
Inferred Ancestral Origin of Cancer Cell Lines Associates with Differential Drug Response

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
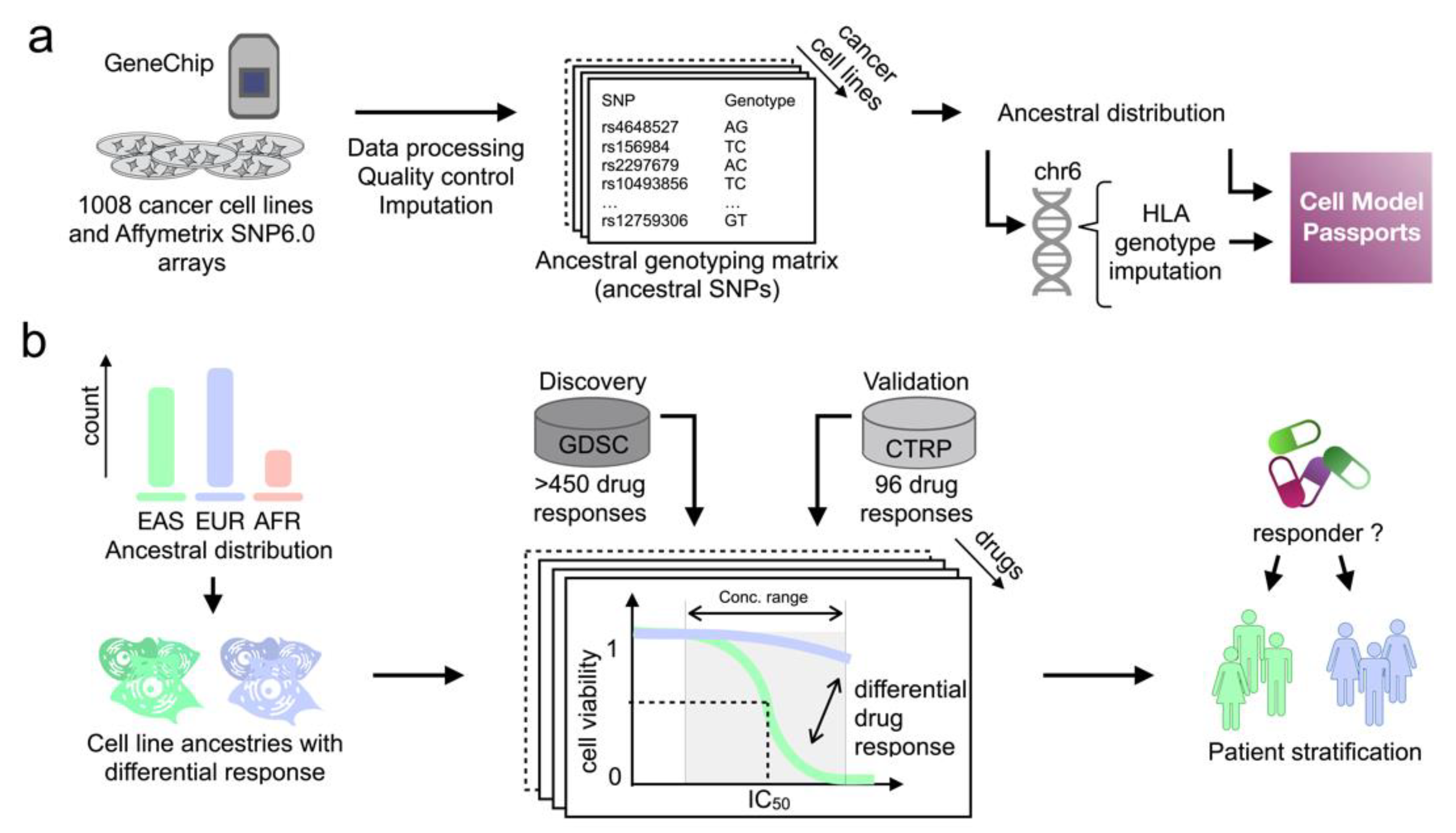
2.1. Inferred Ancestry of Cancer Cell Lines Is Conserved
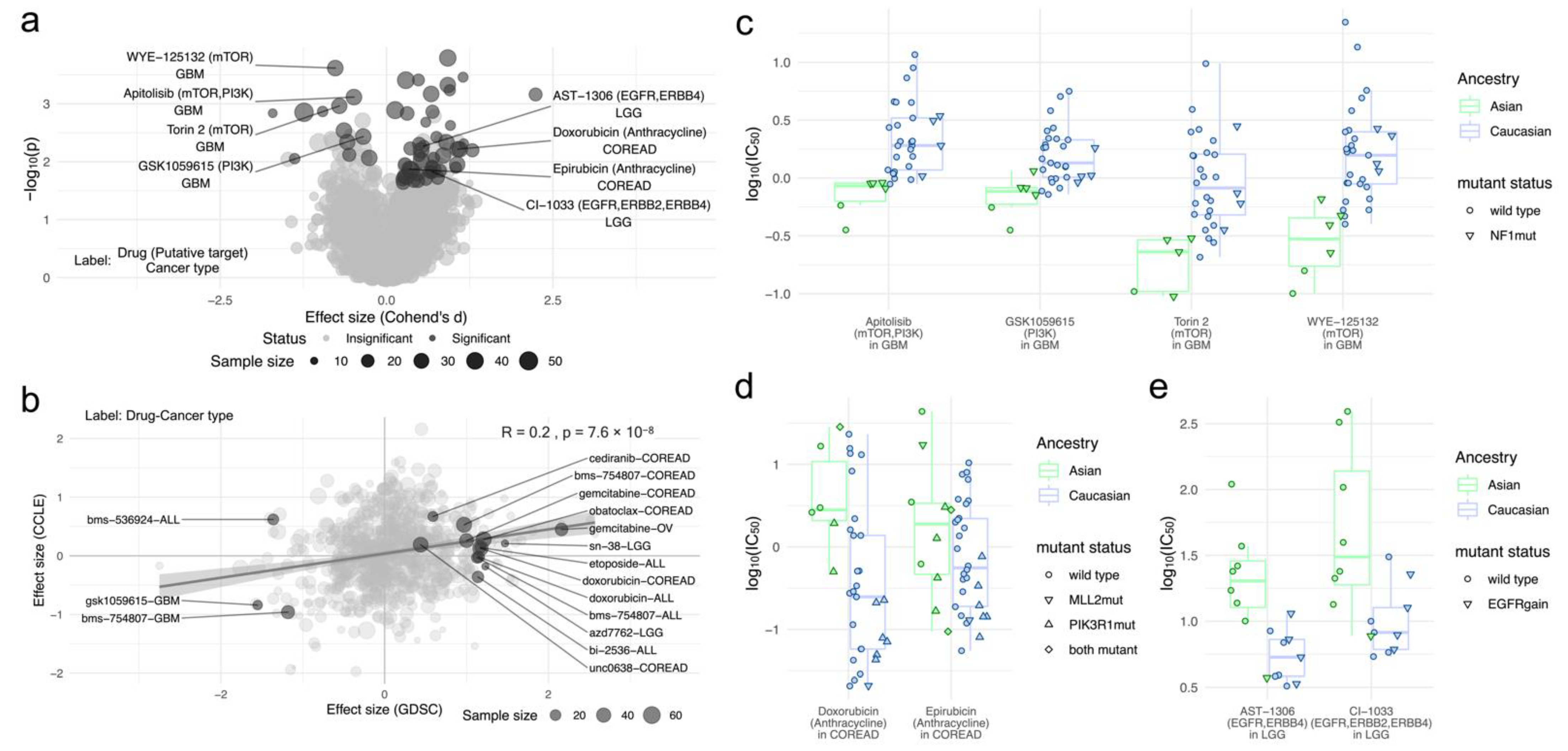
2.2. Differential Drug Responses between Asian and Caucasian Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Data Availability
4.2. Quality Control
4.3. Phasing and Imputation
4.4. Inference of Ancestry
4.5. HLA Prediction
4.6. Ancestry Biomarker Analysis
4.7. Enrichment of Drug Targets
4.8. Enrichment of Somatic Driver Genes in Ancestries
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Code Availability
References
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.K.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barreyina, J.; Gelfand, E.T.; Bielski, G.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Phar-macogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatrath, A.; Ratan, A.; Dutta, A. Germline Variants That Affect Tumor Progression. Trends Genet. 2021, 37, 433–443. [Google Scholar] [CrossRef]
- Chatrath, A.; Przanowska, R.; Kiran, S.; Su, Z.; Saha, S.; Wilson, B.; Tsunematsu, T.; Ahn, J.-H.; Lee, K.Y.; Paulsen, T.; et al. The pan-cancer landscape of prognostic germline variants in 10,582 patients. Genome Med. 2020, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Qing, T.; Mohsen, H.; Marczyk, M.; Ye, Y.; O’Meara, T.; Zhao, H.; Townsend, J.P.; Gerstein, M.; Hatzis, C.; Kluger, Y.; et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 2020, 11, 2438. [Google Scholar] [CrossRef]
- Menden, M.P.; Casale, F.P.; Stephan, J.; Bignell, G.R.; Iorio, F.; McDermott, U.; Garnett, M.J.; Saez-Rodriguez, J.; Stegle, O. The germline genetic component of drug sensitivity in cancer cell lines. Nat. Commun. 2018, 9, 3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Dotto, G.