondrial OXPHOS and MPT Pore Proteins
4.10. Statistical Processing of Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ignatieva, E.; Smolina, N.; Kostareva, A.; Dmitrieva, R. Skeletal muscle mitochondria dysfunction in genetic neuromuscular disorders with cardiac phenotype. Int. J. Mol. Sci. 2021, 22, 7349. [Google Scholar] [CrossRef]
- Bodensteiner, J.B.; Engel, A.G. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology 1978, 28, 439. [Google Scholar] [CrossRef]
- Williams, G.S.B.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [Green Version]
- Rybalka, E.; Timpani, C.; Cooke, M.B.; Williams, A.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.; van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The effect of deflazacort treatment on the functioning of skeletal muscle mitochondria in Duchenne muscular dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochem. Mosc. 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.G.A.; Beutner, G. Cyclophilin D, somehow a master regulator of mitochondrial function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanakis, G.; Murphy, E. Cyclophilin D: An integrator of mitochondrial function. Front. Physiol. 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [Green Version]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Stupka, N.; Gregorevic, P.; Plant, D.R.; Lynch, G.S. The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol. 2004, 107, 299–310. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.O.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmér, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta BBA Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef] [Green Version]
- De Mario, A.; Gherardi, G.; Rizzuto, R.; Mammucari, C. Skeletal muscle mitochondria in health and disease. Cell Calcium 2021, 94, 102357. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Xiao, A.; Gan, X.; Chen, R.; Ren, Y.; Yu, H.; You, C. The cyclophilin D/Drp1 axis regulates mitochondrial fission contributing to oxidative stress-induced mitochondrial dysfunctions in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2017, 483, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Bryson, H.M. Deflazacort. Drugs 1995, 50, 317–333. [Google Scholar] [CrossRef]
- Bylo, M.; Farewell, R.; Coppenrath, V.A.; Yogaratnam, D. A review of deflazacort for patients with Duchenne muscular dystrophy. Ann. Pharmacother. 2020, 54, 788–794. [Google Scholar] [CrossRef]
- Hanaoka, B.Y.; Peterson, C.A.; Horbinski, C.; Crofford, L.J. Implications of glucocorticoid therapy in idiopathic inflammatory myopathies. Nat. Rev. Rheumatol. 2012, 8, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Kalista, S.; Thissen, J. Mechanisms of muscle atrophy induced by glucocorticoids. Horm. Res. 2009, 72, 36–41. [Google Scholar] [CrossRef]
- Morine, K.J.; Bish, L.T.; Pendrak, K.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS ONE 2010, 5, e9176. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy. JAMA Neurol. 2020, 77, 1122. [Google Scholar] [CrossRef]
- Gams, W. Tolypocladiumeine Hyphomycetengattung mit geschwollenen Phialiden. Persoonia 1971, 6, 185–191. [Google Scholar]
- Kirschner, J.; Schessl, J.; Schara, U.; Reitter, B.; Stettner, G.M.; Hobbiebrunken, E.; Wilichowski, E.; Bernert, G.; Weiss, S.; Stehling, F.; et al. Treatment of Duchenne muscular dystrophy with ciclosporin A: A randomised, double-blind, placebo-controlled multicentre trial. Lancet Neurol. 2010, 9, 1053–1059. [Google Scholar] [CrossRef]
- Stepien, G.; Torroni, A.; Chung, A.; Hodge, J.; Wallace, D. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Qi, R.; Wang, D.; Xing, L.; Wu, Z. Cyclosporin A inhibits mitochondrial biogenesis in Hep G2 cells. Biochem. Biophys. Res. Commun. 2018, 496, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Serov, D.A.; Astashev, M.E.; Belosludtsev, M.N.; Dubinin, M.V.; Belosludtsev, K.N. Effect of the MPT pore inhibitor alisporivir on the development of mitochondrial dysfunction in the heart tissue of diabetic mice. Biology 2021, 10, 839. [Google Scholar] [CrossRef]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The effect of S-15176 difumarate salt on ultrastructure and functions of liver mitochondria of c57bl/6 mice with streptozotocin/high-fat diet-induced type 2 diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal (n = 10) | State 2 | State 3 | State 4 | State 3UDNP | RC | ADP/O |
|---|---|---|---|---|---|---|
| nmol O2/min per 1 mg of Protein | Relative Units | |||||
| WT | 19.6 ± 1.0 | 172.7 ± 6.7 | 26.2 ± 1.1 | 213.4 ± 10.7 | 6.6 ± 0.1 | 3.1 ± 0.3 |
| WT+Ali | 17.9 ± 0.8 | 156.4 ± 7.1 | 23.0 ± 0.6 | 202.0 ± 12.5 | 6.8 ± 0.2 | 2.9 ± 0.1 |
| mdx | 17.1 ± 1.3 | 127.9 ± 10.0 * | 25.1 ± 1.3 | 150.4 ± 13.2 * | 5.1 ± 0.2 * | 3.2 ± 0.2 |
| mdx+Ali | 21.0 ± 0.8 | 164.4 ± 3.3 | 28.7 ± 1.3 | 201.9 ± 7.6 | 5.8 ± 0.2 *# | 2.9 ± 0.1 |
| Gene | Forward (5‘ → 3’) | Reverse (5’ → 3’) |
|---|---|---|
| Ant1 | CTATGACACTGCCAAGGGGATG | TCAAACGGATAGGACACCAGC |
| Ant2 | TCTGGACGCAAAGGAACTGA | GACCATGCGCCCTTGAAA |
| Ppif | GCAGATGTCGTGCCAAAGACTG | GCCATTGTGGTTGGTGAAGTCG |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| Nd4 | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gapdh | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. https://doi.org/10.3390/ijms22189780
Dubinin MV, Starinets VS, Talanov EY, Mikheeva IB, Belosludtseva NV, Belosludtsev KN. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. International Journal of Molecular Sciences. 2021; 22(18):9780. https://doi.org/10.3390/ijms22189780
Chicago/Turabian StyleDubinin, Mikhail V., Vlada S. Starinets, Eugeny Yu. Talanov, Irina B. Mikheeva, Natalia V. Belosludtseva, and Konstantin N. Belosludtsev. 2021. "Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis" International Journal of Molecular Sciences 22, no. 18: 9780. https://doi.org/10.3390/ijms22189780
APA StyleDubinin, M. V., Starinets, V. S., Talanov, E. Y., Mikheeva, I. B., Belosludtseva, N. V., & Belosludtsev, K. N. (2021). Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. International Journal of Molecular Sciences, 22(18), 9780. https://doi.org/10.3390/ijms22189780