
Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
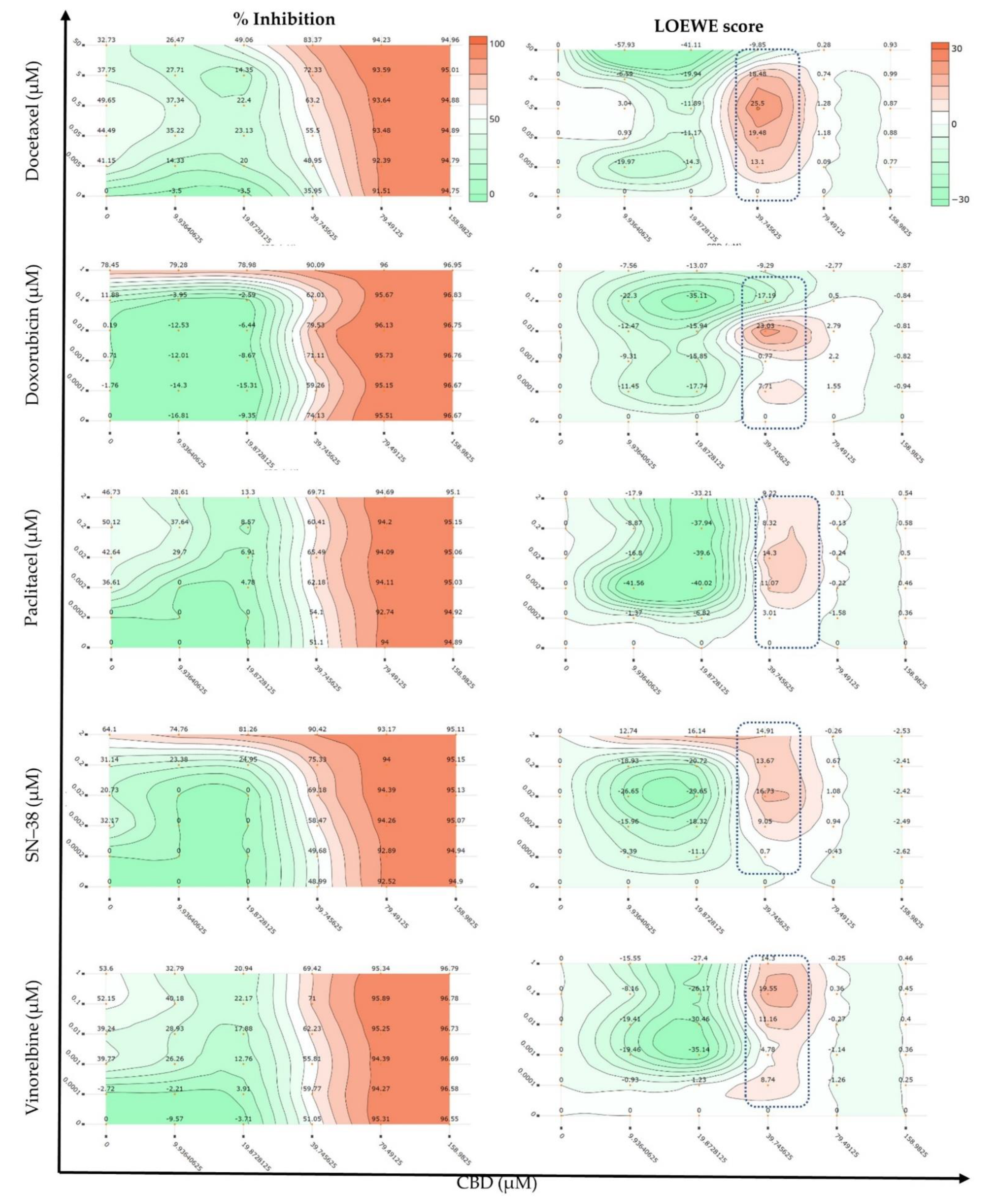
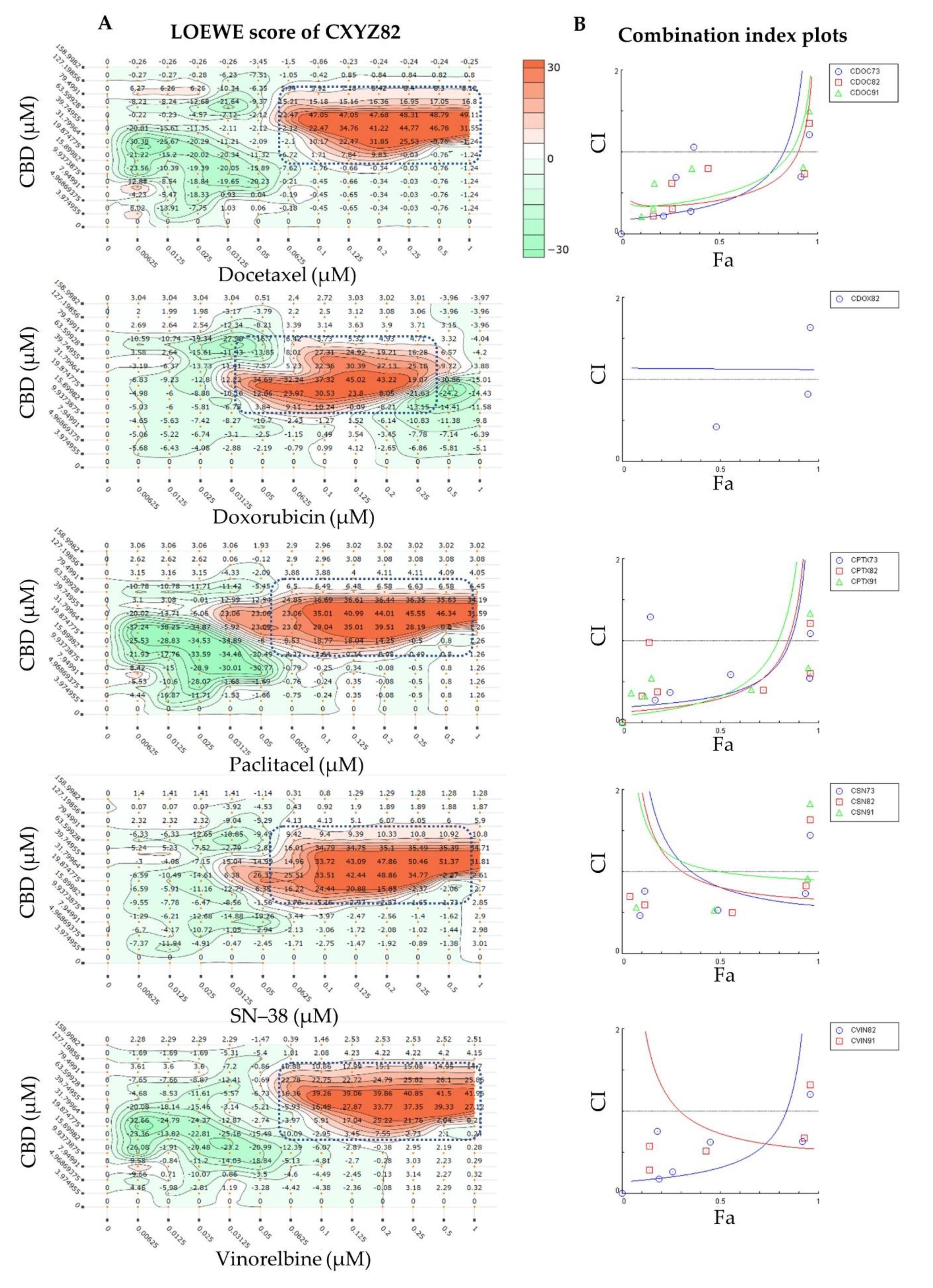
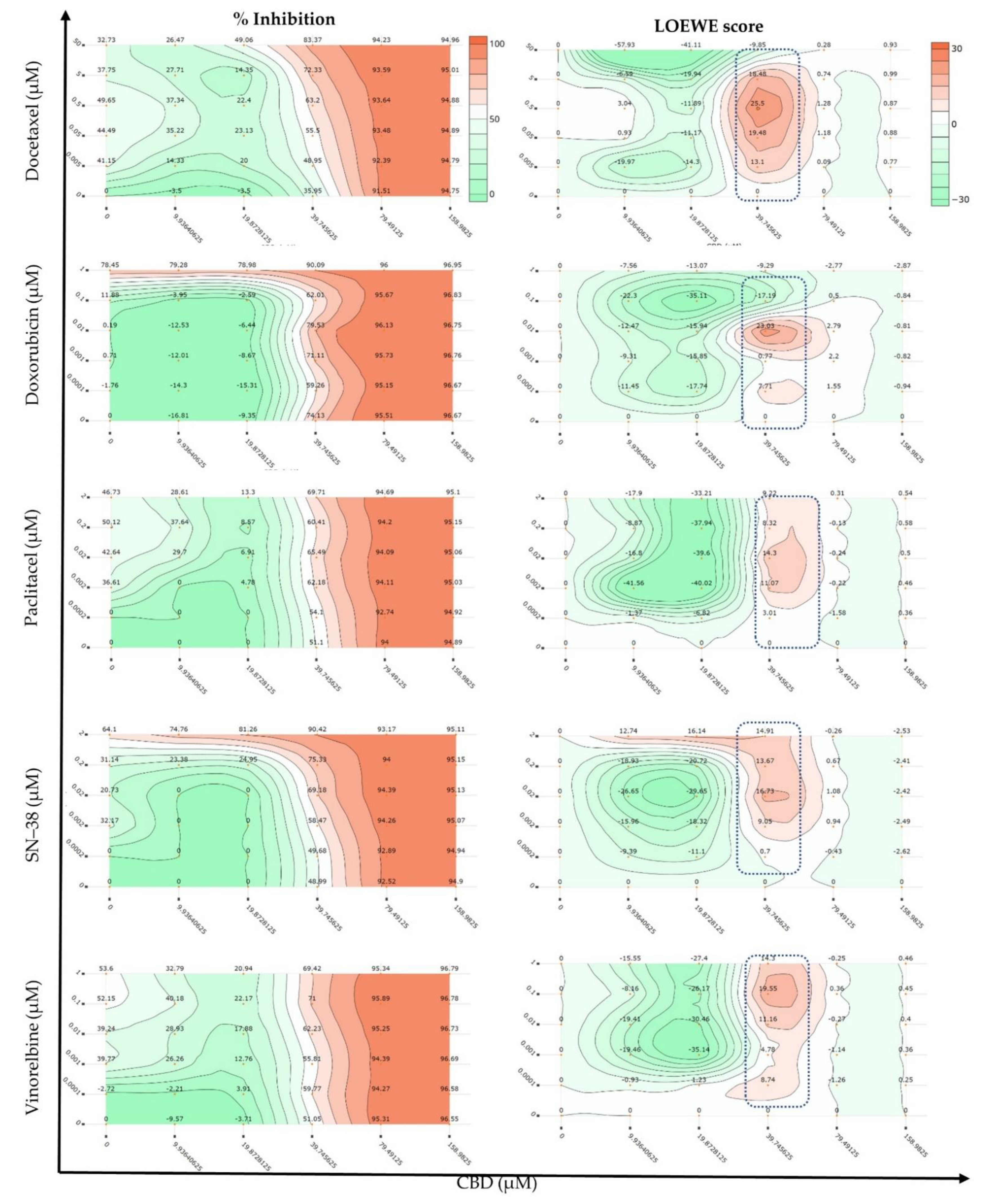
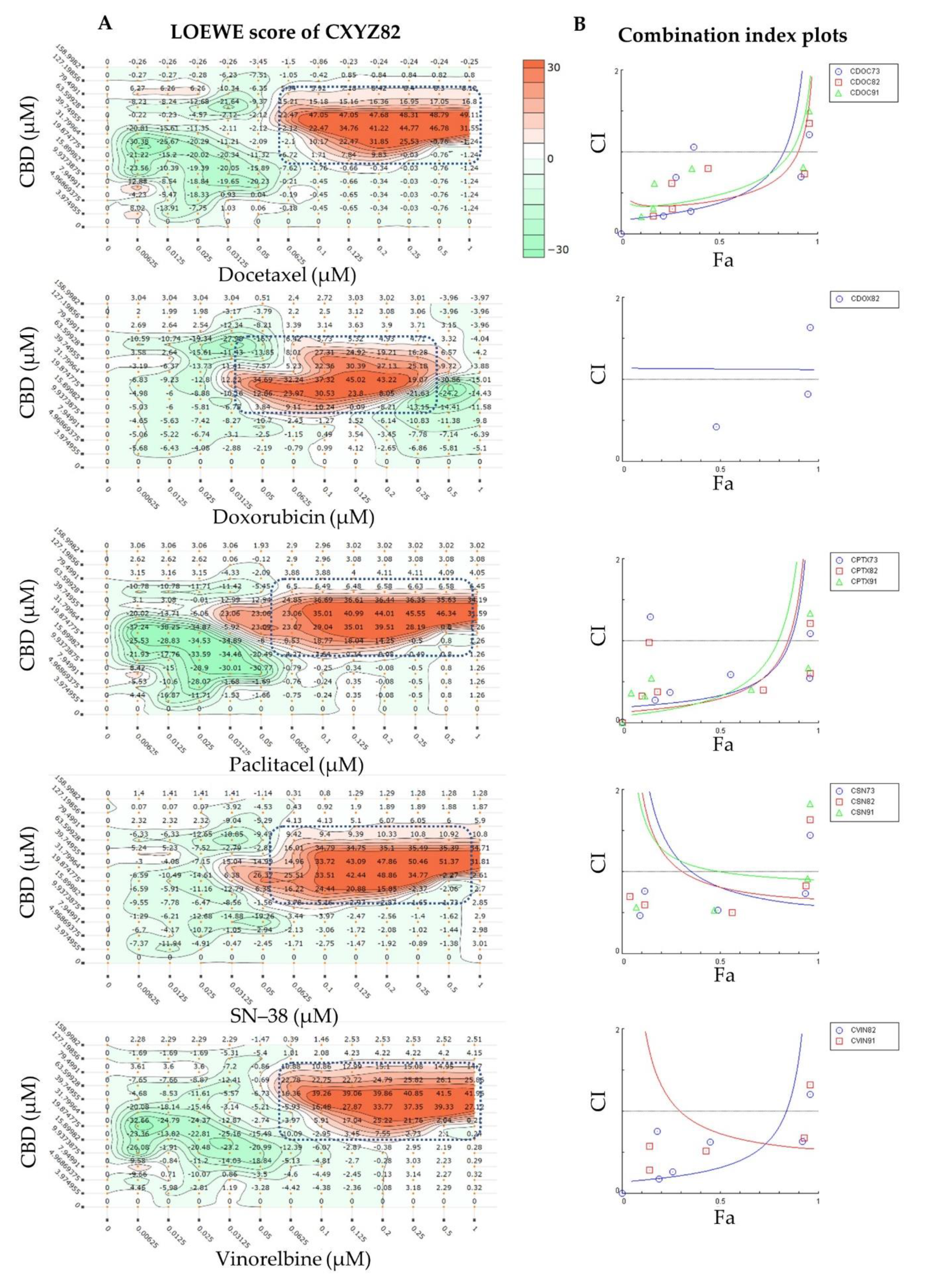
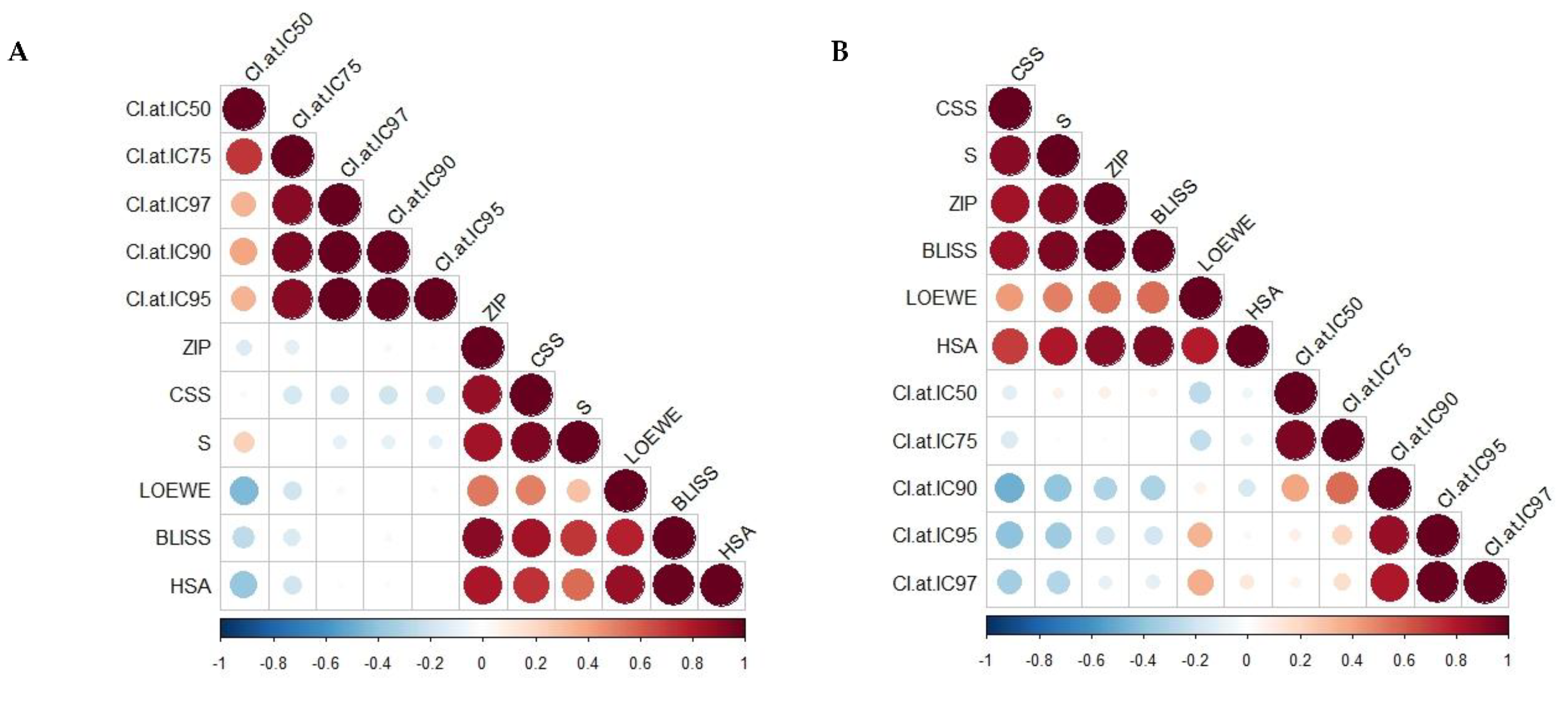
2.1. Synergy Quantification of CBD and Standard Chemotherapeutic Drugs in MCF7 Human Breast Adenocarcinoma Cells
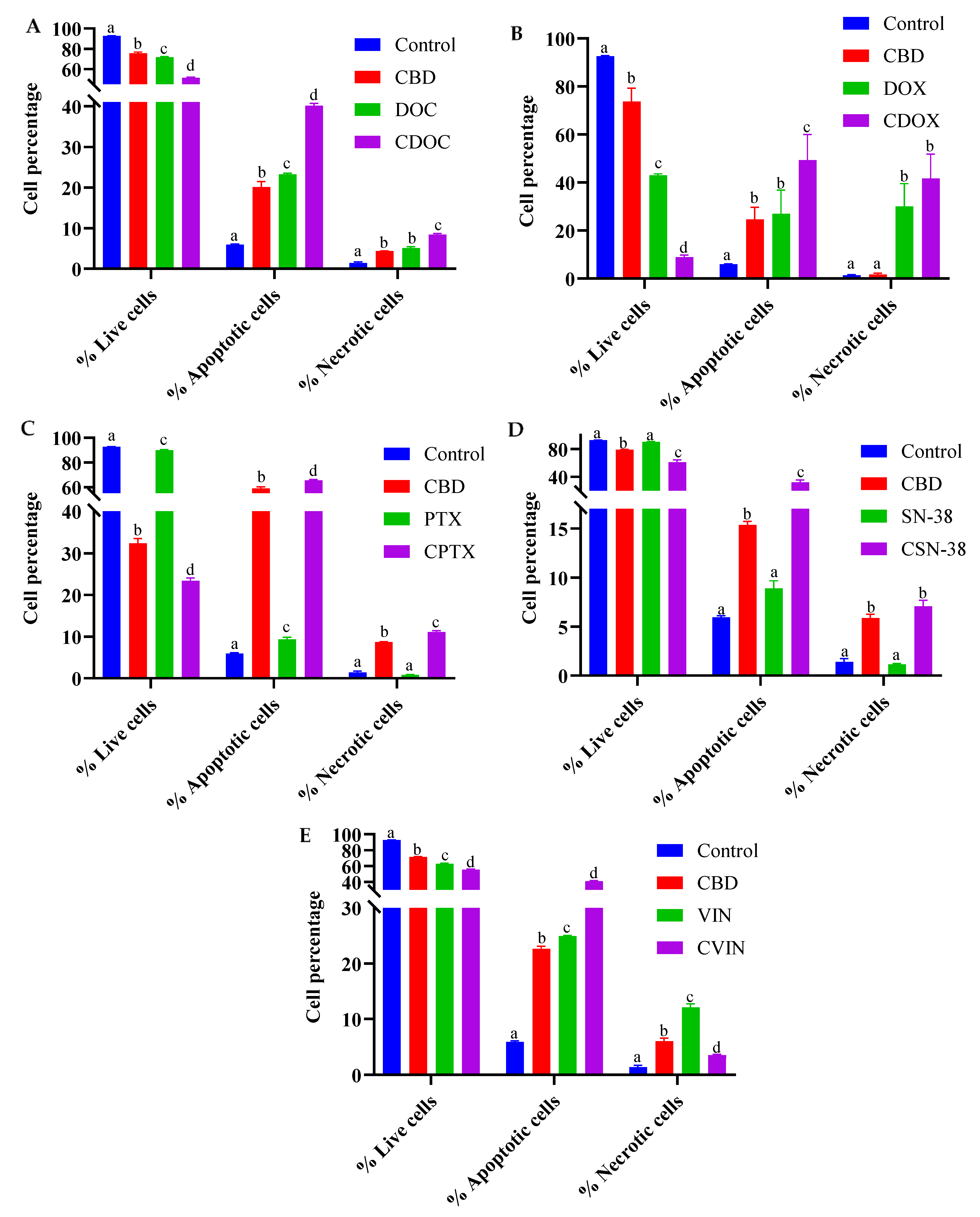
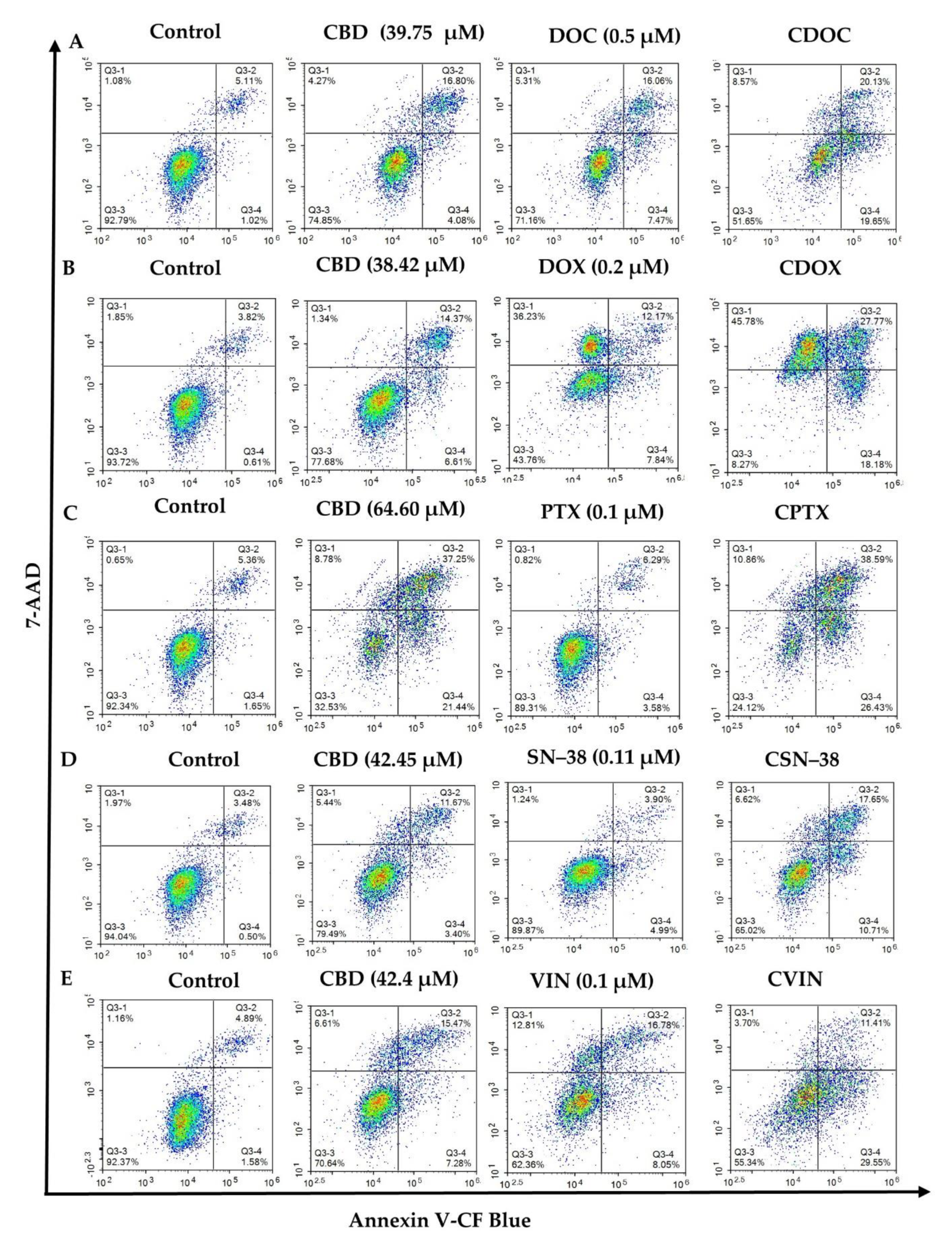
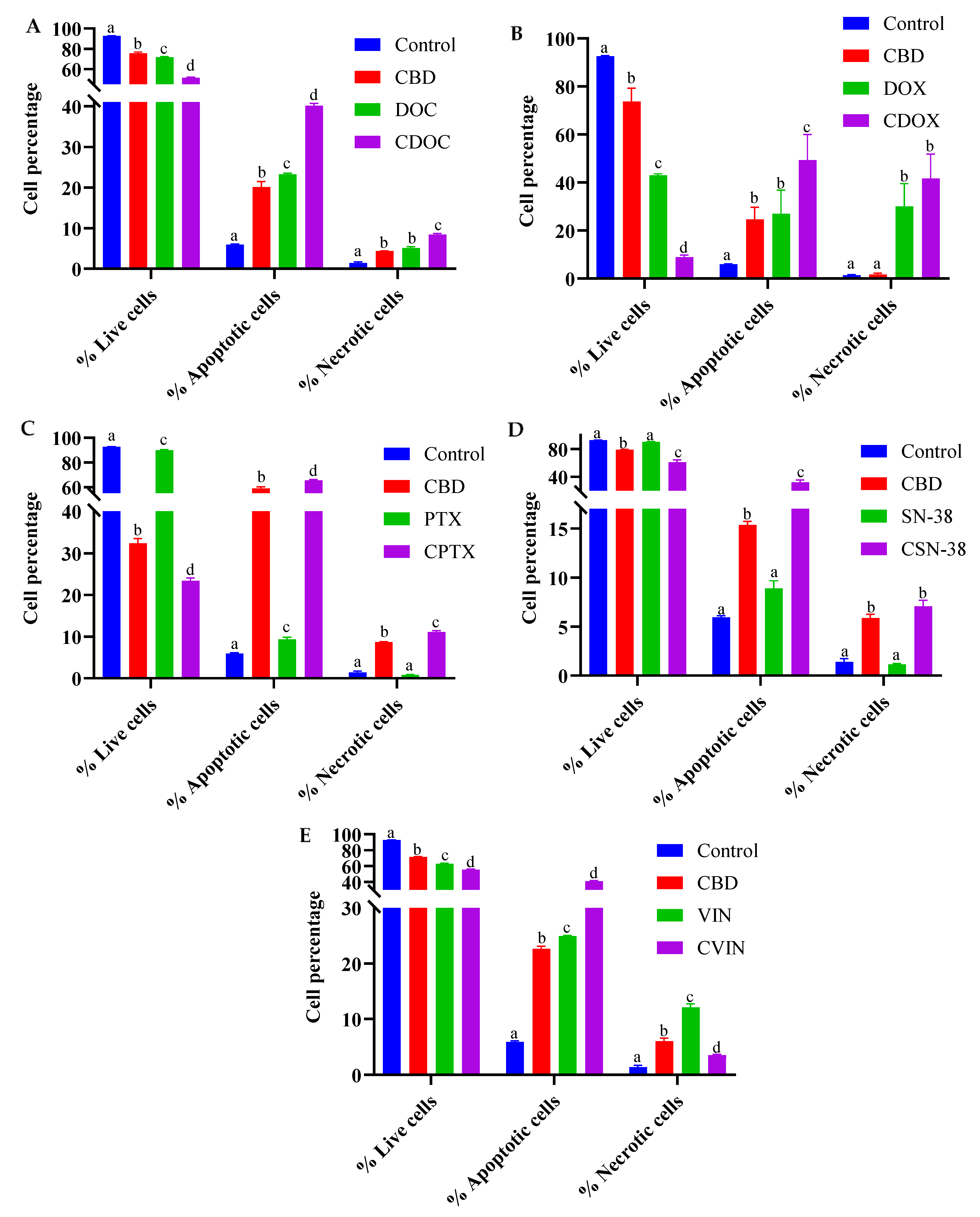
2.2. Flow Cytometric Analyses of Apoptosis in MCF7 Human Breast Adenocarcinoma Cells Using Annexin V-CF Blue and 7-Aminoactinomycin D (7AAD)
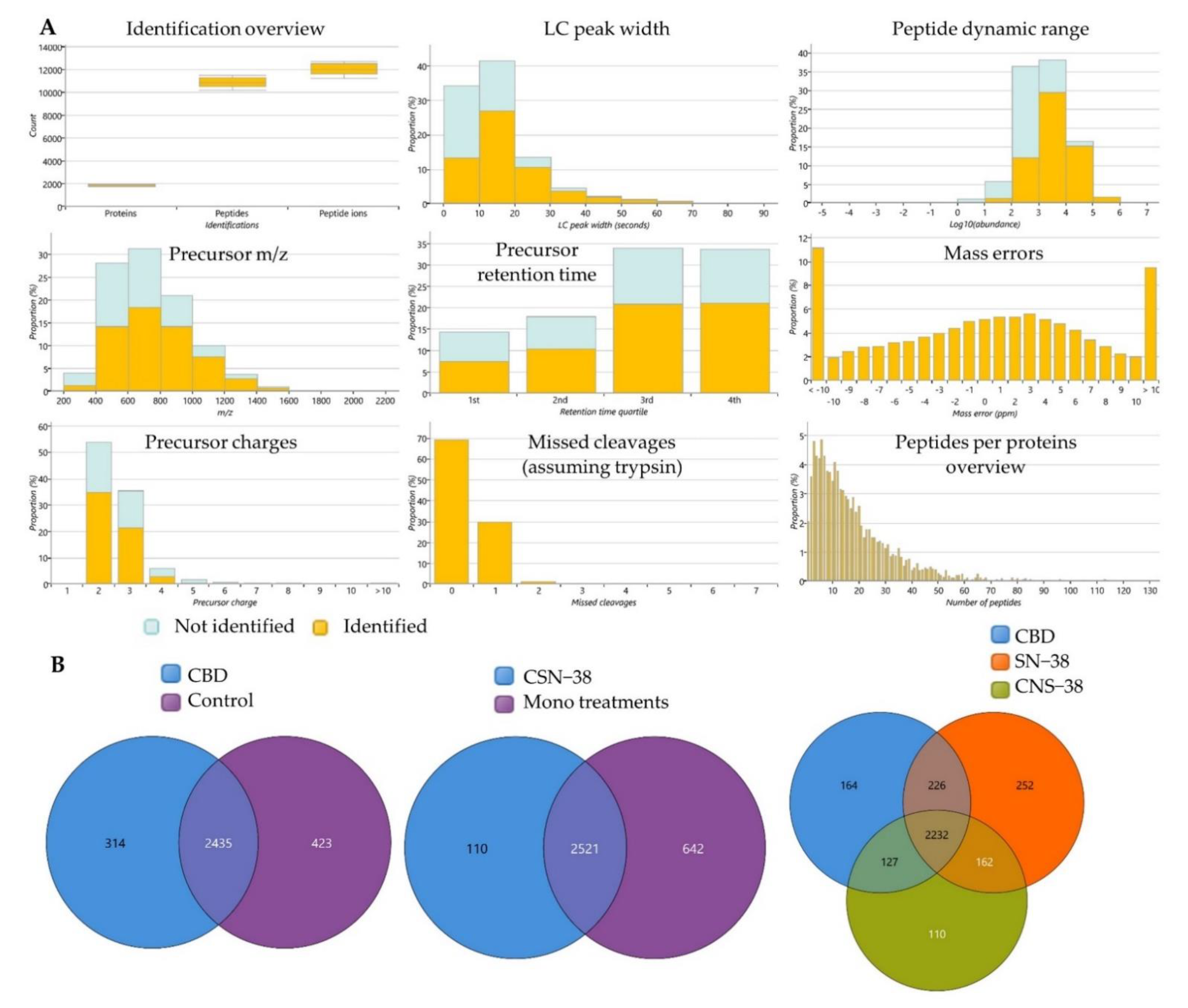
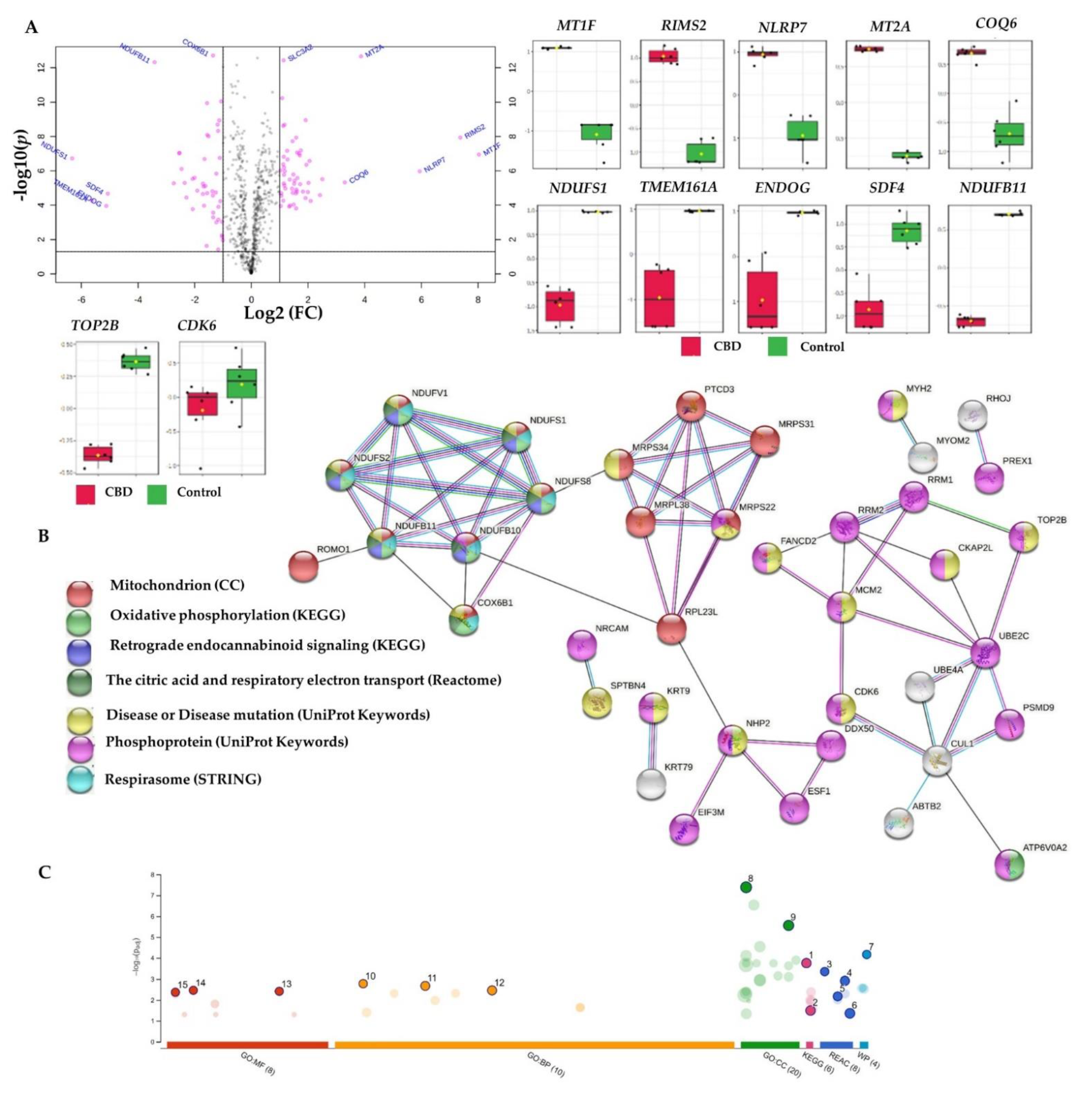
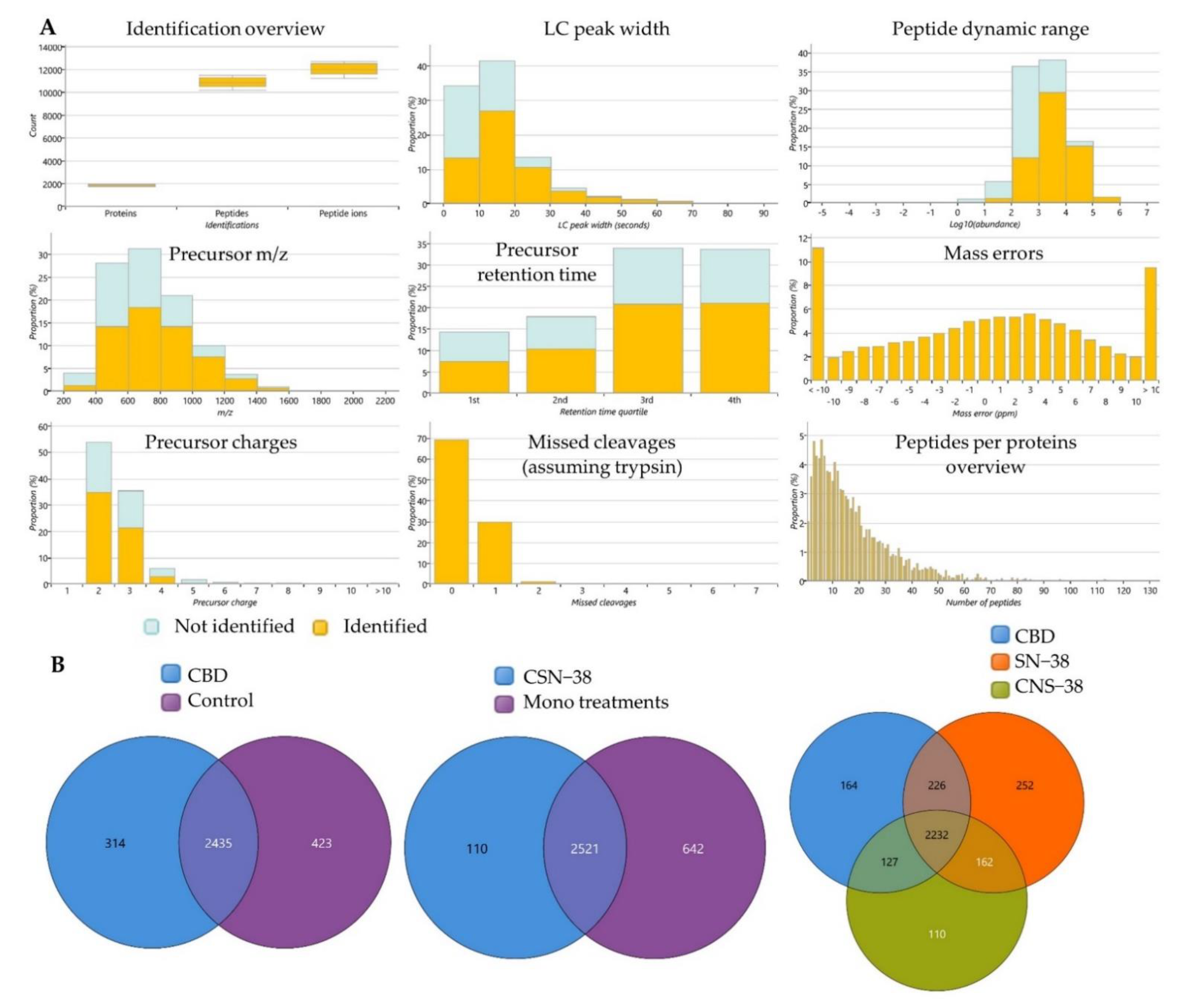
2.3. Bottom-Up Label-Free Quantification Proteomics Study of MCF7 Cell Lysates after Treatment with CBD or Its SN−38 Synergistic Combination
2.3.1. Proteome-Wide Elucidation of the Cytotoxic Effects of CBD in MCF7 Cells: Pilot Shotgun Proteomics Study
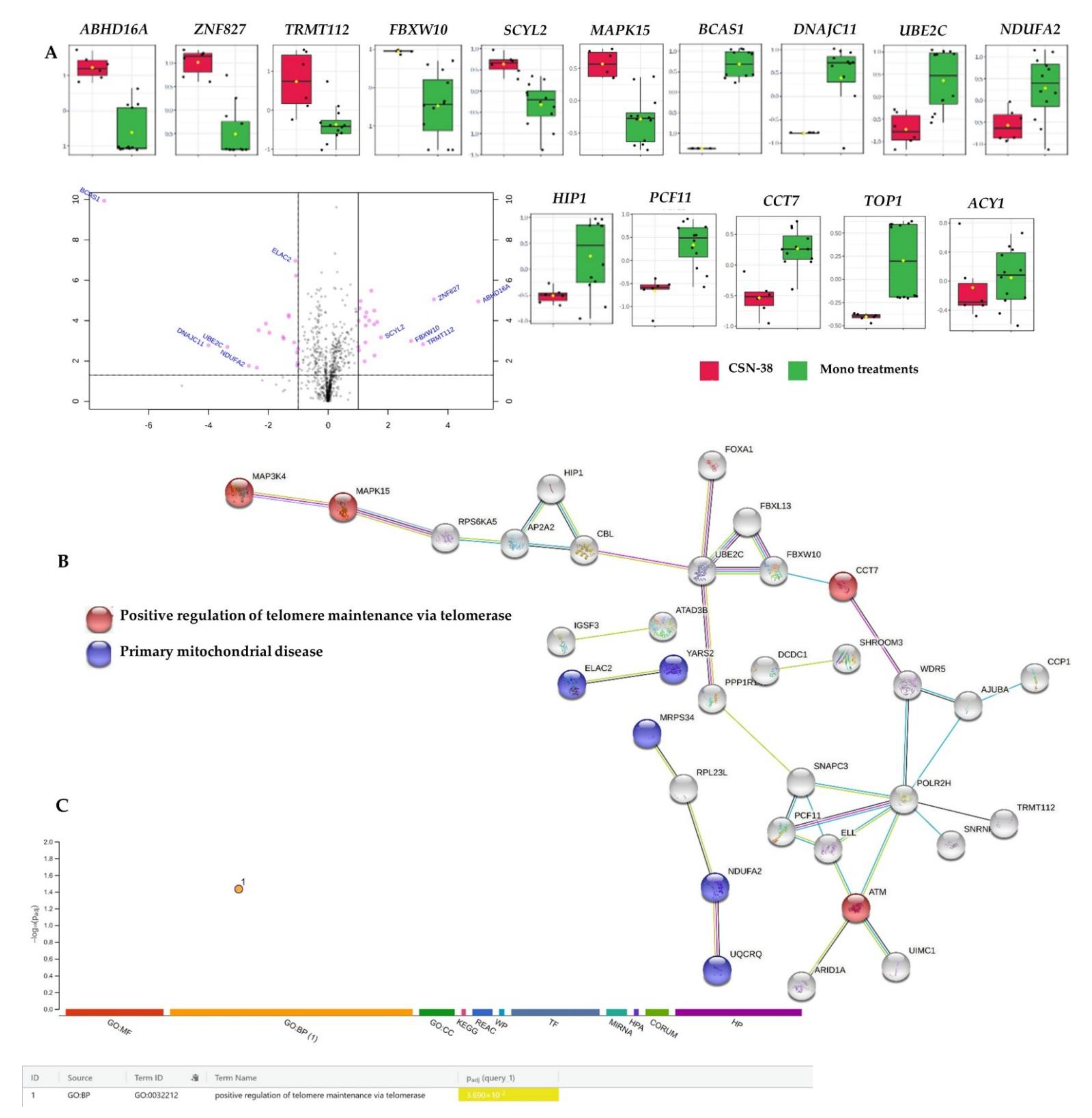
2.3.2. Proteome-Wide Elucidation of Synergistic Mechanisms of SN−38 Synergistic Combination with CBD in MCF7 Cells: Pilot Shotgun Proteomics Study
3. Material and Methods
3.1. Chemicals and Drug Preparation
3.2. Breast Adenocarcinoma Cell Line Culture Conditions
3.3. Cell Viability Determination
3.4. Synergy Quantification of CBD and Standard Chemotherapeutics against MCF7 Human Breast Adenocarcinoma Cells
3.5. Flow Cytometric Analyses of Apoptosis in MCF7 Human Breast Adenocarcinoma Cells Using Annexin V-CF Blue and 7-Aminoactinomycin D (7AAD)
3.6. Bottom-Up Label-Free Quantification Proteomics Study of MCF7 Cell Lysates after Treatment with the Most Synergistic Combination
3.6.1. Cell Culture, Treatment, and Protein Extraction
