
Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring

 ,
,  ,
,  and
and
Abstract
1. Introduction
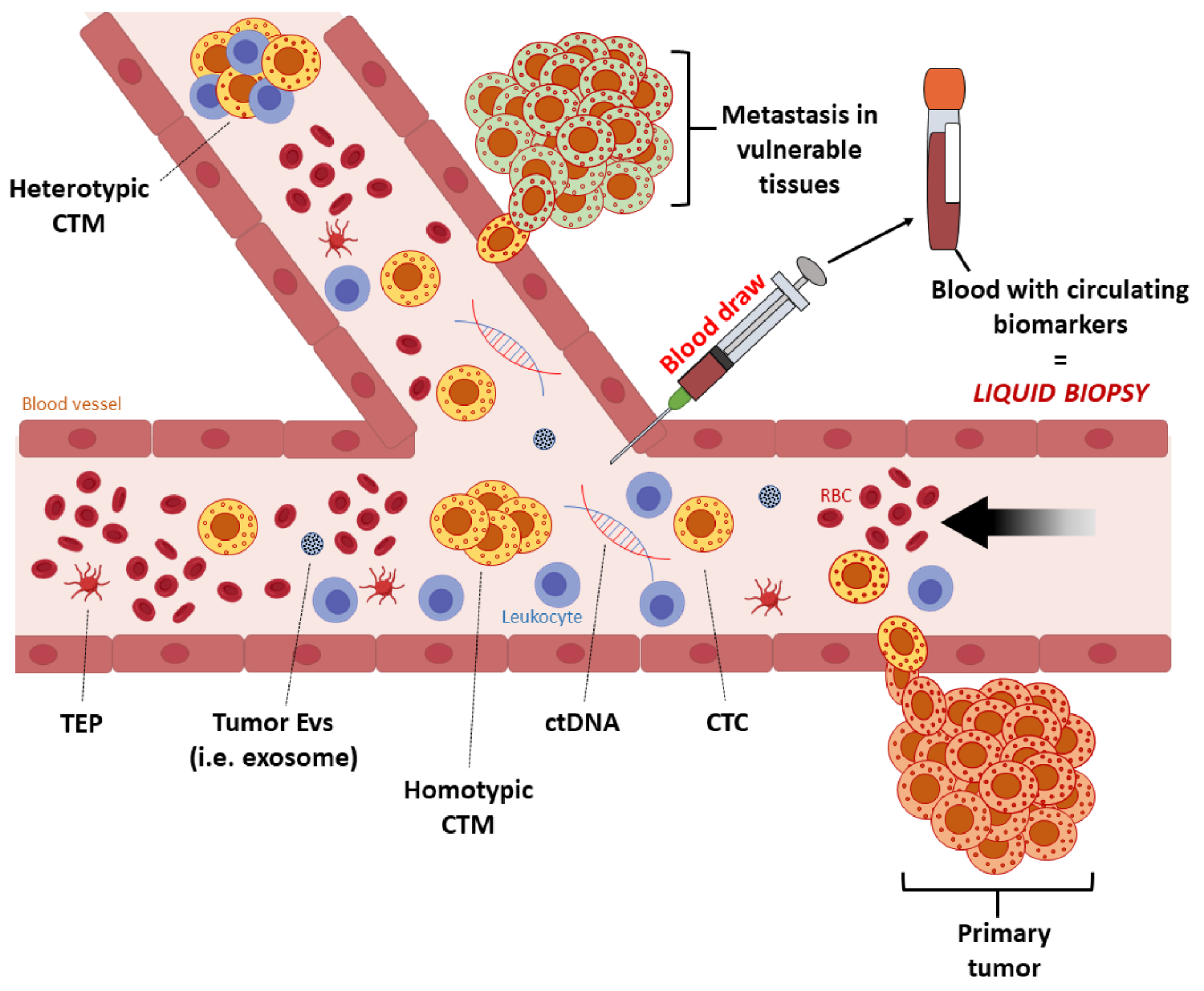
2. Liquid Biopsy in Melanoma
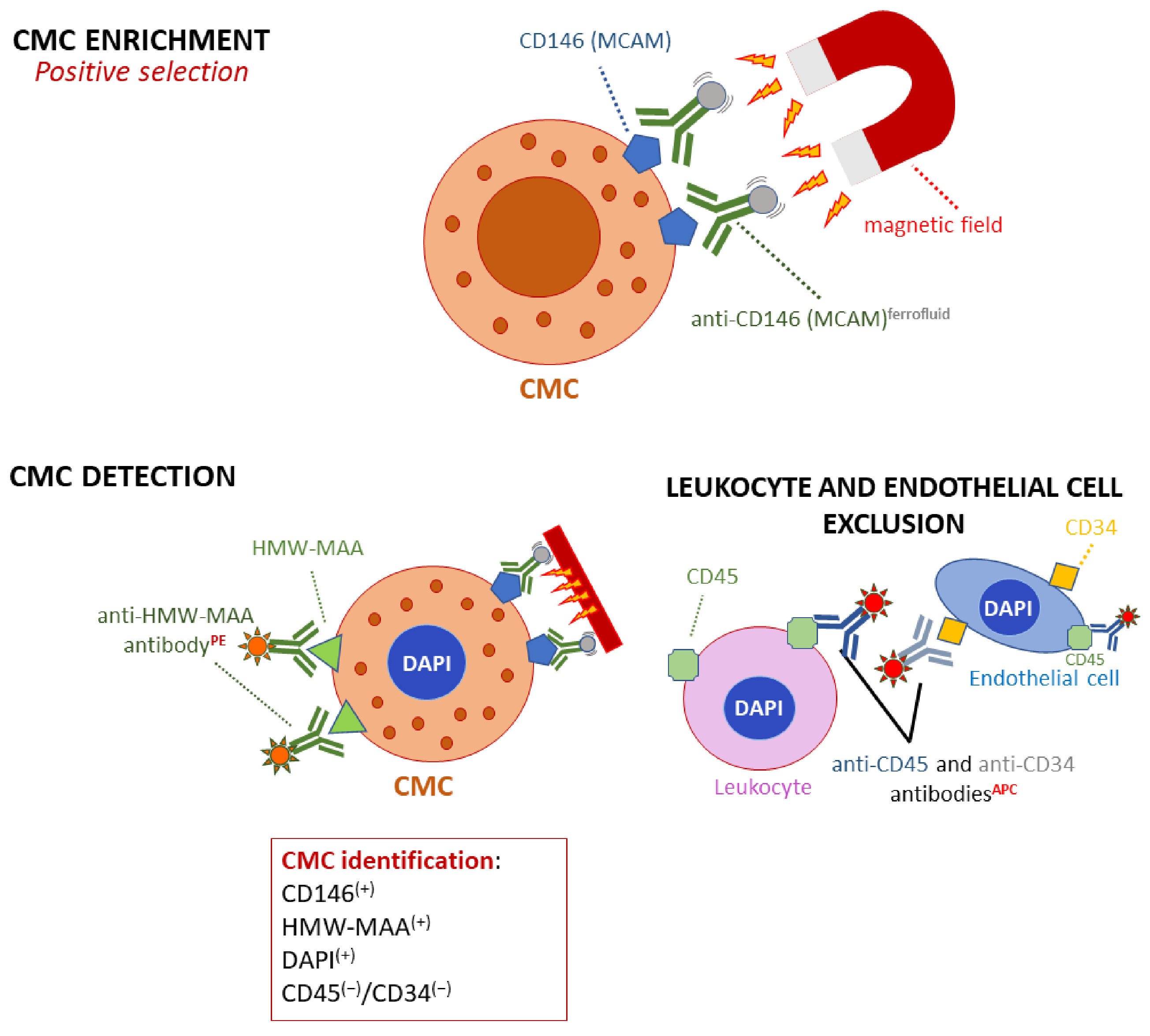
2.1. Circulating Melanoma Cells (CMCs)
2.2. Circulating Tumor DNA (ctDNA)
2.3. Melanoma-Derived Exosomes
3. Technologies for Liquid Biopsy in Melanoma
3.1. CMC Enrichment and Detection
3.2. ctDNA Detection
3.3. Exosome Isolation
4. Clinical Relevance of Liquid Biopsy in Melanoma
4.1. Melanoma Detection and Prognosis
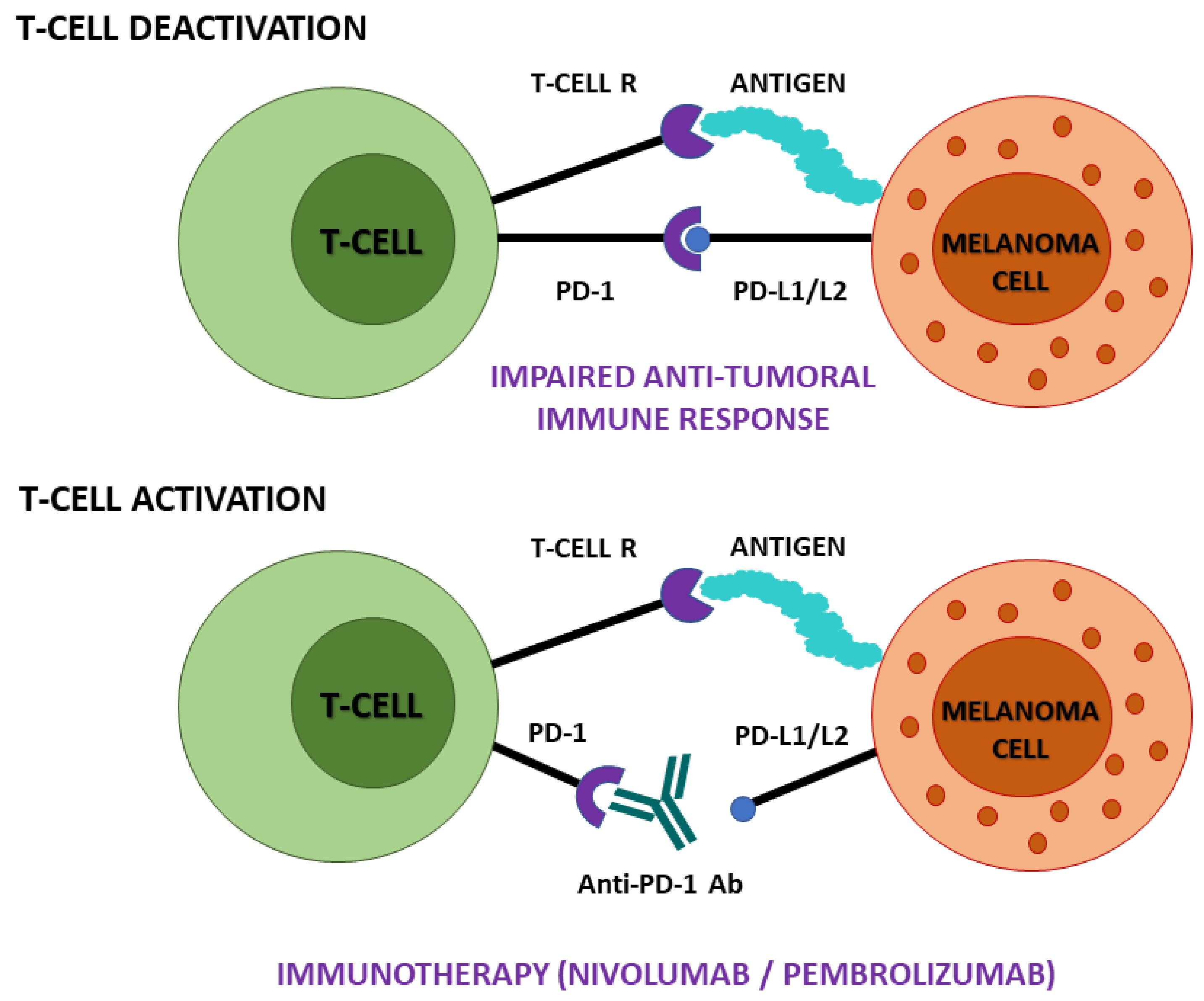
4.2. Assessment of Treatment Efficacy and Acquired Resistance
4.3. Completed and Ongoing Clinical Trials
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, Risk Factors, Pathogenesis, Diagnosis and Classification. In Vivo 2014, 28, 1005–1011. [Google Scholar]
- Joyce, K.M. Surgical Management of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-4-0. [Google Scholar]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of Melanoma. Contemp. Oncol. Poznan Pol. 2018, 22, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.K.; Wolchok, J.D.; Allison, J.P. Anti-CTLA-4 Antibody Therapy: Immune Monitoring during Clinical Development of a Novel Immunotherapy. Semin. Oncol. 2010, 37, 473–484. [Google Scholar] [CrossRef]
- Melero, I.; Grimaldi, A.M.; Perez-Gracia, J.L.; Ascierto, P.A. Clinical Development of Immunostimulatory Monoclonal Antibodies and Opportunities for Combination. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 997–1008. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Mader, S.; Pantel, K. Liquid Biopsy: Current Status and Future Perspectives. Oncol. Res. Treat. 2017, 40, 404–408. [Google Scholar] [CrossRef]
- Blanco, B.A.; Wolfgang, C.L. Liquid Biopsy for the Detection and Management of Surgically Resectable Tumors. Langenbecks Arch. Surg. 2019, 404, 517–525. [Google Scholar] [CrossRef]
- Cabel, L.; Proudhon, C.; Gortais, H.; Loirat, D.; Coussy, F.; Pierga, J.-Y.; Bidard, F.-C. Circulating Tumor Cells: Clinical Validity and Utility. Int. J. Clin. Oncol. 2017, 22, 421–430. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid Biopsy and Minimal Residual Disease—Latest Advances and Implications for Cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Poulet, G.; Massias, J.; Taly, V. Liquid Biopsy: General Concepts. Acta Cytol. 2019, 63, 449–455. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Circulating Tumour Cells in Cancer Patients: Challenges and Perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Hille, C.; Scher, H.I. Circulating Tumor Cells in Prostate Cancer: From Discovery to Clinical Utility. Clin. Chem. 2019, 65, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating Tumor Cells and Circulating Tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Xu, M.J.; Dorsey, J.F.; Amaravadi, R.; Karakousis, G.; Simone, C.B.; Xu, X.; Xu, W.; Carpenter, E.L.; Schuchter, L.; Kao, G.D. Circulating Tumor Cells, DNA, and MRNA: Potential for Clinical Utility in Patients With Melanoma. Oncologist 2016, 21, 84–94. [Google Scholar] [CrossRef]
- Cayrefourcq, L.; De Roeck, A.; Garcia, C.; Stoebner, P.-E.; Fichel, F.; Garima, F.; Perriard, F.; Daures, J.-P.; Meunier, L.; Alix-Panabières, C. S100-EPISPOT: A New Tool to Detect Viable Circulating Melanoma Cells. Cells 2019, 8, 755. [Google Scholar] [CrossRef] [PubMed]
- Weight, R.M.; Viator, J.A. Detection of Circulating Tumor Cells by Photoacoustic Flowmetry. Methods Mol. Biol. Clifton NJ 2014, 1102, 655–663. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Mader, S.; Pantel, K. Epithelial-Mesenchymal Plasticity in Circulating Tumor Cells. J. Mol. Med. Berl. Ger. 2017, 95, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Tayoun, T.; Faugeroux, V.; Oulhen, M.; Aberlenc, A.; Pawlikowska, P.; Farace, F. CTC-Derived Models: A Window into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells 2019, 8, 1145. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Aya-Bonilla, C.A.; Warkiani, M.E.; Gray, E.S.; Ziman, M. Melanoma Circulating Tumor Cells: Benefits and Challenges Required for Clinical Application. Cancer Lett. 2018, 424, 1–8. [Google Scholar] [CrossRef]
- Rapanotti, M.C.; Campione, E.; Spallone, G.; Orlandi, A.; Bernardini, S.; Bianchi, L. Minimal Residual Disease in Melanoma: Circulating Melanoma Cells and Predictive Role of MCAM/MUC18/MelCAM/CD146. Cell Death Discov. 2017, 3, 17005. [Google Scholar] [CrossRef]
- De Giorgi, V.; Pinzani, P.; Salvianti, F.; Panelos, J.; Paglierani, M.; Janowska, A.; Grazzini, M.; Wechsler, J.; Orlando, C.; Santucci, M.; et al. Application of a Filtration- and Isolation-by-Size Technique for the Detection of Circulating Tumor Cells in Cutaneous Melanoma. J. Investig. Dermatol. 2010, 130, 2440–2447. [Google Scholar] [CrossRef]
- Long, E.; Ilie, M.; Bence, C.; Butori, C.; Selva, E.; Lalvée, S.; Bonnetaud, C.; Poissonnet, G.; Lacour, J.-P.; Bahadoran, P.; et al. High Expression of TRF2, SOX10, and CD10 in Circulating Tumor Microemboli Detected in Metastatic Melanoma Patients. A Potential Impact for the Assessment of Disease Aggressiveness. Cancer Med. 2016, 5, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-M.; Krebs, M.; Ward, T.; Sloane, R.; Priest, L.; Hughes, A.; Clack, G.; Ranson, M.; Blackhall, F.; Dive, C. Circulating Tumor Cells as a Window on Metastasis Biology in Lung Cancer. Am. J. Pathol. 2011, 178, 989–996. [Google Scholar] [CrossRef]