-P. Racial Differences in Cancer Susceptibility and Survival: More Than the Color of the Skin? Trends Cancer Res. 2017, 3, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.S.; Galanter, J.; Thakur, N.; Pino-Yanes, M.; Barcelo, N.E.; White, M.J.; de Bruin, D.M.; Greenblatt, R.M.; Bibbins-Domingo, K.; Wu, A.H.B.; et al. Diversity in Clinical and Biomedical Research: A Promise Yet to Be Fulfilled. PLoS Med. 2015, 12, e1001918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, J.N.; Kidd, K.K.; Kidd, J.R.; Zhao, H. Selecting SNPs to identify ancestry. Ann. Hum. Genet. 2011, 75, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Shen, J.; Cox, C.; Wakefield, J.C.; Ehm, M.G.; Nelson, M.R.; Weir, B.S. HIBAG—HLA genotype imputation with attribute bagging. Pharm. J. 2014, 14, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, A.; Knepper, T.C.; Merenda, C.; Mendoza, M.; McLeod, H.L.; Bull, J.; Zhang, L.; Pacanowski, M. Demographic Composition of Select Onco-logic New Molecular Entities Approved by the FDA Between 2008 and 2017. Clin. Pharmacol. Ther. 2018, 104, 940–948. [Google Scholar] [CrossRef] [Green Version]
- Kamineni, A.; Williams, M.A.; Schwartz, S.M.; Cook, L.S.; Weiss, N.S. The incidence of gastric carcinoma in Asian migrants to the United States and their descendants. Cancer Causes Control. 1999, 10, 77–83. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, K.; Yang, L.; Ding, G.; Li, H. Racial Differences in Esophageal Squamous Cell Carcinoma: Incidence and Molecular Features. Biomed. Res. Int. 2017, 2017, 1204082. [Google Scholar] [CrossRef]
- Chen, Z.; Ren, Y.; Du, X.L.; Yang, J.; Shen, Y.; Li, S.; Wu, Y.; Lv, M.; Dong, D.; Li, E.; et al. Incidence and survival differences in esophageal cancer among ethnic groups in the United States. Oncotarget 2017, 8, 47037–47051. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.C.; McKinley, M.; Gupta, S.; Peek, R.M., Jr.; Martinez, M.E.; Gomez, S.L. Population-Based Analysis of Differences in Gastric Cancer Incidence Among Races and Ethnicities in Individuals Age 50 Years and Older. Gastroenterology 2020, 159, 1705–1714.e2. [Google Scholar] [CrossRef]