3.6.2. Protein Quantification
3.6.3. Preparation and Clean-Up of Peptides
3.6.4. Label-Free Bottom-Up Quantification Proteomics Analysis via Nano-Ultra-High-Performance Liquid Chromatography Coupled with Quadruple Time-of-Flight Mass Spectrometry (Nano-UPLC-qTOF-MS)
3.6.5. Data Processing and Availability
3.7. Statistical Analysis
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4. EBP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| 5-HT1A | Serotonin 1A receptor |
| AKT | Protein kinase B |
| CBD | Cannabidiol |
| CB1 and CB2 | Cannabinoid receptor 1 and 2 |
| CCL3 | Chemokine (C-C motif) ligand 3 |
| CDK | Cyclin-dependent kinase |
| CSS | Combination sensitivity score |
| DOC | Docetaxel |
| DOX | Doxorubicin |
| EGFR | Epidermal growth factor receptor |
| ER+/− | Oestrogen receptor-positive/negative |
| ETC | Electron transport chain |
| FC | Fold change |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GPR55 | G protein-coupled receptor 55 |
| Id1 | DNA-binding protein inhibitor ID-1 |
| HER2+ | Human epidermal growth factor receptor 2 positive |
| HER3 | Human epidermal growth factor receptor 3 |
| HGNC | HUGO Gene Nomenclature Committee |
| MCM | Minichromosome maintenance proteins |
| MIP-2 | Macrophage Inflammatory Protein-2 |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ROS | Reactive oxygen species |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PTX | Paclitaxel |
| SN−38 | 7-Ethyl-10-hydroxycamptothecin |
| TAM | Tumour-associated macrophage |
| TRPVs | Family of transient receptor potential (TRP) ion channel |
| TNBC | Triple-negative breast cancer |
| VIN | Vinorelbine |
References
- Jett, J.; Stone, E.; Warren, G.; Cummings, K.M. Cannabis Use, Lung Cancer, and Related Issues. J. Thorac. Oncol. 2018, 13, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. Biomed. Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef] [Green Version]