- Ma, J.; Frank, M.H. Isolation of Circulating Melanoma Cells. Methods Mol. Biol. Clifton NJ 2015. [Google Scholar] [CrossRef]
- Campoli, M.R.; Chang, C.-C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human High Molecular Weight-Melanoma-Associated Antigen (HMW-MAA): A Melanoma Cell Surface Chondroitin Sulfate Proteoglycan (MSCP) with Biological and Clinical Significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef]
- Bennaceur, K.; Popa, I.; Portoukalian, J.; Berthier-Vergnes, O.; Péguet-Navarro, J. Melanoma-Derived Gangliosides Impair Migratory and Antigen-Presenting Function of Human Epidermal Langerhans Cells and Induce Their Apoptosis. Int. Immunol. 2006, 18, 879–886. [Google Scholar] [CrossRef][Green Version]
- Gaiser, M.R.; von Bubnoff, N.; Gebhardt, C.; Utikal, J.S. Liquid Biopsy to Monitor Melanoma Patients. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2018, 16, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 Proteins in Cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The Interplay of Circulating Tumor DNA and Chromatin Modification, Therapeutic Resistance, and Metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the Possible Origin and Mechanism of Circulating DNA Apoptosis and Active DNA Release. Clin. Chim. Acta Int. J. Clin. Chem. 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct Detection of Early-Stage Cancers Using Circulating Tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef] [PubMed]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.-T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor Mutation Burden and Circulating Tumor DNA in Combined CTLA-4 and PD-1 Antibody Therapy in Metastatic Melanoma—Results of a Prospective Biomarker Study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.M.; Tsui, D.W.Y. Circulating Cell-Free DNA for Non-Invasive Cancer Management. Cancer Genet. 2018, 228–229, 169–179. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Butler, T.M.; Spellman, P.T.; Gray, J. Circulating-Tumor DNA as an Early Detection and Diagnostic Tool. Curr. Opin. Genet. Dev. 2017, 42, 14–21. [Google Scholar] [CrossRef]
- Calapre, L.; Warburton, L.; Millward, M.; Ziman, M.; Gray, E.S. Circulating Tumour DNA (CtDNA) as a Liquid Biopsy for Melanoma. Cancer Lett. 2017, 404, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.J.; Scolyer, R.A.; et al. Circulating Tumor DNA to Monitor Treatment Response and Detect Acquired Resistance in Patients with Metastatic Melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A. Circulating Tumor DNA Analysis as a Real-Time Method for Monitoring Tumor Burden in Melanoma Patients Undergoing Treatment with Immune Checkpoint Blockade. J. Immunother. Cancer 2014, 2, 42. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Wellings, R.; Orr, M.C.M.; McCormack, R.; Malone, M.; Board, R.E.; Cantarini, M.V. Circulating Tumour-Derived Predictive Biomarkers in Oncology. Drug Discov. Today 2010, 15, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Cayrefourcq, L.; Alix-Panabières, C. The Different Facets of Liquid Biopsy: A Kaleidoscopic View. Cold Spring Harb. Perspect. Med. 2020, 10, a037333. [Google Scholar] [CrossRef]
- Sacco, A.; Forgione, L.; Carotenuto, M.; Luca, A.D.; Ascierto, P.A.; Botti, G.; Normanno, N. Circulating Tumor DNA Testing Opens New Perspectives in Melanoma Management. Cancers 2020, 12, 2914. [Google Scholar] [CrossRef]
- Hood, J.L. Natural Melanoma-Derived Extracellular Vesicles. Semin. Cancer Biol. 2019, 59, 251–265. [Google Scholar] [CrossRef]
- Hood, J.L.; Wickline, S.A. A Systematic Approach to Exosome-Based Translational Nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Exosomes in Cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Zhang, W.; Xia, W.; Lv, Z.; Ni, C.; Xin, Y.; Yang, L. Liquid Biopsy for Cancer: Circulating Tumor Cells, Circulating Free DNA or Exosomes? Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 755–768. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current Knowledge of Their Composition, Biological Functions, and Diagnostic and Therapeutic Potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Isola, A.L.; Eddy, K.; Chen, S. Biology, Therapy and Implications of Tumor Exosomes in the Progression of Melanoma. Cancers 2016, 8, 110. [Google Scholar] [CrossRef]
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel Biomarkers for Clinical Diagnosis. Sci. World J. 2015, 2015, 657086. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Manian, M.; Jazayeri, M.H.; Ranjbar, M.; Salehi, R.; Sharifi, M.; Mohaghegh, F.; Goli, M.; Jahednia, S.H.; Avan, A.; et al. Circulating Exosomes and Exosomal MicroRNAs as Biomarkers in Gastrointestinal Cancer. Cancer Gene Ther. 2017, 24, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma Cell-Derived Exosomes Promote Epithelial-Mesenchymal Transition in Primary Melanocytes through Paracrine/Autocrine Signaling in the Tumor Microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef]