- Aggarwal, R.; Grabowsky, J.; Strait, N.; Cockerill, A.; Munster, P. Impact of patient ethnicity on the metabolic and immunologic effects of PI3K–mTOR pathway inhibition in patients with solid tumor malignancies. Cancer Chemother. Pharmacol. 2014, 74, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Chong, D.Q.; Toh, X.Y.; Ho, I.A.W.; Sia, K.C.; Newman, J.P.; Yulyana, Y.; Ng, W.-H.; Lai, S.H.; Ho, M.M.F.; Dinesh, N.; et al. Combined treatment of Nimotuzumab and rapamycin is effective against temozolomide-resistant human gliomas regardless of the EGFR mutation status. BMC Cancer 2015, 15, 255. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, P.H.; Dolan, M.E. Cancer pharmacoethnicity: Ethnic differences in susceptibility to the effects of chemotherapy. Clin. Cancer Res. 2009, 15, 4806–4814. [Google Scholar] [CrossRef] [Green Version]
- Calvo, E.; Baselga, J. Ethnic differences in response to epidermal growth factor receptor tyrosine kinase inhibitors. J. Clin. Oncol. 2006, 24, 2158–2163. [Google Scholar] [CrossRef]
- Zavala, V.A.; Bracci, P.M.; Carethers, J.M.; Carvajal-Carmona, L.; Coggins, N.B.; Cruz-Correa, M.R.; Davis, M.; de Smith, A.J.; Dutil, J.; Figueiredo, J.C.; et al. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer 2021, 124, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.L.; Davis, K.J.; Wingo, P.A.; Ries, L.A.; Heath, C.W., Jr. Cancer statistics by race and ethnicity. CA Cancer J. Clin. 1998, 48, 31–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, D.G.; Cassidy, J.; Clarke, S.J.; Cunningham, D.; Van Cutsem, E.; Hoff, P.M.; Rothenberg, M.L.; Saltz, L.B.; Schmoll, H.