- Good, P.; Haywood, A.; Gogna, G.; Martin, J.; Yates, P.; Greer, R.; Hardy, J. Oral medicinal cannabinoids to relieve symptom burden in the palliative care of patients with advanced cancer: A double-blind, placebo controlled, randomised clinical trial of efficacy and safety of cannabidiol (CBD). BMC Palliat. Care 2019, 18, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgeman, I.; Bar-Sela, G. Cannabis for cancer-illusion or the tip of an iceberg: A review of the evidence for the use of Cannabis and synthetic cannabinoids in oncology. Expert Opin. Investig. Drugs 2019, 28, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Kim, H.K.; Abdi, S. Update on cannabis and cannabinoids for cancer pain. Curr. Opin. Anaesthesiol. 2020, 33, 825–831. [Google Scholar] [CrossRef]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, D.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a Promising Anti-Cancer Drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Kis, B.; Ifrim, F.C.; Buda, V.; Avram, S.; Pavel, I.Z.; Antal, D.; Paunescu, V.; Dehelean, C.A.; Ardelean, F.; Diaconeasa, Z.; et al. Cannabidiol-from Plant to Human Body: A Promising Bioactive Molecule with Multi-Target Effects in Cancer. Int. J. Mol. Sci. 2019, 20, 5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Kovalchuk, O.; Kovalchuk, I. Cannabinoids as anticancer therapeutic agents. Cell Cycle 2020, 19, 961–989. [Google Scholar] [CrossRef]
- Schoeman, R.; Beukes, N.; Frost, C. Cannabinoid Combination Induces Cytoplasmic Vacuolation in MCF7 Breast Cancer Cells. Molecules 2020, 25, 4682. [Google Scholar] [CrossRef]
- Franco, V.; Perucca, E. Pharmacological and Therapeutic Properties of Cannabidiol for Epilepsy. Drugs 2019, 79, 1435–1454. [Google Scholar] [CrossRef]