- Xiao, D.; Ohlendorf, J.; Chen, Y.; Taylor, D.D.; Rai, S.N.; Waigel, S.; Zacharias, W.; Hao, H.; McMasters, K.M. Identifying MRNA, MicroRNA and Protein Profiles of Melanoma Exosomes. PLoS ONE 2012, 7, e46874. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells toward a Pro-Metastatic Phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Namee, N.M.; O’Driscoll, L. Extracellular Vesicles and Anti-Cancer Drug Resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Cesi, G.; Philippidou, D.; Kozar, I.; Kim, Y.J.; Bernardin, F.; Van Niel, G.; Wienecke-Baldacchino, A.; Felten, P.; Letellier, E.; Dengler, S.; et al. A New ALK Isoform Transported by Extracellular Vesicles Confers Drug Resistance to Melanoma Cells. Mol. Cancer 2018, 17, 145. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Behren, A.; Coleman, B.; Greening, D.W.; Hill, A.F.; Cebon, J. Intercellular Resistance to BRAF Inhibition Can Be Mediated by Extracellular Vesicle-Associated PDGFRβ. Neoplasia 2017, 19, 932–940. [Google Scholar] [CrossRef]
- Eisenstein, A.; Gonzalez, E.C.; Raghunathan, R.; Xu, X.; Wu, M.; McLean, E.O.; McGee, J.; Ryu, B.; Alani, R.M. Emerging Biomarkers in Cutaneous Melanoma. Mol. Diagn. Ther. 2018, 22, 203–218. [Google Scholar] [CrossRef]
- Joshi, P.; Jacobs, B.; Derakhshan, A.; Moore, L.R.; Elson, P.; Triozzi, P.L.; Borden, E.; Zborowski, M. Enrichment of Circulating Melanoma Cells (CMCs) Using Negative Selection from Patients with Metastatic Melanoma. Oncotarget 2014, 5, 2450–2461. [Google Scholar] [CrossRef][Green Version]
- Rodic, S.; Mihalcioiu, C.; Saleh, R.R. Detection Methods of Circulating Tumor Cells in Cutaneous Melanoma: A Systematic Review. Crit. Rev. Oncol. Hematol. 2014, 91, 74–92. [Google Scholar] [CrossRef]
- Aya-Bonilla, C.A.; Morici, M.; Hong, X.; McEvoy, A.C.; Sullivan, R.J.; Freeman, J.; Calapre, L.; Khattak, M.A.; Meniawy, T.; Millward, M.; et al. Detection and Prognostic Role of Heterogeneous Populations of Melanoma Circulating Tumour Cells. Br. J. Cancer 2020, 122, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.B.; Gray, E.S.; Millward, M.; Pearce, R.; Ziman, M. Evaluation of a Multi-Marker Immunomagnetic Enrichment Assay for the Quantification of Circulating Melanoma Cells. J. Transl. Med. 2012, 10, 192. [Google Scholar] [CrossRef]
- Roland, C.L.; Ross, M.I.; Hall, C.S.; Laubacher, B.; Upshaw, J.; Anderson, A.E.; Lucci, A. Detection of Circulating Melanoma Cells in the Blood of Melanoma Patients: A Preliminary Study. Melanoma Res. 2015, 25, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Fusi, A.; Reichelt, U.; Busse, A.; Ochsenreither, S.; Rietz, A.; Maisel, M.; Keilholz, U. Expression of the Stem Cell Markers Nestin and CD133 on Circulating Melanoma Cells. J. Investig. Dermatol. 2011, 131, 487–494. [Google Scholar] [CrossRef]
- Siewert, C.; Herber, M.; Hunzelmann, N.; Fodstad, O.; Miltenyi, S.; Assenmacher, M.; Schmitz, J. Rapid Enrichment and Detection of Melanoma Cells from Peripheral Blood Mononuclear Cells by a New Assay Combining Immunomagnetic Cell Sorting and Immunocytochemical Staining. Minimal Residual Dis. Melanoma 2001, 158, 51–60. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation Analysis of BRAF and KIT in Circulating Melanoma Cells at the Single Cell Level. Br. J. Cancer 2012, 106, 939–946. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker Utility of Circulating Tumor Cells in Metastatic Cutaneous Melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef]
- Rao, C.; Bui, T.; Connelly, M.; Doyle, G.; Karydis, I.; Middleton, M.R.; Clack, G.; Malone, M.; Coumans, F.A.W.; Terstappen, L.W.M.M. Circulating Melanoma Cells and Survival in Metastatic Melanoma. Int. J. Oncol. 2011, 38, 755–760. [Google Scholar] [CrossRef]
- Halse, H.; Colebatch, A.J.; Petrone, P.; Henderson, M.A.; Mills, J.K.; Snow, H.; Westwood, J.A.; Sandhu, S.; Raleigh, J.M.; Behren, A.; et al. Multiplex Immunohistochemistry Accurately Defines the Immune Context of Metastatic Melanoma. Sci. Rep. 2018, 8, 11158. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, B.S.; Viator, J.A. Capture and Isolation of Circulating Melanoma Cells Using Photoacoustic Flowmetry. Methods Mol. Biol. Clifton NJ 2015, 1–9. [Google Scholar] [CrossRef]
- Alix-Panabières, C. EPISPOT Assay: Detection of Viable DTCs/CTCs in Solid Tumor Patients. Recent Results Cancer Res. Fortschr. Krebsforsch. Progres Dans Rech. Sur Cancer 2012, 195, 69–76. [Google Scholar] [CrossRef]
- Hasselmann, D.O.; Rappl, G.; Rössler, M.; Ugurel, S.; Tilgen, W.; Reinhold, U. Detection of Tumor-Associated Circulating MRNA in Serum, Plasma and Blood Cells from Patients with Disseminated Malignant Melanoma. Oncol. Rep. 2001, 8, 115–118. [Google Scholar] [CrossRef]
- Stevens, G.L.; Scheer, W.D.; Levine, E.A. Detection of Tyrosinase MRNA from the Blood of Melanoma Patients. Cancer Epidemiol. Biomark. 1996, 5, 293–296. [Google Scholar]
- Smith, B.; Selby, P.; Southgate, J.; Pittman, K.; Bradley, C.; Blair, G.E. Detection of Melanoma Cells in Peripheral Blood by Means of Reverse Transcriptase and Polymerase Chain Reaction. Lancet Lond. Engl. 1991, 338, 1227–1229. [Google Scholar] [CrossRef]
- Forthun, R.B.; Hovland, R.; Schuster, C.; Puntervoll, H.; Brodal, H.P.; Namløs, H.M.; Aasheim, L.B.; Meza-Zepeda, L.A.; Gjertsen, B.T.; Knappskog, S.; et al. CtDNA Detected by DdPCR Reveals Changes in Tumour Load in Metastatic Malignant Melanoma Treated with Bevacizumab. Sci. Rep. 2019, 9, 17471. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Lu, H.; Garlan, F.; Taly, V. Droplet-Based Digital PCR: Application in Cancer Research. Adv. Clin. Chem. 2017, 79, 43–91. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.H.; Rapini, R.P. Acral Lentiginous Melanoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.-F. Droplet-Based Digital PCR and next Generation Sequencing for Monitoring Circulating Tumor DNA: A Cancer Diagnostic Perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Knuever, J.; Weiss, J.; Persa, O.-D.; Kreuzer, K.; Mauch, C.; Hallek, M.; Schlaak, M. The Use of Circulating Cell-Free Tumor DNA in Routine Diagnostics of Metastatic Melanoma Patients. Sci. Rep. 2020, 10, 4940. [Google Scholar] [CrossRef]