-J.; Allegra, C.; et al. Potential regional differences for the tolera-bility profiles of fluoropyrimidines. J. Clin. Oncol. 2008, 26, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.; Chua, D.; Yao, Y.; Soo, R.A.; Garrett, K.; Zeps, N.; Platell, C.; Minamoto, T.; Kawakami, K.; Iacopetta, B.; et al. Can population differences in chemotherapy outcomes be inferred from differences in pharmacogenetic frequencies? Pharm. J. 2013, 13, 423–429. [Google Scholar] [CrossRef]
- Koual, M.; Tomkiewicz, C.; Cano-Sancho, G.; Antignac, J.-P.; Bats, A.-S.; Coumoul, X. Environmental chemicals, breast cancer progression and drug resistance. Environ. Health 2020, 19, 117. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Associ-ation and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplo-type Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, P.B.H.; Ohnmacht, A.J.; Sharifli, S.; Garnett, M.J.; Menden, M.P. Inferred Ancestral Origin of Cancer Cell Lines Associates with Differential Drug Response. Int. J. Mol. Sci. 2021, 22, 10135. https://doi.org/10.3390/ijms221810135
Nguyen PBH, Ohnmacht AJ, Sharifli S, Garnett MJ, Menden MP. Inferred Ancestral Origin of Cancer Cell Lines Associates with Differential Drug Response. International Journal of Molecular Sciences. 2021; 22(18):10135. https://doi.org/10.3390/ijms221810135
Chicago/Turabian StyleNguyen, Phong B. H., Alexander J. Ohnmacht, Samir Sharifli, Mathew J. Garnett, and Michael P. Menden. 2021. "Inferred Ancestral Origin of Cancer Cell Lines Associates with Differential Drug Response" International Journal of Molecular Sciences 22, no. 18: 10135. https://doi.org/10.3390/ijms221810135
APA StyleNguyen, P. B. H., Ohnmacht, A. J., Sharifli, S., Garnett, M. J., & Menden, M. P. (2021). Inferred Ancestral Origin of Cancer Cell Lines Associates with Differential Drug Response. International Journal of Molecular Sciences, 22(18), 10135. https://doi.org/10.3390/ijms221810135