- Samanta, D. Cannabidiol: A Review of Clinical Efficacy and Safety in Epilepsy. Pediatr. Neurol. 2019, 96, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Mammana, S.; Cavalli, E.; Bramanti, P.; Mazzon, E. Use of Cannabidiol in the Treatment of Epilepsy: Efficacy and Security in Clinical Trials. Molecules 2019, 24, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzimanoglou, A.; Brandl, U.; Cross, J.H.; Gil-Nagel, A.; Lagae, L.; Landmark, C.J.; Specchio, N.; Nabbout, R.; Thiele, E.A.; Gubbay, O.; et al. Epilepsy and cannabidiol: A guide to treatment. Epileptic Disord. 2020, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Fogaca, M.V.; Sonego, A.B.; Guimaraes, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef]
- Premoli, M.; Aria, F.; Bonini, S.A.; Maccarinelli, G.; Gianoncelli, A.; Pina, S.D.; Tambaro, S.; Memo, M.; Mastinu, A. Cannabidiol: Recent advances and new insights for neuropsychiatric disorders treatment. Life Sci. 2019, 224, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Tian, D.; Tian, L.; Ju, X.; Qi, L.; Wang, Y.; Liang, C. Overview of cannabidiol (CBD) and its analogues: Structures, biological activities, and neuroprotective mechanisms in epilepsy and Alzheimer’s disease. Eur. J. Med. Chem 2020, 192, 112163. [Google Scholar] [CrossRef]
- Pacher, P.; Kogan, N.M.; Mechoulam, R. Beyond THC and Endocannabinoids. Annu Rev. Pharmacol. Toxicol. 2020, 60, 637–659. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves First Drug Comprised of an Active Ingredient Derived from Marijuana to Treat Rare, Severe Forms of Epilepsy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-comprised-active-ingredient-derived-marijuana-treat-rare-severe-forms (accessed on 12 August 2021).