- Mosko, M.J.; Nakorchevsky, A.A.; Flores, E.; Metzler, H.; Ehrich, M.; van den Boom, D.J.; Sherwood, J.L.; Nygren, A.O.H. Ultrasensitive Detection of Multiplexed Somatic Mutations Using MALDI-TOF Mass Spectrometry. J. Mol. Diagn. JMD 2016, 18, 23–31. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Hrebien, S.; Beaney, M.; Fribbens, C.; Garcia-Murillas, I.; Jiang, J.; Li, Y.; Huang Bartlett, C.; André, F.; Loibl, S.; et al. Comparison of BEAMing and Droplet Digital PCR for Circulating Tumor DNA Analysis. Clin. Chem. 2019, 65, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring Melanoma Using Circulating Free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1–14. [Google Scholar] [CrossRef]
- Váraljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.-M.; Horn, P.A.; von Neuhoff, N.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2020, 3, PO.18.00229. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.C.; Calapre, L.; Pereira, M.R.; Giardina, T.; Robinson, C.; Khattak, M.A.; Meniawy, T.M.; Pritchard, A.L.; Hayward, N.K.; Amanuel, B.; et al. Sensitive Droplet Digital PCR Method for Detection of TERT Promoter Mutations in Cell Free DNA from Patients with Metastatic Melanoma. Oncotarget 2017, 8, 78890–78900. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhao, H. Next-Generation Sequencing in Liquid Biopsy: Cancer Screening and Early Detection. Hum. Genomics 2019, 13, 34. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Strbenac, D.; Yang, J.Y.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Spillane, A.J.; Stretch, J.R.; Saw, R.P.M.; et al. Analysis of the Whole-Exome Sequencing of Tumor and Circulating Tumor DNA in Metastatic Melanoma. Cancers 2019, 11, 1905. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Nazer, M.; Ahmed, Z.; Kam, A.E.; Pluard, T.J.; Bahaj, W.; Levy, M.; Kuzel, T.M.; Hayden, D.M.; Pappas, S.G.; et al. Matched Whole-Genome Sequencing (WGS) and Whole-Exome Sequencing (WES) of Tumor Tissue with Circulating Tumor DNA (CtDNA) Analysis: Complementary Modalities in Clinical Practice. Cancers 2019, 11, 1399. [Google Scholar] [CrossRef]
- Wee, E.J.H.; Wang, Y.; Tsao, S.C.-H.; Trau, M. Simple, Sensitive and Accurate Multiplex Detection of Clinically Important Melanoma DNA Mutations in Circulating Tumour DNA with SERS Nanotags. Theranostics 2016, 6, 1506–1513. [Google Scholar] [CrossRef]
- Gorges, K.; Wiltfang, L.; Gorges, T.M.; Sartori, A.; Hildebrandt, L.; Keller, L.; Volkmer, B.; Peine, S.; Babayan, A.; Moll, I.; et al. Intra-Patient Heterogeneity of Circulating Tumor Cells and Circulating Tumor DNA in Blood of Melanoma Patients. Cancers 2019, 11, 1685. [Google Scholar] [CrossRef]
- Salvianti, F.; Massi, D.; De Giorgi, V.; Gori, A.; Pazzagli, M.; Pinzani, P. Evaluation of the Liquid Biopsy for the Detection of BRAFV600E Mutation in Metastatic Melanoma Patients. Cancer Biomark. Sect. Dis. Markers 2019, 26, 271–279. [Google Scholar] [CrossRef]
- Nanou, A.; Miller, M.C.; Zeune, L.L.; de Wit, S.; Punt, C.J.A.; Groen, H.J.M.; Hayes, D.F.; de Bono, J.S.; Terstappen, L.W.M.M. Tumour-Derived Extracellular Vesicles in Blood of Metastatic Cancer Patients Associate with Overall Survival. Br. J. Cancer 2020, 122, 801–811. [Google Scholar] [CrossRef]
- Shu, S.L.; Yang, Y.; Allen, C.L.; Hurley, E.; Tung, K.H.; Minderman, H.; Wu, Y.; Ernstoff, M.S. Purity and Yield of Melanoma Exosomes Are Dependent on Isolation Method. J. Extracell. Vesicles 2020, 9, 1692401. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Xu, R.; Ji, H.; Tauro, B.J.; Simpson, R.J. A Protocol for Exosome Isolation and Characterization: Evaluation of Ultracentrifugation, Density-Gradient Separation, and Immunoaffinity Capture Methods. Methods Mol. Biol. Clifton NJ 2015, 1295, 179–209. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Sidhom, K.; Obi, P.O.; Saleem, A. A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-Step Isolation of Extracellular Vesicles by Size-Exclusion Chromatography. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef]
- Vergauwen, G.; Dhondt, B.; Van Deun, J.; De Smedt, E.; Berx, G.; Timmerman, E.; Gevaert, K.; Miinalainen, I.; Cocquyt, V.; Braems, G.; et al. Confounding Factors of Ultrafiltration and Protein Analysis in Extracellular Vesicle Research. Sci. Rep. 2017, 7, 2704. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-T.; Purcell, E.; Palacios-Rolston, C.; Lo, T.-W.; Ramnath, N.; Jolly, S.; Nagrath, S. Isolation and Profiling of Circulating Tumor-Associated Exosomes Using Extracellular Vesicular Lipid-Protein Binding Affinity Based Microfluidic Device. Small Weinh. Bergstr. Ger. 2019, 15, e1903600. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High Levels of Exosomes Expressing CD63 and Caveolin-1 in Plasma of Melanoma Patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-Based Isolation of Melanoma Cell-Derived Exosomes from Plasma of Patients with Melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef]
- Ulmer, A.; Schmidt-Kittler, O.; Fischer, J.; Ellwanger, U.; Rassner, G.; Riethmüller, G.; Fierlbeck, G.; Klein, C.A. Immunomagnetic Enrichment, Genomic Characterization, and Prognostic Impact of Circulating Melanoma Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 531–537. [Google Scholar] [CrossRef]
- Visús, C.; Andres, R.; Mayordomo, J.I.; Martinez-Lorenzo, M.J.; Murillo, L.; Sáez-Gutiérrez, B.; Diestre, C.; Marcos, I.; Astier, P.; Godino, J.; et al. Prognostic Role of Circulating Melanoma Cells Detected by Reverse Transcriptase-Polymerase Chain Reaction for Tyrosinase MRNA in Patients with Melanoma. Melanoma Res. 2007, 17, 83–89. [Google Scholar] [CrossRef]
- Koyanagi, K.; Kuo, C.; Nakagawa, T.; Mori, T.; Ueno, H.; Lorico, A.R.; Wang, H.-J.; Hseuh, E.; O’Day, S.J.; Hoon, D.S.B. Multimarker Quantitative Real-Time PCR Detection of Circulating Melanoma Cells in Peripheral Blood: Relation to Disease Stage in Melanoma Patients. Clin. Chem. 2005, 51, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Leiter, U.; Ellwanger, U.; Blaheta, H.-J.; Meier, F.; Rassner, G.; Schittek, B. Diagnostic Value and Prognostic Significance of Protein S-100beta, Melanoma-Inhibitory Activity, and Tyrosinase/MART-1 Reverse Transcription-Polymerase Chain Reaction in the Follow-up of High-Risk Melanoma Patients. Cancer 2003, 97, 1737–1745. [Google Scholar] [CrossRef]
- Brownbridge, G.G.; Gold, J.; Edward, M.; MacKie, R.M. Evaluation of the Use of Tyrosinase-Specific and MelanA/MART-1-Specific Reverse Transcriptase-Coupled--Polymerase Chain Reaction to Detect Melanoma Cells in Peripheral Blood Samples from 299 Patients with Malignant Melanoma. Br. J. Dermatol. 2001, 144, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Khoja, L.; Lorigan, P.; Dive, C.; Keilholz, U.; Fusi, A. Circulating Tumour Cells as Tumour Biomarkers in Melanoma: Detection Methods and Clinical Relevance. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hoshimoto, S.; Faries, M.B.; Morton, D.L.; Shingai, T.; Kuo, C.; Wang, H.-J.; Elashoff, R.; Mozzillo, N.; Kelley, M.C.; Thompson, J.F.; et al. Assessment of Prognostic Circulating Tumor Cells in a Phase III Trial of Adjuvant Immunotherapy after Complete Resection of Stage IV Melanoma. Ann. Surg. 2012, 255, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, P.; Osella-Abate, S.; Cappello, N.; Ortoncelli, M.; Nardò, T.; Fierro, M.T.; Cavallo, F.; Savoia, P.; Bernengo, M.G. Prognostic Relevance of Baseline and Sequential Peripheral Blood Tyrosinase Expression in 200 Consecutive Advanced Metastatic Melanoma Patients. Melanoma Res. 2007, 17, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Boldin, I.; Langmann, G.; Richtig, E.; Schwantzer, G.; Ardjomand, N.; Wegscheider, B.; El-Shabrawi, Y. Five-Year Results of Prognostic Value of Tyrosinase in Peripheral Blood of Uveal Melanoma Patients. Melanoma Res. 2005, 15, 503–507. [Google Scholar] [CrossRef]
- Mazzini, C.; Pinzani, P.; Salvianti, F.; Scatena, C.; Paglierani, M.; Ucci, F.; Pazzagli, M.; Massi, D. Circulating Tumor Cells Detection and Counting in Uveal Melanomas by a Filtration-Based Method. Cancers 2014, 6, 323–332. [Google Scholar] [CrossRef]
- Pinzani, P.; Mazzini, C.; Salvianti, F.; Massi, D.; Grifoni, R.; Paoletti, C.; Ucci, F.; Molinara, E.; Orlando, C.; Pazzagli, M.; et al. Tyrosinase MRNA Levels in the Blood of Uveal Melanoma Patients: Correlation with the Number of Circulating Tumor Cells and Tumor Progression. Melanoma Res. 2010, 20, 303–310. [Google Scholar] [CrossRef]
- Schuster, R.; Bechrakis, N.E.; Stroux, A.; Busse, A.; Schmittel, A.; Thiel, E.; Foerster, M.H.; Keilholz, U. Prognostic Relevance of Circulating Tumor Cells in Metastatic Uveal Melanoma. Oncology 2011, 80, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Bechrakis, N.E.; Stroux, A.; Busse, A.; Schmittel, A.; Scheibenbogen, C.; Thiel, E.; Foerster, M.H.; Keilholz, U. Circulating Tumor Cells as Prognostic Factor for Distant Metastases and Survival in Patients with Primary Uveal Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Del Fiore, P.; Guarnieri, L.; Scalerta, R.; Foletto, M.; Chiarion, V.; Pilati, P.; Nitti, D.; Lise, M.; Rossi, C.R. Molecular Detection of Circulating Tumor Cells Is an Independent Prognostic Factor in Patients with High-Risk Cutaneous Melanoma. Int. J. Cancer 2004, 111, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Reid, A.L.; Bowyer, S.; Calapre, L.; Siew, K.; Pearce, R.; Cowell, L.; Frank, M.H.; Millward, M.; Ziman, M. Circulating Melanoma Cell Subpopulations: Their Heterogeneity and Differential Responses to Treatment. J. Investig. Dermatol. 2015, 135, 2040–2048. [Google Scholar] [CrossRef]
- Kupas, V.; Weishaupt, C.; Siepmann, D.; Kaserer, M.-L.; Eickelmann, M.; Metze, D.; Luger, T.A.; Beissert, S.; Loser, K. RANK Is Expressed in Metastatic Melanoma and Highly Upregulated on Melanoma-Initiating Cells. J. Investig. Dermatol. 2011, 131, 944–955. [Google Scholar] [CrossRef]
- Mocellin, S.; Hoon, D.; Ambrosi, A.; Nitti, D.; Rossi, C.R. The Prognostic Value of Circulating Tumor Cells in Patients with Melanoma: A Systematic Review and Meta-Analysis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 4605–4613. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Mitra, D.; Sullivan, R.J.; Wittner, B.S.; Kimura, A.M.; Pan, S.; Hoang, M.P.; Brannigan, B.W.; Lawrence, D.P.; Flaherty, K.T.; et al. Isolation and Molecular Characterization of Circulating Melanoma Cells. Cell Rep. 2014, 7, 645–653. [Google Scholar] [CrossRef]
- Xu, X.; Zhong, J.F. Circulating Tumor Cells and Melanoma Progression. J. Investig. Dermatol. 2010, 130, 2349–2351. [Google Scholar] [CrossRef]