- Drug Enforcement Administration, D.o.J. Schedules of controlled substances: Placement in Schedule V of certain FDA-approved drugs containing cannabidiol; corresponding change to permit requirements. Final order. Fed. Regist. 2018, 83, 48950–48953. [Google Scholar]
- Brunetti, P.; Faro, A.F.L.; Pirani, F.; Berretta, P.; Pacifici, R.; Pichini, S.; Busardò, F.P. Pharmacology and legal status of cannabidiol. Annali dell’Istituto Superiore di Sanità 2020, 56, 285–291. [Google Scholar]
- Brunetti, P.; Pichini, S.; Pacifici, R.; Busardò, F.P.; del Rio, A. Herbal Preparations of Medical Cannabis: A Vademecum for Prescribing Doctors. Medicina 2020, 56, 237. [Google Scholar] [CrossRef]
- Abd-Elsalam, W.H.; Alsherbiny, M.A.; Kung, J.Y.; Pate, D.W.; Lobenberg, R. LC-MS/MS quantitation of phytocannabinoids and their metabolites in biological matrices. Talanta 2019, 204, 846–867. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, C.; Aikins, J.; Warshal, D.; Ostrovsky, O. Can Cannabidiol Affect the Efficacy of Chemotherapy and Epigenetic Treatments in Cancer? Biomolecules 2021, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Ross, R.A.; Craib, S.J.; Thomas, A. (-)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur. J. Pharmacol. 2002, 456, 99–106. [Google Scholar] [CrossRef]
- Ibeas Bih, C.; Chen, T.; Nunn, A.V.; Bazelot, M.; Dallas, M.; Whalley, B.J. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Harpe, A.; Beukes, N.; Frost, C.L. CBD activation of TRPV1 induces oxidative signaling and subsequent ER stress in breast cancer cell lines. Biotechnol. Appl. Biochem. 2021, 1–11. [Google Scholar] [CrossRef]
- Jeong, S.; Jo, M.J.; Yun, H.K.; Kim, D.Y.; Kim, B.R.; Kim, J.L.; Park, S.H.; Na, Y.J.; Jeong, Y.A.; Kim, B.G.; et al. Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 2019, 10, 846. [Google Scholar] [CrossRef] [Green Version]
- Mould, R.R.; Botchway, S.W.; Parkinson, J.R.C.; Thomas, E.L.; Guy, G.W.; Bell, J.D.; Nunn, A.V.W. Cannabidiol Modulates Mitochondrial Redox and Dynamics in MCF7 Cancer Cells: A Study Using Fluorescence Lifetime Imaging Microscopy of NAD(P)H. Front. Mol. Biosci. 2021, 8, 630107. [Google Scholar] [CrossRef] [PubMed]
- Trac, J.; Keck, J.M.; Deweese, J.E. Cannabidiol oxidation product HU-331 is a potential anticancer cannabinoid-quinone: A narrative review. J. Cannabis Res. 2021, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Fraguas-Sanchez, A.I.; Fernandez-Carballido, A.; Simancas-Herbada, R.; Martin-Sabroso, C.; Torres-Suarez, A.I. CBD loaded microparticles as a potential formulation to improve paclitaxel and doxorubicin-based chemotherapy in breast cancer. Int. J. Pharm. 2020, 574, 118916. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. The Endocannabinoid System as a Target. for Treatment of Breast Cancer; Virginia Commonwealth University: Richmond, VA, USA, 2010. [Google Scholar]
- Bergamaschi, M.M.; Queiroz, R.H.; Chagas, M.H.; de Oliveira, D.C.; De Martinis, B.S.; Kapczinski, F.; Quevedo, J.; Roesler, R.; Schröder, N.; Nardi, A.E.; et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology 2011, 36, 1219–1226. [Google Scholar] [CrossRef]
- Crippa, J.A.; Derenusson, G.N.; Ferrari, T.B.; Wichert-Ana, L.; Duran, F.L.; Martin-Santos, R.; Simões, M.V.; Bhattacharyya, S.; Fusar-Poli, P.; Atakan, Z.; et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: A preliminary report. J. Psychopharmacol. 2011, 25, 121–130. [Google Scholar] [CrossRef]
- Crippa, J.A.; Zuardi, A.W.; Garrido, G.E.; Wichert-Ana, L.; Guarnieri, R.; Ferrari, L.; Azevedo-Marques, P.M.; Hallak, J.E.; McGuire, P.K.; Filho Busatto, G. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology 2004, 29, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Zuardi, A.W.; Rodrigues, N.P.; Silva, A.L.; Bernardo, S.A.; Hallak, J.E.C.; Guimarães, F.S.; Crippa, J.A.S. Inverted U-Shaped Dose-Response Curve of the Anxiolytic Effect of Cannabidiol during Public Speaking in Real Life. Front. Pharmacol. 2017, 8, 259. [Google Scholar] [CrossRef] [Green Version]
- Martin-Santos, R.; Crippa, J.A.; Batalla, A.; Bhattacharyya, S.; Atakan, Z.; Borgwardt, S.; Allen, P.; Seal, M.; Langohr, K.; Farré, M.; et al. Acute effects of a single, oral dose of d9-tetrahydrocannabinol (THC) and cannabidiol (CBD) administration in healthy volunteers. Curr Pharm Des. 2012, 18, 4966–4979. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Morrison, P.D.; Fusar-Poli, P.; Martin-Santos, R.; Borgwardt, S.; Winton-Brown, T.; Nosarti, C.; CM, O.C.; Seal, M.; Allen, P.; et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 2010, 35, 764–774. [Google Scholar] [CrossRef]
- Hallak, J.E.; Machado-de-Sousa, J.P.; Crippa, J.A.; Sanches, R.F.; Trzesniak, C.; Chaves, C.; Bernardo, S.A.; Regalo, S.C.; Zuardi, A.W. Performance of schizophrenic patients in the Stroop Color Word Test and electrodermal responsiveness after acute administration of cannabidiol (CBD). Br. J. Psychiatry 2010, 32, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linares, I.M.; Zuardi, A.W.; Pereira, L.C.; Queiroz, R.H.; Mechoulam, R.; Guimarães, F.S.; Crippa, J.A. Cannabidiol presents an inverted U-shaped dose-response curve in a simulated public speaking test. Br. J. Psychiatry 2019, 41, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundal, H.; Lister, R.; Evans, N.; Antley, A.; Englund, A.; Murray, R.M.; Freeman, D.; Morrison, P.D. The effects of cannabidiol on persecutory ideation and anxiety in a high trait paranoid group. J. Psychopharmacol. 2018, 32, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Boggs, D.L.; Surti, T.; Gupta, A.; Gupta, S.; Niciu, M.; Pittman, B.; Schnakenberg Martin, A.M.; Thurnauer, H.; Davies, A.; D’Souza, D.C.; et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology 2018, 235, 1923–1932. [Google Scholar] [CrossRef]
- McGuire, P.; Robson, P.; Cubala, W.J.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Irving, P.M.; Iqbal, T.; Nwokolo, C.; Subramanian, S.; Bloom, S.; Prasad, N.; Hart, A.; Murray, C.; Lindsay, J.O.; Taylor, A.; et al. A Randomized, Double-blind, Placebo-controlled, Parallel-group, Pilot Study of Cannabidiol-rich Botanical Extract in the Symptomatic Treatment of Ulcerative Colitis. Inflamm. Bowel Dis. 2018, 24, 714–724. [Google Scholar] [CrossRef]
- Jadoon, K.A.; Ratcliffe, S.H.; Barrett, D.A.; Thomas, E.L.; Stott, C.; Bell, J.D.; O’Sullivan, S.E.; Tan, G.D. Efficacy and Safety of Cannabidiol and Tetrahydrocannabivarin on Glycemic and Lipid Parameters in Patients With Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled, Parallel Group Pilot Study. Diabetes Care 2016, 39, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, C.; Shahinas, J. Dosage, Efficacy and Safety of Cannabidiol Administration in Adults: A Systematic Review of Human Trials. J. Clin. Med. Res. 2020, 12, 129–141. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murase, R.; Kawamura, R.; Singer, E.; Pakdel, A.; Sarma, P.; Judkins, J.; Elwakeel, E.; Dayal, S.; Martinez-Martinez, E.; Amere, M.; et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br. J. Pharmacol. 2014, 171, 4464–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosgodage, U.S.; Mould, R.; Henley, A.B.; Nunn, A.V.; Guy, G.W.; Thomas, E.L.; Inal, J.M.; Bell, J.D.; Lange, S. Cannabidiol (CBD) Is a Novel Inhibitor for Exosome and Microvesicle (EMV) Release in Cancer. Front. Pharmacol. 2018, 9, 889. [Google Scholar] [CrossRef] [Green Version]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef]
- Yeh, P.J.; Hegreness, M.J.; Aiden, A.P.; Kishony, R. Drug interactions and the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Bhuyan, D.J.; Perera, S.; Kaur, K.; Alsherbiny, M.A.; Low, M.; Seto, S.-W.; Li, C.-G.; Zhou, X. Synergistic Effects of Chinese Herbal Medicine and Biological Networks. Approaching Complex. Dis. 2020, 393–436. [Google Scholar]
- Ward, S.J.; McAllister, S.D.; Kawamura, R.; Murase, R.; Neelakantan, H.; Walker, E.A. Cannabidiol inhibits paclitaxel-induced neuropathic pain through 5-HT(1A) receptors without diminishing nervous system function or chemotherapy efficacy. Br. J. Pharmacol. 2014, 171, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Hao, E.; Mukhopadhyay, P.; Cao, Z.; Erdelyi, K.; Holovac, E.; Liaudet, L.; Lee, W.S.; Hasko, G.; Mechoulam, R.; Pacher, P. Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol. Med. 2015, 21, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Alsherbiny, M.A.; Li, C.G. Medicinal Cannabis—Potential Drug Interactions. Medicines 2019, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Huestis, M.A.; Solimini, R.; Pichini, S.; Pacifici, R.; Carlier, J.; Busardò, F.P. Cannabidiol adverse effects and toxicity. Curr. Neuropharmacol. 2019, 17, 974–989. [Google Scholar] [CrossRef]
- Meyer, C.T.; Wooten, D.J.; Lopez, C.F.; Quaranta, V. Charting the Fragmented Landscape of Drug Synergy. Trends Pharmacol. Sci. 2020, 41, 266–280. [Google Scholar] [CrossRef]
- Vlot, A.H.; Aniceto, N.; Menden, M.P.; Ulrich-Merzenich, G.; Bender, A. Applying drug synergy metrics to oncology combination screening data: Agreements, disagreements and pitfalls. Drug Discov. Today 2019, 24, 2286–2298. [Google Scholar] [CrossRef]
- Alsherbiny, M.A.; Bhuyan, D.J.; Radwan, I.; Chang, D.; Li, C.G. Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells. Int. J. Mol. Sci. 2021, 22, 7840. [Google Scholar] [CrossRef]
- Gilvary, C.; Dry, J.R.; Elemento, O. Multi-task learning predicts drug combination synergy in cells and in the clinic. bioRxiv 2019, 576017. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF7 Cells—Changing the Course of Breast Cancer Research and Care for 45 Years. JNCI J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- NCI. Stages of Breast Cancer. Available online: https://www.cancer.gov/types/breast/patient/breast-treatment-pdq#link/_148 (accessed on 11 September 2018).
- Chou, T.-C. The combination index (CI<1) as the definition of synergism and of synergy claims. Synergy 2018, 7, 49–50. [Google Scholar]
- O’Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doroshow, J.H.; Simon, R.M. On the Design of Combination Cancer Therapy. Cell 2017, 171, 1476–1478. [Google Scholar] [CrossRef] [Green Version]
- Zagidullin, B.; Aldahdooh, J.; Zheng, S.; Wang, W.; Wang, Y.; Saad, J.; Malyutina, A.; Jafari, M.; Tanoli, Z.; Pessia, A.; et al. DrugComb: An integrative cancer drug combination data portal. Nucleic Acids Res. 2019, 47, W43–W51. [Google Scholar] [CrossRef] [PubMed]
- Fouad, A.A.; Albuali, W.H.; Al-Mulhim, A.S.; Jresat, I. Cardioprotective effect of cannabidiol in rats exposed to doxorubicin toxicity. Environ. Toxicol. Pharmacol. 2013, 36, 347–357. [Google Scholar] [CrossRef]
- King, K.M.; Myers, A.M.; Soroka-Monzo, A.J.; Tuma, R.F.; Tallarida, R.J.; Walker, E.A.; Ward, S.J. Single and combined effects of Delta(9) -tetrahydrocannabinol and cannabidiol in a mouse model of chemotherapy-induced neuropathic pain. Br. J. Pharmacol. 2017, 174, 2832–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Petrocellis, L.; Ligresti, A.; Schiano Moriello, A.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growth in vitro and in vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2013, 168, 79–102. [Google Scholar] [CrossRef] [Green Version]
- Engels, F.K.; de Jong, F.A.; Sparreboom, A.; Mathot, R.A.; Loos, W.J.; Kitzen, J.J.; de Bruijn, P.; Verweij, J.; Mathijssen, R.H. Medicinal cannabis does not influence the clinical pharmacokinetics of irinotecan and docetaxel. Oncologist 2007, 12, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, gkw937. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef] [PubMed]
- Bertram, R.; Gram Pedersen, M.; Luciani, D.S.; Sherman, A. A simplified model for mitochondrial ATP production. J. Theor. Biol. 2006, 243, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Urra, F.A.; Munoz, F.; Lovy, A.; Cardenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7, 118. [Google Scholar] [CrossRef]
- Chan, J.Z.; Duncan, R.E. Regulatory Effects of Cannabidiol on Mitochondrial Functions: A Review. Cells 2021, 10, 1251. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Perez-Sayans, M.; Somoza-Martin, J.M.; Barros-Angueira, F.; Rey, J.M.; Garcia-Garcia, A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 2009, 35, 707–713. [Google Scholar] [CrossRef]
- Hu, B.; Guo, Y. Inhibition of mitochondrial translation as a therapeutic strategy for human ovarian cancer to overcome chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Yong, H.M.; Chen, F.F.; Mei, P.J.; Liu, H.; Li, C.; Pan, Z.Q.; Wu, Y.P.; Zheng, J.N. Cullin1 is a novel marker of poor prognosis and a potential therapeutic target in human breast cancer. Ann. Oncol. 2013, 24, 2016–2022. [Google Scholar] [CrossRef]
- Ren, Z.Q.; Yan, W.J.; Zhang, X.Z.; Zhang, P.B.; Zhang, C.; Chen, S.K. CUL1 Knockdown Attenuates the Adhesion, Invasion, and Migration of Triple-Negative Breast Cancer Cells via Inhibition of Epithelial-Mesenchymal Transition. Pathol. Oncol. Res. 2020, 26, 1153–1163. [Google Scholar] [CrossRef]
- Huang, Y.F.; Zhang, Z.; Zhang, M.; Chen, Y.S.; Song, J.; Hou, P.F.; Yong, H.M.; Zheng, J.N.; Bai, J. CUL1 promotes breast cancer metastasis through regulating EZH2-induced the autocrine expression of the cytokines CXCL8 and IL11. Cell Death Dis. 2018, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Gou, K.; Liu, J.; Feng, X.; Li, H.; Yuan, Y.; Xing, C. Expression of Minichromosome Maintenance Proteins (MCM) and Cancer Prognosis: A meta-analysis. J. Cancer 2018, 9, 1518–1526. [Google Scholar] [CrossRef]
- Qin, T.; Huang, G.; Chi, L.; Sui, S.; Song, C.; Li, N.; Sun, S.; Li, N.; Zhang, M.; Zhao, Z.; et al. Exceptionally high UBE2C expression is a unique phenomenon in basal-like type breast cancer and is regulated by BRCA1. Biomed. Pharmacother. 2017, 95, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.; Gopal, G.; Selvaluxmy, G.; Rajkumar, T. Inhibition of ubiquitin conjugating enzyme UBE2C reduces proliferation and sensitizes breast cancer cells to radiation, doxorubicin, tamoxifen and letrozole. Cell Oncol. 2013, 36, 459–467. [Google Scholar] [CrossRef]
- Hande, K.R. Topoisomerase II inhibitors. Update Cancer Ther. 2008, 3, 13–26. [Google Scholar] [CrossRef]
- Xu, W.X.; Song, W.; Jiang, M.P.; Yang, S.J.; Zhang, J.; Wang, D.D.; Tang, J.H. Systematic Characterization of Expression Profiles and Prognostic Values of the Eight Subunits of the Chaperonin TRiC in Breast Cancer. Front. Genet. 2021, 12, 637887. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Kastan, M.B. ATM: Genome stability, neuronal development, and cancer cross paths. Adv. Cancer Res. 2001, 83, 209–254. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Tanic, M.; Krivokuca, A.; Cavic, M.; Mladenovic, J.; Plesinac Karapandzic, V.; Beck, S.; Radulovic, S.; Susnjar, S.; Jankovic, R. Molecular signature of response to preoperative radiotherapy in locally advanced breast cancer. Radiat. Oncol. 2018, 13, 193. [Google Scholar] [CrossRef]
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.; Elhassan, G.O.; Ibrahim, M.E.; David Polo Orozco, J.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef]
- Garcia-Flores, A.E.; Sollome, J.J.; Thavathiru, E.; Bower, J.L.; Vaillancourt, R.R. HER2/HER3 regulates lactate secretion and expression of lactate receptor mRNA through the MAP3K4 associated protein GIT1. Sci. Rep. 2019, 9, 10823. [Google Scholar] [CrossRef]
- Chia, J.; Tham, K.M.; Gill, D.J.; Bard-Chapeau, E.A.; Bard, F.A. ERK8 is a negative regulator of O-GalNAc glycosylation and cell migration. eLife 2014, 3, e01828. [Google Scholar] [CrossRef]
- Gill, D.J.; Tham, K.M.; Chia, J.; Wang, S.C.; Steentoft, C.; Clausen, H.; Bard-Chapeau, E.A.; Bard, F.A. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2013, 110, E3152–E3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Salvia, M.; Simo-Riudalbas, L.; Llinas-Arias, P.; Roa, L.; Setien, F.; Soler, M.; de Moura, M.C.; Bradner, J.E.; Gonzalez-Suarez, E.; Moutinho, C.; et al. Bromodomain inhibition shows antitumoral activity in mice and human luminal breast cancer. Oncotarget 2017, 8, 51621–51629. [Google Scholar] [CrossRef] [Green Version]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttacavoli, M.; Di Cara, G.; D’Amico, C.; Geraci, F.; Pucci-Minafra, I.; Feo, S.; Cancemi, P. Prognostic and Functional Significant of Heat Shock Proteins (HSPs) in Breast Cancer Unveiled by Multi-Omics Approaches. Biology 2021, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Lin, J.; Lv, C.; Qiao, G. miR-146a Enhances the Sensitivity of Breast Cancer Cells to Paclitaxel by Downregulating IRAK1. Cancer Biother. Radiopharm. 2020. [Google Scholar] [CrossRef]
- Wee, Z.N.; Yatim, S.M.; Kohlbauer, V.K.; Feng, M.; Goh, J.Y.; Bao, Y.; Lee, P.L.; Zhang, S.; Wang, P.P.; Lim, E.; et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat. Commun. 2015, 6, 8746. [Google Scholar] [CrossRef]
- Yang, M.; Qin, X.; Qin, G.; Zheng, X. The role of IRAK1 in breast cancer patients treated with neoadjuvant chemotherapy. Onco. Targets Ther. 2019, 12, 2171–2180. [Google Scholar] [CrossRef] [Green Version]
- Dillon, L.M.; Bean, J.R.; Yang, W.; Shee, K.; Symonds, L.K.; Balko, J.M.; McDonald, W.H.; Liu, S.; Gonzalez-Angulo, A.M.; Mills, G.B.; et al. P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer. Oncogene 2015, 34, 3968–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.C.; Seoane, S.; Ocana, A.; Pandiella, A. P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 2011, 30, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Minard, M.E.; Kim, L.S.; Price, J.E.; Gallick, G.E. The role of the guanine nucleotide exchange factor Tiam1 in cellular migration, invasion, adhesion and tumor progression. Breast Cancer Res. Treat. 2004, 84, 21–32. [Google Scholar] [CrossRef]
- Ogorodnikov, A.; Levin, M.; Tattikota, S.; Tokalov, S.; Hoque, M.; Scherzinger, D.; Marini, F.; Poetsch, A.; Binder, H.; Macher-Goppinger, S.; et al. Transcriptome 3′ end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018, 9, 5331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Liu, X.; Cao, X.; Zhang, M.; Chang, H. Study of the expression and function of ACY1 in patients with colorectal cancer. Oncol. Lett. 2017, 13, 2459–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hayes, M.T.; Kirana, C.; Miller, R.J.; Keating, J.P.; Stubbs, R.S. Overexpression of aminoacylase 1 is associated with colorectal cancer progression. Hum. Pathol. 2013, 44, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.M.; Franklin, W.A.; Moore, M.D.; Johnson, B.E.; Miller, Y.E. Mutational inactivation of aminoacylase-1 in a small cell lung cancer cell line. Genes Chromosomes Cancer 1998, 21, 320–325. [Google Scholar] [CrossRef]
- Zhong, Y.; Onuki, J.; Yamasaki, T.; Ogawa, O.; Akatsuka, S.; Toyokuni, S. Genome-wide analysis identifies a tumor suppressor role for aminoacylase 1 in iron-induced rat renal cell carcinoma. Carcinogenesis 2009, 30, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Li, J.; Xie, H.; Ling, Q.; Wang, J.; Lu, D.; Zhou, L.; Xu, X.; Zheng, S. Proteomics-based identification of the tumor suppressor role of aminoacylase 1 in hepatocellular carcinoma. Cancer Lett. 2014, 351, 117–125. [Google Scholar] [CrossRef]
- Lee, S.U.; Kim, B.T.; Min, Y.K.; Kim, S.H. Protein profiling and transcript expression levels of heat shock proteins in 17beta-estradiol-treated human MCF7 breast cancer cells. Cell Biol. Int. 2006, 30, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Mamoor, S. ITGAL Is Differentially Expressed in Lymph Node Metastasis in Human Breast Cancer. OSF Prepr. 2021. [Google Scholar]
- Bersini, S.; Lytle, N.K.; Schulte, R.; Huang, L.; Wahl, G.M.; Hetzer, M.W. Nup93 regulates breast tumor growth by modulating cell proliferation and actin cytoskeleton remodeling. Life Sci. Alliance 2020, 3, e201900623. [Google Scholar] [CrossRef] [Green Version]
- Nataraj, N.; Noronha, A.; Lee, J.; Ghosh, S.; Raju, H.R.M.; Sekar, A.; Zuckerman, B.; Lindzen, M.; Srivastava, S.; Selitrennik, M. Nucleoporin-93 Overexpression Overcomes Multiple Nucleocytoplsamic Trafficking Bottlenecks to Permit Robust Metastasis; 2020. [Google Scholar]