- Spindler, K.-L.G.; Appelt, A.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Cell-Free DNA in Healthy Individuals, Noncancerous Disease and Strong Prognostic Value in Colorectal Cancer. Int. J. Cancer 2014, 135, 2984–2991. [Google Scholar] [CrossRef]
- Haselmann, V.; Gebhardt, C.; Brechtel, I.; Duda, A.; Czerwinski, C.; Sucker, A.; Holland-Letz, T.; Utikal, J.; Schadendorf, D.; Neumaier, M. Liquid Profiling of Circulating Tumor DNA in Plasma of Melanoma Patients for Companion Diagnostics and Monitoring of BRAF Inhibitor Therapy. Clin. Chem. 2018, 64, 830–842. [Google Scholar] [CrossRef]
- Schreuer, M.; Meersseman, G.; Van Den Herrewegen, S.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative Assessment of BRAF V600 Mutant Circulating Cell-Free Tumor DNA as a Tool for Therapeutic Monitoring in Metastatic Melanoma Patients Treated with BRAF/MEK Inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative Cell-Free Circulating BRAFV600E Mutation Analysis by Use of Droplet Digital PCR in the Follow-up of Patients with Melanoma Being Treated with BRAF Inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Menzies, A.M.; Carlino, M.S.; McEvoy, A.C.; Sandhu, S.; Weppler, A.M.; Diefenbach, R.J.; Dawson, S.-J.; Kefford, R.F.; Millward, M.J.; et al. Longitudinal Monitoring of CtDNA in Patients with Melanoma and Brain Metastases Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 4064–4071. [Google Scholar] [CrossRef] [PubMed]
- Váraljai, R.; Elouali, S.; Lueong, S.S.; Wistuba-Hamprecht, K.; Seremet, T.; Siveke, J.T.; Becker, J.C.; Sucker, A.; Paschen, A.; Horn, P.A.; et al. The Predictive and Prognostic Significance of Cell-Free DNA Concentration in Melanoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2021, 35, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Methylated Circulating Tumor DNA as a Biomarker in Cutaneous Melanoma. Melanoma Manag. 2020, 7, MMT46. [Google Scholar] [CrossRef]
- Hoon, D.S.B.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling Epigenetic Inactivation of Tumor Suppressor Genes in Tumors and Plasma from Cutaneous Melanoma Patients. Oncogene 2004, 23, 4014–4022. [Google Scholar] [CrossRef]
- Marini, A.; Mirmohammadsadegh, A.; Nambiar, S.; Gustrau, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Inactivation of Tumor Suppressor Genes in Serum of Patients with Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Salvianti, F.; Orlando, C.; Massi, D.; De Giorgi, V.; Grazzini, M.; Pazzagli, M.; Pinzani, P. Tumor-Related Methylated Cell-Free DNA and Circulating Tumor Cells in Melanoma. Front. Mol. Biosci. 2015, 2, 76. [Google Scholar] [CrossRef]
- Mori, T.; O’Day, S.J.; Umetani, N.; Martinez, S.R.; Kitago, M.; Koyanagi, K.; Kuo, C.; Takeshima, T.-L.; Milford, R.; Wang, H.-J.; et al. Predictive Utility of Circulating Methylated DNA in Serum of Melanoma Patients Receiving Biochemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 9351–9358. [Google Scholar] [CrossRef]
- Koyanagi, K.; Mori, T.; O’Day, S.J.; Martinez, S.R.; Wang, H.-J.; Hoon, D.S.B. Association of Circulating Tumor Cells with Serum Tumor-Related Methylated DNA in Peripheral Blood of Melanoma Patients. Cancer Res. 2006, 66, 6111–6117. [Google Scholar] [CrossRef]
- Hoshimoto, S.; Kuo, C.T.; Chong, K.K.; Takeshima, T.-L.; Takei, Y.; Li, M.W.; Huang, S.K.; Sim, M.-S.; Morton, D.L.; Hoon, D.S.B. AIM1 and LINE-1 Epigenetic Aberrations in Tumor and Serum Relate to Melanoma Progression and Disease Outcome. J. Investig. Dermatol. 2012, 132, 1689–1697. [Google Scholar] [CrossRef]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma Cell-Derived Exosomes in Plasma of Melanoma Patients Suppress Functions of Immune Effector Cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.-P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the Evolution of Circulating Exosomal-PD-L1 to Monitor Melanoma Patients. J. Extracell. Vesicles 2020, 9, 1710899. [Google Scholar] [CrossRef]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating Melanoma Exosomes as Diagnostic and Prognosis Biomarkers. Clin. Chim. Acta Int. J. Clin. Chem. 2016, 454, 28–32. [Google Scholar] [CrossRef]
- Margue, C.; Reinsbach, S.; Philippidou, D.; Beaume, N.; Walters, C.; Schneider, J.G.; Nashan, D.; Behrmann, I.; Kreis, S. Comparison of a Healthy MiRNome with Melanoma Patient MiRNomes: Are MicroRNAs Suitable Serum Biomarkers for Cancer? Oncotarget 2015, 6, 12110–12127. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Grossmann, K.F.; Cassidy, P.B.; Yang, C.H.; Fan, M.; Kopelovich, L.; Leachman, S.A.; Pfeffer, L.M. Detection of Exosomal MiRNAs in the Plasma of Melanoma Patients. J. Clin. Med. 2015, 4, 2012–2027. [Google Scholar] [CrossRef]