- Hsu, F.F.; Chou, Y.T.; Chiang, M.T.; Li, F.A.; Yeh, C.T.; Lee, W.H.; Chau, L.Y. Signal peptide peptidase promotes tumor progression via facilitating FKBP8 degradation. Oncogene 2019, 38, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Ravipati, A.S.; Zhang, L.; Koyyalamudi, S.R.; Jeong, S.C.; Reddy, N.; Bartlett, J.; Smith, P.T.; Shanmugam, K.; Munch, G.; Wu, M.J.; et al. Antioxidant and anti-inflammatory activities of selected Chinese medicinal plants and their relation with antioxidant content. BMC Complement. Altern. Med. 2012, 12, 173. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.A.; Gogal, R.M.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H]thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Bhuyan, D.J.; Alsherbiny, M.A.; Low, M.N.; Zhou, X.; Kaur, K.; Li, G.; Li, C.G. Broad-spectrum pharmacological activity of Australian propolis and metabolomic-driven identification of marker metabolites of propolis samples from three continents. Food Funct. 2021, 12, 2498–2519. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.; Martin, N. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program. for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values; ComboSyn: Paramus, NJ, USA, 2005. [Google Scholar]
- Chou, T.-C.; Martin, N. The mass-action law-based new computer software, CompuSyn, for automated simulation of synergism and antagonism in drug combination studies. Exp. Mol. Ther. 2007. [Google Scholar]
- Kumar, R.; Saneja, A.; Panda, A.K. An Annexin V-FITC—Propidium Iodide-Based Method for Detecting Apoptosis in a Non-Small Cell Lung Cancer Cell Line. In Lung Cancer: Methods and Protocols; Santiago-Cardona, P.G., Ed.; Springer US: New York, NY, USA, 2021; pp. 213–223. [Google Scholar]
- Wadkins, R.M.; Jovin, T.M. Actinomycin D and 7-aminoactinomycin D binding to single-stranded DNA. Biochemistry 1991, 30, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Schmit, T.; Klomp, M.; Khan, M.N. An Overview of Flow Cytometry: Its Principles and Applications in Allergic Disease Research. Methods Mol. Biol. 2021, 2223, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combo ID, Molar Ratio | CI Values at | CSS | S | ZIP | BLISS | LOEWE | HSA | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | IC75 | IC90 | IC95 | IC97 | |||||||
| CBD: Docetaxel | |||||||||||
| CDOC19, 18:1 | 1.23 | 1.35 | 1.64 | 2.07 | 2.60 | 35.5 | 0.33 | −11.81 | −19.12 | −6.84 | −15.76 |
| CDOC28, 40:1 | 0.96 | 1.14 | 1.62 | 2.32 | 3.18 | 54.6 | 18.07 | −10.9 | −13.6 | 14.29 | −9.42 |
| CDOC37, 68:1 | 0.70 | 0.91 | 1.46 | 2.27 | 3.27 | 41.33 | 3.96 | −9.74 | −11.35 | −8.14 | −7.08 |
| CDOC46, 106:1 | 0.49 | 0.70 | 1.23 | 2.04 | 3.03 | 49.82 | 13.09 | −3.4 | −5.61 | −2.36 | −1.38 |
| CDOC55, 159:1 | 0.45 | 0.70 | 1.35 | 2.33 | 3.54 | 52.65 | 14.33 | −3.34 | −4.58 | −2.05 | −0.8 |
| CDOC64, 238:1 | 0.50 | 0.86 | 1.80 | 3.20 | 4.94 | 51.66 | 12.22 | −3.04 | −4.2 | −1.74 | −0.29 |
| CDOC73, 371:1 | 0.38 | 0.72 | 1.61 | 2.95 | 4.61 | 52.44 | 13.36 | −2.36 | −3.69 | −1.35 | 0.07 |
| CDOC82, 636:1 | 0.42 | 0.61 | 1.00 | 1.46 | 1.93 | 56.64 | 20.16 | 1.94 | −0.1 | 1.86 | 3.4 |
| CDOC91, 1431:1 | 0.48 | 0.73 | 1.17 | 1.65 | 2.12 | 56.04 | 20.19 | 1.96 | −1.08 | 0.16 | 2.06 |
| CI to DC | - | - | - | - | - | 55.50 | 18.89 | −8.41 | −7.93 | −1.46 | −0.65 |
| Checkerboard | - | - | - | - | - | 72.91 | −11.77 | 1.76 | −5.07 | −4.17 | −0.66 |
| Selected dose (39.75, 0.5 µM) | - | - | - | - | - | - | - | 28.12 | 21.45 | 24.27 | 46.02 |
| CBD: Doxorubicin | |||||||||||
| CDOX19, 18:1 | 1.17 | 1.16 | 1.15 | 1.14 | 1.14 | 64.78 | 46.12 | 6.07 | 4.52 | −5.24 | 1.62 |
| CDOX28, 40:1 | 1.12 | 1.11 | 1.10 | 1.10 | 1.09 | 66.93 | 46.23 | 3.32 | 1.97 | −9.04 | 0 |
| CDOX37, 68:1 | 1.07 | 1.06 | 1.05 | 1.05 | 1.05 | 66.94 | 43.69 | 0.18 | −1.53 | −10.92 | −2.66 |
| CDOX46, 106:1 | 1.42 | 1.41 | 1.40 | 1.39 | 1.38 | 65.5 | 42.45 | 0.61 | −1.64 | −11.27 | −2.78 |
| CDOX55, 159:1 | 1.34 | 1.33 | 1.32 | 1.31 | 1.31 | 24.4 | 1.7 | −13.69 | −46.05 | −55.66 | −48.18 |
| CDOX64, 238:1 | 1.27 | 1.26 | 1.25 | 1.25 | 1.25 | 69.85 | 45.41 | 4.15 | 2.24 | −6.37 | 0.99 |
| CDOX73, 371:1 | 1.20 | 1.19 | 1.19 | 1.18 | 1.18 | 73.33 | 48.85 | 6.67 | 5.38 | −2.87 | 4.15 |
| CDOX82, 636:1 | 1.13 | 1.13 | 1.12 | 1.12 | 1.12 | 75.33 | 52.37 | 10.19 | 9.06 | 0.67 | 7.38 |
| CDOX91, 1431:1 | 1.07 | 1.06 | 1.06 | 1.06 | 1.06 | 76.85 | 55.09 | 9.89 | 8.72 | −2.69 | 6.75 |
| CI to DC | - | - | - | - | - | 70.24 | 53.04 | 4.96 | 4.05 | −5.43 | 2.21 |
| Checkerboard | - | - | - | - | - | 54.90 | −5.31 | 1.04 | −1.05 | −9.15 | −4.31 |
| Selected dose (38.42, 0.2 µM) | - | - | - | - | - | - | - | 32.52 | 25.65 | 15.16 | 31.53 |
| CBD: Paclitaxel | |||||||||||
| CPTX19, 18:1 | 1.50 | 1.61 | 1.89 | 2.35 | 2.95 | 35.94 | 0.09 | −12.32 | −20.86 | −20.59 | −19.3 |
| CPTX28, 40:1 | 0.91 | 1.04 | 1.40 | 2.00 | 2.78 | 42.63 | 5.16 | −7.42 | −12.28 | −11 | −9.74 |
| CPTX37, 68:1 | 0.70 | 0.85 | 1.31 | 2.05 | 3.03 | 40.73 | 2.11 | −8.38 | −11.49 | −10.07 | −8.88 |
| CPTX46, 106:1 | 0.48 | 0.63 | 1.09 | 1.83 | 2.82 | 50.48 | 12.66 | −2.18 | −5.93 | −4.42 | −3.42 |
| CPTX55, 159:1 | 0.37 | 0.54 | 1.04 | 1.85 | 2.93 | 55.54 | 16.97 | −0.14 | −3.26 | −2.93 | −1.55 |
| CPTX64, 238:1 | 0.37 | 0.59 | 1.24 | 2.32 | 3.75 | 51.98 | 11.68 | −1.04 | −4.26 | −3.37 | −1.96 |
| CPTX73, 371:1 | 0.34 | 0.61 | 1.41 | 2.73 | 4.49 | 52.76 | 12.71 | −0.76 | −3.19 | −2.35 | −1.01 |
| CPTX82, 636:1 * | 0.30 | 0.62 | 1.57 | 3.14 | 5.24 | 59.16 | 21.67 | 4.4 | 2.03 | 2.33 | 3.84 |
| CPTX91, 1431:1 * | 0.33 | 0.80 | 2.21 | 4.56 | 7.68 | 59.28 | 23.01 | 3.81 | 1.2 | 1.2 | 2.66 |
| CI to DC | - | - | - | - | - | 57.70 | 21.51 | −6.44 | −8.30 | −3.69 | −3.53 |
| Checkerboard | - | - | - | - | - | 53.78 | −31.41 | −5.35 | −10.85 | −7.90 | −6.63 |
| Selected dose (64.6, 0.1 µM) | - | - | 0.68 | - | - | - | - | ~2.98 | ~−1.69 | ~6.1 | ~1.95 |
| CBD:SN38 | |||||||||||
| CSN19, 18:1 | 1.77 | 5.14 | 18.44 | 51.56 | 115.29 | 40.24 | 13.73 | −0.43 | −3.87 | −3 | −1.38 |
| CSN28, 40:1 | 0.65 | 0.86 | 1.49 | 2.52 | 3.84 | 48.26 | 20.38 | 1.13 | −0.58 | 1.23 | 2.95 |
| CSN37, 68:1 | 0.57 | 0.76 | 1.33 | 2.19 | 3.23 | 45.18 | 15.31 | −2.32 | −4.31 | −2.18 | −0.62 |
| CSN46, 106:1 * | 0.47 | 0.59 | 0.95 | 1.44 | 1.98 | 50.63 | 21.35 | 1.96 | 0.13 | 2.04 | 3.69 |
| CSN55, 159:1 | 0.47 | 0.60 | 0.94 | 1.37 | 1.82 | 50.13 | 19.75 | 1.08 | −0.83 | −0.05 | 1.7 |
| CSN64, 238:1 | 0.45 | 0.60 | 0.97 | 1.40 | 1.85 | 49.6 | 18.07 | 0.23 | −1.72 | 0.05 | 1.64 |
| CSN73, 371:1 ** | 0.81 | 0.66 | 0.61 | 0.60 | 0.59 | 52.98 | 21.84 | 1.83 | 0.38 | 1.99 | 3.57 |
| CSN82, 636:1 ** | 0.81 | 0.71 | 0.68 | 0.67 | 0.67 | 59.38 | 29.68 | 5.05 | 3.67 | 4.45 | 6.4 |
| CSN91, 1431:1 ** | 0.92 | 0.93 | 0.91 | 0.91 | 0.90 | 58.73 | 29.9 | 3.43 | 2.21 | 2.79 | 4.66 |
| CI to DC | - | - | - | - | - | 48.39 | 23.86 | 2.37 | 1.35 | 3.58 | 4.46 |
| Checkerboard | - | - | - | - | - | 75.94 | 1.36 | 0.31 | −3.24 | −3.09 | −0.08 |
| Selected dose (42.45, 0.11 µM) | - | 0.66 | - | - | - | - | - | 14.55 | 23.86 | 31.38 | 33.57 |
| CBD: Vinorelbine | |||||||||||
| CVIN19, 18:1 | 1.30 | 1.39 | 1.64 | 2.02 | 2.53 | 31.94 | −1.9 | −9.19 | −16.9 | −16.72 | −15.15 |
| CVIN28, 40:1 | 1.03 | 1.18 | 1.58 | 2.25 | 3.12 | 37.98 | 2.98 | −8.44 | −12.37 | −12.2 | −10.12 |
| CVIN37, 68:1 | 0.71 | 0.86 | 1.30 | 2.02 | 2.96 | 41.71 | 5.92 | −6.9 | −8.99 | −7.59 | −5.91 |
| CVIN46, 106:1 | 0.56 | 0.74 | 1.26 | 2.12 | 3.25 | 44.84 | 9.79 | −4.31 | −6.29 | −5.35 | −3.24 |
| CVIN55, 159:1 | 0.46 | 0.67 | 1.27 | 2.26 | 3.58 | 50.93 | 14.65 | −1.54 | −3.63 | −3.13 | −1.06 |
| CVIN64, 238:1 | 0.45 | 0.72 | 1.52 | 2.84 | 4.58 | 49.75 | 12.98 | −2.21 | −5.08 | −3.64 | −1.84 |
| CVIN73, 371:1 | 0.33 | 0.59 | 1.36 | 2.63 | 4.32 | 52.5 | 16.13 | 0.16 | -2.16 | −1.01 | 1.06 |
| CVIN82, 636:1 * | 0.32 | 0.65 | 1.65 | 3.30 | 5.49 | 55.57 | 21.19 | 3.31 | 1.6 | 1.94 | 4.38 |
| CVIN91, 1431:1 ** | 0.73 | 0.60 | 0.56 | 0.55 | 0.55 | 56.32 | 25.34 | 5.55 | 3.36 | 2.56 | 5.09 |
| CI to DC | - | - | - | - | - | 57.45 | 23.54 | −5.89 | −6.49 | −1.64 | −0.70 |
| Checkerboard | - | - | - | - | - | 56.45 | −30.43 | −1.79 | −6.39 | −4.94 | −4.07 |
| Selected dose (42.4, 0.1 µM) | - | ~0.62- | - | - | - | - | - | 18.29 | 28.44 | 43.26 | 48.43 |
| Uniprot ID * | HGNC Gene ID | Protein Name | Log2 Fold Change |
|---|---|---|---|
| P04733 | MT1F | Metallothionein-1F | 7.03 |
| A0A0J9YWD4 | RIMS2 | Regulating synaptic membrane exocytosis protein 2 | 5.80 |
| Q8WX94 | NLRP7 | NACHT_ LRR and PYD domains-containing protein 7 | 5.75 |
| P02795 | MT2A | Metallothionein-2 | 3.78 |
| Q9Y2Z9 | COQ6 | Ubiquinone biosynthesis monooxygenase COQ6 | 2.60 |
| A6NCE7 | MAP1LC3B2 | Microtubule-associated proteins 1A/1B light chain 3 beta 2 | 1.68 |
| E9PRY0 | EIF3M | Eukaryotic translation initiation factor 3 subunit M | 1.66 |
| Q9NX24 | NHP2 | H/ACA ribonucleoprotein complex subunit 2 | 1.51 |
| Q5JPF3 | ANKRD36C | Ankyrin repeat domain-containing protein 36C | 1.50 |
| A0A0B4J1X2 | CACNA1G | Voltage-dependent T-type calcium channel subunit α-1G | 1.48 |
| P12645 | BMP3 | Bone morphogenetic protein 3 | 1.45 |
| Q9BQ39 | DDX50 | ATP-dependent RNA helicase DDX50 | 1.44 |
| P08910 | ABHD2 | Monoacylglycerol lipase ABHD2 | 1.40 |
| Q8TCU6 | PREX1 | Phosphatidylinositol 3_4_5-trisphosphate-dependent Rac exchanger 1 protein | 1.35 |
| Q3KQV9 | UAP1L1 | UDP-N-acetylhexosamine pyrophosphorylase-like protein 1 | 1.34 |
| Q14139 | UBE4A | Ubiquitin conjugation factor E4 A | 1.26 |
| M0QZQ3 | SPTBN4 | Spectrin beta chain | 1.26 |
| F5GZS6 | SLC3A2 | 4F2 cell-surface antigen heavy chain | 1.25 |
| P35527 | KRT9 | Keratin_ type I cytoskeletal 9 | 1.23 |
| F5GX23 | PSMD9 | 26S proteasome non-ATPase regulatory subunit 9 | 1.22 |
| Q5XKE5 | KRT79 | Keratin_ type II cytoskeletal 79 | 1.07 |
| Q9NRV9 | HEBP1 | Heme-binding protein 1 | 1.05 |
| H0YMI4 | USP3 | Ubiquitin carboxyl-terminal hydrolase | 1.01 |
| B4DIG0 | CDC25B | Protein-tyrosine-phosphatase | 1.00 |
| P29992 | GNA11 | Guanine nucleotide-binding protein subunit alpha-11 | 1.00 |
| Uniprot ID * | HGNC Gene ID | Protein Name | Log2 Fold Change |
|---|---|---|---|
| P02538 | KRT6A | Keratin 6A | −28.33 |
| E9PC85 | LRRK2 | Leucine-rich repeat kinase 2 | −30.08 |