- Tengda, L.; Shuping, L.; Mingli, G.; Jie, G.; Yun, L.; Weiwei, Z.; Anmei, D. Serum Exosomal MicroRNAs as Potent Circulating Biomarkers for Melanoma. Melanoma Res. 2018, 28, 295–303. [Google Scholar] [CrossRef]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; González, A. Study of Circulating MicroRNA-125b Levels in Serum Exosomes in Advanced Melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.; Barbagallo, C.; Statello, L.; Caltabiano, R.; Russo, A.; Puzzo, L.; Avitabile, T.; Longo, A.; Toro, M.D.; Barbagallo, D.; et al. MiRNA Profiling in Vitreous Humor, Vitreal Exosomes and Serum from Uveal Melanoma Patients: Pathological and Diagnostic Implications. Cancer Biol. Ther. 2015, 16, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- García-Silva, S.; Benito-Martín, A.; Sánchez-Redondo, S.; Hernández-Barranco, A.; Ximénez-Embún, P.; Nogués, L.; Mazariegos, M.S.; Brinkmann, K.; Amor López, A.; Meyer, L.; et al. Use of Extracellular Vesicles from Lymphatic Drainage as Surrogate Markers of Melanoma Progression and BRAF V600E Mutation. J. Exp. Med. 2019, 216, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Klinac, D.; Gray, E.S.; Freeman, J.B.; Reid, A.; Bowyer, S.; Millward, M.; Ziman, M. Monitoring Changes in Circulating Tumour Cells as a Prognostic Indicator of Overall Survival and Treatment Response in Patients with Metastatic Melanoma. BMC Cancer 2014, 14, 423. [Google Scholar] [CrossRef]
- Boyer, M.; Cayrefourcq, L.; Dereure, O.; Meunier, L.; Becquart, O.; Alix-Panabières, C. Clinical Relevance of Liquid Biopsy in Melanoma and Merkel Cell Carcinoma. Cancers 2020, 12, 960. [Google Scholar] [CrossRef]
- Koyanagi, K.; O’Day, S.J.; Gonzalez, R.; Lewis, K.; Robinson, W.A.; Amatruda, T.T.; Wang, H.-J.; Elashoff, R.M.; Takeuchi, H.; Umetani, N.; et al. Serial Monitoring of Circulating Melanoma Cells during Neoadjuvant Biochemotherapy for Stage III Melanoma: Outcome Prediction in a Multicenter Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8057–8064. [Google Scholar] [CrossRef]
- Reid, A.L.; Millward, M.; Pearce, R.; Lee, M.; Frank, M.H.; Ireland, A.; Monshizadeh, L.; Rai, T.; Heenan, P.; Medic, S.; et al. Markers of Circulating Tumour Cells in the Peripheral Blood of Patients with Melanoma Correlate with Disease Recurrence and Progression. Br. J. Dermatol. 2013, 168, 85–92. [Google Scholar] [CrossRef]
- Khattak, M.A.; Reid, A.; Freeman, J.; Pereira, M.; McEvoy, A.; Lo, J.; Frank, M.H.; Meniawy, T.; Didan, A.; Spencer, I.; et al. PD-L1 Expression on Circulating Tumor Cells May Be Predictive of Response to Pembrolizumab in Advanced Melanoma: Results from a Pilot Study. The Oncologist 2020, 25, e520–e527. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.M.; Millward, M.; Ziman, M.; et al. Correlation between Circulating Tumour DNA and Metabolic Tumour Burden in Metastatic Melanoma Patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef] [PubMed]
- Braune, J.; Keller, L.; Schiller, F.; Graf, E.; Rafei-Shamsabadi, D.; Wehrle, J.; Follo, M.; Philipp, U.; Hussung, S.; Pfeifer, D.; et al. Circulating Tumor DNA Allows Early Treatment Monitoring in BRAF- and NRAS-Mutant Malignant Melanoma. JCO Precis. Oncol. 2020, 20–31. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK Reactivation in Early Resistance to Dabrafenib/Trametinib Combination Therapy of BRAF-Mutant Metastatic Melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.-J.; et al. Phase II Trial (BREAK-2) of the BRAF Inhibitor Dabrafenib (GSK2118436) in Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.R.; Lee, R.J.; Garner, G.; Dhomen, N.; Lorigan, P.C.; et al. Plasma Total Cell-Free DNA (CfDNA) Is a Surrogate Biomarker for Tumour Burden and a Prognostic Biomarker for Survival in Metastatic Melanoma Patients. Eur. J. Cancer Oxf. Engl. 2018, 88, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating Tumour DNA Predicts Response to Anti-PD1 Antibodies in Metastatic Melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable Circulating Tumor DNA (CtDNA) Levels Correlate with Favorable Outcome in Metastatic Melanoma Patients Treated with Anti-PD1 Therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated With Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.A.; Tadepalli, J.S.; Shao, Y.; Zhang, Y.; Weiss, S.; Robinson, E.; Spittle, C.; Furtado, M.; Shelton, D.N.; Karlin-Neumann, G.; et al. Sensitivity of Plasma BRAFmutant and NRASmutant Cell-Free DNA Assays to Detect Metastatic Melanoma in Patients with Low RECIST Scores and Non-RECIST Disease Progression. Mol. Oncol. 2016, 10, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 Contributes to Immunosuppression and Is Associated with Anti-PD-1 Response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Del Re, M.; Marconcini, R.; Pasquini, G.; Rofi, E.; Vivaldi, C.; Bloise, F.; Restante, G.; Arrigoni, E.; Caparello, C.; Bianco, M.G.; et al. PD-L1 MRNA Expression in Plasma-Derived Exosomes Is Associated with Response to Anti-PD-1 Antibodies in Melanoma and NSCLC. Br. J. Cancer 2018, 118, 820–824. [Google Scholar] [CrossRef]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Stucci, L.S.; Ascierto, P.A.; Capone, M.; Madonna, G.; Lopalco, P.; Silvestris, F. Serum Exosomes as Predictors of Clinical Response to Ipilimumab in Metastatic Melanoma. Oncoimmunology 2018, 7, e1387706. [Google Scholar] [CrossRef]
- Svedman, F.C.; Lohcharoenkal, W.; Bottai, M.; Brage, S.E.; Sonkoly, E.; Hansson, J.; Pivarcsi, A.; Eriksson, H. Extracellular Microvesicle MicroRNAs as Predictive Biomarkers for Targeted Therapy in Metastastic Cutaneous Malignant Melanoma. PLoS ONE 2018, 13, e0206942. [Google Scholar] [CrossRef] [PubMed]
- Lunavat, T.R.; Cheng, L.; Einarsdottir, B.O.; Olofsson Bagge, R.; Veppil Muralidharan, S.; Sharples, R.A.; Lässer, C.; Gho, Y.S.; Hill, A.F.; Nilsson, J.A.; et al. BRAFV600 Inhibition Alters the MicroRNA Cargo in the Vesicular Secretome of Malignant Melanoma Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E5930–E5939. [Google Scholar] [CrossRef] [PubMed]
- Home—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 6 July 2021).
- Lianidou, E.S.; Markou, A.; Strati, A. The Role of CTCs as Tumor Biomarkers. Adv. Exp. Med. Biol. 2015, 867, 341–367. [Google Scholar] [CrossRef]
- Batth, I.S.; Mitra, A.; Manier, S.; Ghobrial, I.M.; Menter, D.; Kopetz, S.; Li, S. Circulating Tumor Markers: Harmonizing the Yin and Yang of CTCs and CtDNA for Precision Medicine. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.-F.; Pan, F.-Q.; Li, D. Expression and Clinical Significance of S100 Family Genes in Patients with Melanoma. Melanoma Res. 2019, 29, 23–29. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | NCT No. | Study Title | Cancer | Location | Participants (Active/Original) |
|---|---|---|---|---|---|
| 1. | NCT01573494 | Study of Circulating Tumor Cells Before and After Treatment in Patients With Metastatic Melanoma | Metastatic melanoma | CHU of Nice, Nice, France | 30/30 |
| 2. | NCT01558349 | Circulating Tumor Cells and Melanoma: Comparing the EPISPOT and CellSearch Techniques | Metastatic melanoma | CHU of Montpellier, Montpellier, France CHU of Nîmes, Nîmes, France | 73/82 |
| 3. | NCT01528774 | Culture and Characterization of Circulating Tumor Cells (CTC) in Melanoma and Other Cancers | Melanoma and other cancers | Comprehensive Cancer Centers of Nevada Las Vegas, Nevada, United States | 150/1000 |
| 4. | NCT02133222 | Circulating Cell-free DNA in Metastatic Melanoma Patient: Mutational Analyses in Consecutive Measurement Before and After Chemotherapy | Metastatic melanoma | CHU of Nice Nice, France | 22/20 |
| 5. | NCT03007823 | High-Activity Natural Killer Immunotherapy for Small Metastases of Melanoma | Metastatic melanoma | Fuda Cancer Institute of Fuda Cancer Hospital Guangzhou, Guangdong, China | 20/20 |
| 6. | NCT02768207 | A Study to Detect V-Raf Murine Sarcoma Viral Oncogene Homolog B1 (BRAF) V600 Mutation on Cell-Free Deoxyribonucleic Acid (cfDNA) from Plasma in Participants with Advanced Melanoma | Metastatic melanoma | UZ Brussel, Brussel, Belgium Institute Jules Bordet, Brussel, Belgium CHIREC Edith Cavell, Brussel, Belgium (and 11 more…) | 40/208 |
| 7. | NCT02251314 | Use of Exome Sequence Analysis and Circulating Tumor in Assessing Tumor Heterogeneity in BRAF Mutant Melanoma | BRAF-mutated melanoma | Princess Margaret Cancer Centre Toronto, Ontario, Canada | 12/6 |
| No. | NCT No. | Study Title | Cancer | Location | Participants (Estimated) |
|---|---|---|---|---|---|
| 1. | NCT01776905 | In Vivo Real-Time Detection of Circulating Melanoma Cells | Melanoma stage I–IV | University of Arkansas for Medical Sciences Little Rock, Arkansas, United States | 75 |
| 2. | NCT03808441 | CAcTUS—Circulating Tumour DNA Guided Switch | Metastatic melanoma | The Christie NHS Foundation Trust Manchester, United Kingdom | 40 |
| 3. | NCT03416933 | Therapeutic Drug Monitoring of BRAF-Mutated Advanced Melanoma | Metastatic melanoma | Hôpital de Mercy, Ars-Laquenexy, Fr CHRU Nancy, Vandœuvre-lès-Nancy, Fr Institut de Cancérologie de Lorraine (ICL), Vandœuvre-lès-Nancy, Fr | 35 |
| 4. | NCT03797053 | Ex Vivo Expansion of Circulating Tumor Cells as a Model for Cancer Predictive Pharmacology | Melanoma stage III–IV | Saint-Louis Hospital Paris, France | 450 |
| 5. | NCT01565837 | Concurrent Ipilimumab and Stereotactic Ablative Radiation Therapy (SART) for Oligometastatic but Unresectable Melanoma | Melanoma stage III–IV | Comprehensive Cancer Centers of Nevada Las Vegas, Nevada, United States | 50 |
| 6. | NCT02862743 | Molecular Characterization of Advanced Stage Melanoma by Blood Sampling | Metastatic melanoma | CHU of Reims Reims, France | 80 |
| 7. | NCT01878396 | Circulating Melanoma Cells in Metastatic Patients Treated with Selective BRAF Inhibitors | Metastatic melanoma | Istituto Oncologico Veneto IRCCS Padova, Italy | 200 |
| 8. | NCT02583516 | Clinical Trial to Evaluate the Efficacy of Vemurafenib in Combination with Cobimetinib (Continuous and Intermittent) in BRAFV600-Mutation-Positive Patients With Unresectable Locally Advanced or Metastatic Melanoma | Melanoma stage III–IV | Hospital Universitario Donostia, San Sebastián, Guipuzcoa, Spain Hospital General Universitario Santa Lucía, Cartagena, Murcia, Spain Hospital Clínic de Barcelona, Barcelona, Spain (and 15 more…) | 70 |
| 9. | NCT03175432 | Bevacizumab and Atezolizumab with or without Cobimetinib in Treating Patients with Untreated Melanoma Brain Metastases | Metastatic melanoma | MD Anderson Cancer Center Houston, Texas, United States | 60 |
| 10. | NCT03873818 | Low-Dose Ipilimumab With Pembrolizumab in Treating Patients with Melanoma that has Spread to the Brain | Metastatic melanoma and other cancers | MD Anderson Cancer Center Houston, Texas, United States | 30 |
| 11. | NCT02537600 | Vemurafenib and Cobimetinib Combination in BRAF Mutated Melanoma with Brain Metastasis | Metastatic melanoma | CHU of Bordeaux, Bordeaux, France CHU Ambroise Paré, Boulogne, France CHU Brest Hôpital Morvan, Brest, France (and 14 more…) | 43 |
| 12. | NCT02673970 | Biomarkers for the Activity of Immune Checkpoint Inhibitor Therapy in Patients with Advanced Melanoma | Metastatic melanoma | UZ Brussel Jette, Brabant, Belgium | 200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. https://doi.org/10.3390/ijms22189714
Kamińska P, Buszka K, Zabel M, Nowicki M, Alix-Panabières C, Budna-Tukan J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. International Journal of Molecular Sciences. 2021; 22(18):9714. https://doi.org/10.3390/ijms22189714
Chicago/Turabian StyleKamińska, Paula, Karolina Buszka, Maciej Zabel, Michał Nowicki, Catherine Alix-Panabières, and Joanna Budna-Tukan. 2021. "Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring" International Journal of Molecular Sciences 22, no. 18: 9714. https://doi.org/10.3390/ijms22189714
APA StyleKamińska, P., Buszka, K., Zabel, M., Nowicki, M., Alix-Panabières, C., & Budna-Tukan, J. (2021). Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. International Journal of Molecular Sciences, 22(18), 9714. https://doi.org/10.3390/ijms22189714