| Q9BRK5 | SDF4 | 45 kDa calcium-binding protein | −10.86 |
| P28331 | NDUFS1 | NADH-ubiquinone oxidoreductase 75 kDa subunit_ mitochondrial | −8.44 |
| K7EPA3 | TMEM161A | Transmembrane protein 161A | −7.83 |
| Q14249 | ENDOG | Endonuclease G_ mitochondrial | −7.53 |
| Q9H501 | ESF1 | ESF1 homologue | −4.39 |
| Q9NRY5 | FAM114A2 | Protein FAM114A2 | −4.24 |
| C9J2C7 | NT5C1B-RDH14 | NT5C1B-RDH14 readthrough | −3.95 |
| Q9NX14 | NDUFB11 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 11_ mitochondrial | −3.49 |
| A0A3B3ISF0 | LARP1B | La-related protein 1B | −3.35 |
| Q9UP79 | ADAMTS8 | A disintegrin and metalloproteinase with thrombospondin motifs 8 | −3.34 |
| Q8IYA6 | CKAP2L | Cytoskeleton-associated protein 2-like | −3.31 |
| P60602 | ROMO1 | Reactive oxygen species modulator 1 | −3.26 |
| P55317 | FOXA1 | Hepatocyte nuclear factor 3-alpha | −3.25 |
| Q96G01 | BICD1 | Protein bicaudal D homologue 1 | −3.23 |
| O43291 | SPINT2 | Kunitz-type protease inhibitor 2 | −3.21 |
| O00762 | UBE2C | Ubiquitin-conjugating enzyme E2 C | −3.04 |
| O00291 | HIP1 | Huntingtin-interacting protein 1 | −2.83 |
| Q02818 | NUCB1 | Nucleobindin-1 | −2.81 |
| E9PJC6 | AHNAK | Neuroblast differentiation-associated protein AHNAK | −2.58 |
| Q2TV78 | MST1L | Putative macrophage stimulating 1-like protein | −2.51 |
| A0A6Q8PFY3 | FANCD2 | Fanconi anemia group D2 protein | −2.45 |
| A0A0U1RR07 | SYTL2 | Synaptotagmin-like protein 2 | −2.43 |
| Q8IVV2 | LOXHD1 | Lipoxygenase homologuey domain-containing protein 1 | −2.31 |
| Q9BUB7 | TMEM70 | Transmembrane protein 70_ mitochondrial | −2.28 |
| Q8TCT8 | SPPL2A | Signal peptide peptidase-like 2A | −2.27 |
| Q96DV4 | MRPL38 | 39S ribosomal protein L38_ mitochondrial | −2.27 |
| Q9Y646 | CPQ | Carboxypeptidase Q | −2.23 |
| Q15036 | SNX17 | Sorting nexin-17 | −2.22 |
| O75306 | NDUFS2 | NADH dehydrogenase [ubiquinone] iron–sulfur protein 2_ mitochondrial | −2.17 |
| G3V0I5 | NDUFV1 | NADH dehydrogenase [ubiquinone] flavoprotein 1_ mitochondrial | −2.12 |
| A0A590UJ21 | CUL1 | Cullin-1 | −1.97 |
| Q00534 | CDK6 | Cyclin-dependent kinase 6 | −1.96 |
| Q9P1A6 | DLGAP2 | Disks large-associated protein 2 | −1.95 |
| K7EJV0 | SEPTIN9 | Septin-9 | −1.92 |
| E7EN73 | KIAA0319L | Dyslexia-associated protein KIAA0319-like protein | −1.92 |
| C9JJ19 | MRPS34 | 28S ribosomal protein S34_ mitochondrial | −1.92 |
| Q92665 | MRPS31 | 28S ribosomal protein S31_ mitochondrial | −1.88 |
| Q96C01 | FAM136A | Protein FAM136A | −1.86 |
| Q16540 | MRPL23 | 39S ribosomal protein L23_ mitochondrial | −1.83 |
| A0A2R8YGD3 | RAPGEF2 | Cyclic nucleotide ras GEF | −1.81 |
| G8JLA1 | RDH13 | Retinol dehydrogenase 13 | −1.80 |
| P82650 | MRPS22 | 28S ribosomal protein S22_ mitochondrial | −1.76 |
| O00217 | NDUFS8 | NADH dehydrogenase [ubiquinone] iron–sulfur protein 8_ mitochondrial | −1.75 |
| Q9Y2Z2 | MTO1 | Protein MTO1 homologue_ mitochondrial | −1.68 |
| A0A0A0MRM2 | NRAP | Nebulin-related-anchoring protein | −1.64 |
| Q9UKX2 | MYH2 | Myosin-2 | −1.64 |
| Q6X4W1 | NSMF | NMDA receptor synaptonuclear signaling and neuronal migration factor | −1.62 |
| O75363 | BCAS1 | Breast carcinoma-amplified sequence 1 | −1.62 |
| O94913 | PCF11 | Pre-mRNA cleavage complex 2 protein Pcf11 | −1.62 |
| Q92823 | NRCAM | Neuronal cell adhesion molecule | −1.60 |
| Q96EY7 | PTCD3 | Pentatricopeptide repeat domain-containing protein 3_ mitochondrial | −1.57 |
| Q6ZXV5 | TMTC3 | Protein O-mannosyl-transferase TMTC3 | −1.53 |
| Q6IBS0 | TWF2 | Twinfilin-2 | −1.50 |
| Q9NY74 | ETAA1 | Ewing’s tumour-associated antigen 1 | −1.50 |
| Q8N961 | ABTB2 | Ankyrin repeat and BTB/POZ domain-containing protein 2 | −1.42 |
| A0A1B0GU86 | ACY1 | N-acyl-L-amino-acid amidohydrolase | −1.42 |
| P55011 | SLC12A2 | Solute carrier family 12 member 2 | −1.42 |
| H7BYU6 | ZNF521 | Zinc finger protein 521 | −1.38 |
| P04181 | OAT | Ornithine aminotransferase_ mitochondrial | −1.36 |
| Q8N565 | MREG | Melanoregulin | −1.33 |
| Q14802 | FXYD3 | FXYD domain-containing ion transport regulator 3 | −1.33 |
| Q7L2E3 | DHX30 | ATP-dependent RNA helicase DHX30 | −1.32 |
| Q12767 | TMEM94 | Transmembrane protein 94 | −1.31 |
| Q8N0X2 | SPAG16 | Sperm-associated antigen 16 protein | −1.30 |
| P14854 | COX6B1 | Cytochrome c oxidase subunit 6B1 | −1.30 |
| Q92820 | GGH | Gamma-glutamyl hydrolase | −1.28 |
| M0QY24 | ZNF546 | Zinc finger protein 546 | −1.26 |
| Q02880 | TOP2B | DNA topoisomerase 2-beta | −1.25 |
| P50579 | METAP2 | Methionine aminopeptidase 2 | −1.23 |
| Q8NEZ3 | WDR19 | WD repeat-containing protein 19 | −1.23 |
| G5E9Z9 | LRP2BP | LRP2 binding protein_ isoform CRA_a | −1.22 |
| Q8TDY2 | RB1CC1 | RB1-inducible coiled-coil protein 1 | −1.22 |
| H0Y5K5 | ERGIC3 | Endoplasmic reticulum-Golgi intermediate compartment protein 3 | −1.20 |
| Q9H869 | YY1AP1 | YY1-associated protein 1 | −1.20 |
| A7XYQ1 | SOBP | Sine oculis-binding protein homologue | −1.18 |
| P49006 | MARCKSL1 | MARCKS-related protein | −1.18 |
| H3BLV9 | SRPK1 | SRSF protein kinase 1 | −1.17 |
| Q9Y487 | ATP6V0A2 | V-type proton ATPase 116 kDa subunit a2 | −1.16 |
| Q9BZG8 | DPH1 | 2-(3-amino-3-carboxypropyl)histidine synthase subunit 1 | −1.15 |
| H3BV16 | NDUFB10 | Complex I-PDSW | −1.15 |
| Q99797 | MIPEP | Mitochondrial intermediate peptidase | −1.15 |
| O95294 | RASAL1 | RasGAP-activating-like protein 1 | −1.14 |
| P23921 | RRM1 | Ribonucleoside-diphosphate reductase large subunit | −1.14 |
| Q8IVG5 | SAMD9L | Sterile alpha motif domain-containing protein 9-like | −1.13 |
| P31350 | RRM2 | Ribonucleoside-diphosphate reductase subunit M2 | −1.13 |
| Q8WVV9 | HNRNPLL | Heterogeneous nuclear ribonucleoprotein L-like | −1.08 |
| A0A2R8YET2 | PRRC2C | Protein PRRC2C | −1.07 |
| Q9HCM1 | RESF1 | Retroelement silencing factor 1 | −1.07 |
| F8VWW7 | SPRYD3 | SPRY domain-containing protein 3 | −1.06 |
| P54296 | MYOM2 | Myomesin-2 | −1.03 |
| Q86XE3 | MICU3 | Calcium uptake protein 3_ mitochondrial | −1.02 |
| P49736 | MCM2 | DNA replication licensing factor MCM2 | −1.01 |
| Q9H4E5 | RHOJ | Rho-related GTP-binding protein RhoJ | −1.01 |
| Q9HAZ2 | PRDM16 | Histone-lysine N-methyltransferase PRDM16 | −1.00 |
| UniProt ID | HGNC Gene ID | Description | Log2 Fold Change * |
|---|---|---|---|
| Downregulated proteins | |||
| O75363 | BCAS1 | Breast carcinoma-amplified sequence 1 | −27.75 |
| Q9NVH1 | DNAJC11 | DnaJ homologue subfamily C member 11 | −24.68 |
| O00762 | UBE2C | Ubiquitin-conjugating enzyme E2 C | −4.84 |
| O94913 | PCF11 | Pre-mRNA cleavage complex 2 protein Pcf11 | −4.32 |
| A0A1B0GU86 | ACY1 | N-acyl-L-amino-acid amidohydrolase | −4.16 |
| A0A2R8YGD3 | RAPGEF2 | Cyclic nucleotide ras GEF | −2.48 |
| Q9P1V8 | SAMD15 | Sterile alpha motif domain-containing protein 15 | −2.44 |
| Q8TDI0 | CHD5 | Chromodomain-helicase-DNA-binding protein 5 | −2.42 |
| K7ER88 | ACAA2 | 3-Ketoacyl-CoA thiolase_ mitochondrial (Fragment) | −2.05 |
| P20701 | ITGAL | Integrin alpha-L | −2.01 |
| A0A0D9SG95 | CCT7 | T-complex protein 1 subunit eta | −1.78 |
| P03952 | KLKB1 | Plasma kallikrein | −1.72 |
| Q92823 | NRCAM | Neuronal cell adhesion molecule | −1.63 |
| O75582 | RPS6KA5 | Ribosomal protein S6 kinase alpha-5 | −1.56 |
| Q6UXG2 | ELAPOR1 | Endosome/lysosome-associated apoptosis and autophagy regulator 1 | −1.56 |
| Q5H9M0 | PWWP3B | PWWP domain-containing DNA repair factor 3B | −1.47 |
| P55199 | ELL | RNA polymerase II elongation factor ELL | −1.46 |
| Q9NPB8 | GPCPD1 | Glycerophosphocholine phosphodiesterase GPCPD1 | −1.46 |
| Q2TV78 | MST1L | Putative macrophage stimulating 1-like protein | −1.42 |
| A0A0U1RQX8 | CBL | E3 ubiquitin-protein ligase CBL | −1.35 |
| Q6P4H8 | ATPSCKMT | ATP synthase subunit C lysine N-methyltransferase | −1.34 |
| O14513 | NCKAP5 | Nck-associated protein 5 | −1.33 |
| Q8WVV9 | HNRNPLL | Heterogeneous nuclear ribonucleoprotein L-like | −1.32 |
| A0A2R8Y5P9 | SHROOM3 | Protein Shroom3 | −1.22 |
| O43303 | CCP110 | Centriolar coiled-coil protein of 110 kDa | −1.18 |
| Q9C091 | GREB1L | GREB1-like protein | −1.13 |
| Q9BQ52 | ELAC2 | Zinc phosphodiesterase ELAC protein 2 | −1.12 |
| A0A075B757 | NBPF14 | Neuroblastoma breakpoint family member 14 | −1.11 |
| O14497 | ARID1A | AT-rich interactive domain-containing protein 1A | −1.10 |
| Q96C90 | PPP1R14B | Protein phosphatase 1 regulatory subunit 14B | −1.04 |
| Q6IEG0 | SNRNP48 | U11/U12 small nuclear ribonucleoprotein 48 kDa protein | −1.00 |
| Upregulated proteins | |||
| O94973 | AP2A2 | AP-2 complex subunit alpha-2 | 1.20 |
| O14949 | UQCRQ | Cytochrome b-c1 complex subunit 8 | 1.11 |
| A0A1C7CYZ1 | MAPK15 | Mitogen-activated protein kinase 15 (Fragment) | 1.10 |
| Q5T9A4 | ATAD3B | ATPase family AAA domain-containing protein 3B | 1.03 |
| P49189 | ALDH9A1 | 4-Trimethylaminobutyraldehyde dehydrogenase | 1.01 |
| Combination Code * | Highest Concentration (µM) | Molar Ratio (CBD:Drug) | |
|---|---|---|---|
| CBD | Drug | ||
| CXYZ19 | 15.9 | 0.9 | 18:1 |
| CXYZ28 | 31.8 | 0.8 | 40:1 |
| CXYZ37 | 47.7 | 0.7 | 68:1 |
| CXYZ46 | 63.6 | 0.6 | 106:1 |
| CXYZ55 | 79.5 | 0.5 | 159:1 |
| CXYZ64 | 95.4 | 0.4 | 238:1 |
| CXYZ73 | 111.3 | 0.3 | 371:1 |
| CXYZ82 | 127.2 | 0.2 | 636:1 |
| CXYZ91 | 143.1 | 0.1 | 1431:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsherbiny, M.A.; Bhuyan, D.J.; Low, M.N.; Chang, D.; Li, C.G. Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms. Int. J. Mol. Sci. 2021, 22, 10103. https://doi.org/10.3390/ijms221810103
Alsherbiny MA, Bhuyan DJ, Low MN, Chang D, Li CG. Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms. International Journal of Molecular Sciences. 2021; 22(18):10103. https://doi.org/10.3390/ijms221810103
Chicago/Turabian StyleAlsherbiny, Muhammad A., Deep J. Bhuyan, Mitchell N. Low, Dennis Chang, and Chun Guang Li. 2021. "Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms" International Journal of Molecular Sciences 22, no. 18: 10103. https://doi.org/10.3390/ijms221810103
APA StyleAlsherbiny, M. A., Bhuyan, D. J., Low, M. N., Chang, D., & Li, C. G. (2021). Synergistic Interactions of Cannabidiol with Chemotherapeutic Drugs in MCF7 Cells: Mode of Interaction and Proteomics Analysis of Mechanisms. International Journal of Molecular Sciences, 22(18), 10103. https://doi.org/10.3390/ijms221810103