
Fusion with Promiscuous Gα16 Subunit Reveals Signaling Bias at Muscarinic Receptors


Abstract
1. Introduction
2. Results
2.1. Fusion Proteins
2.1.1. Description of Fusion Proteins
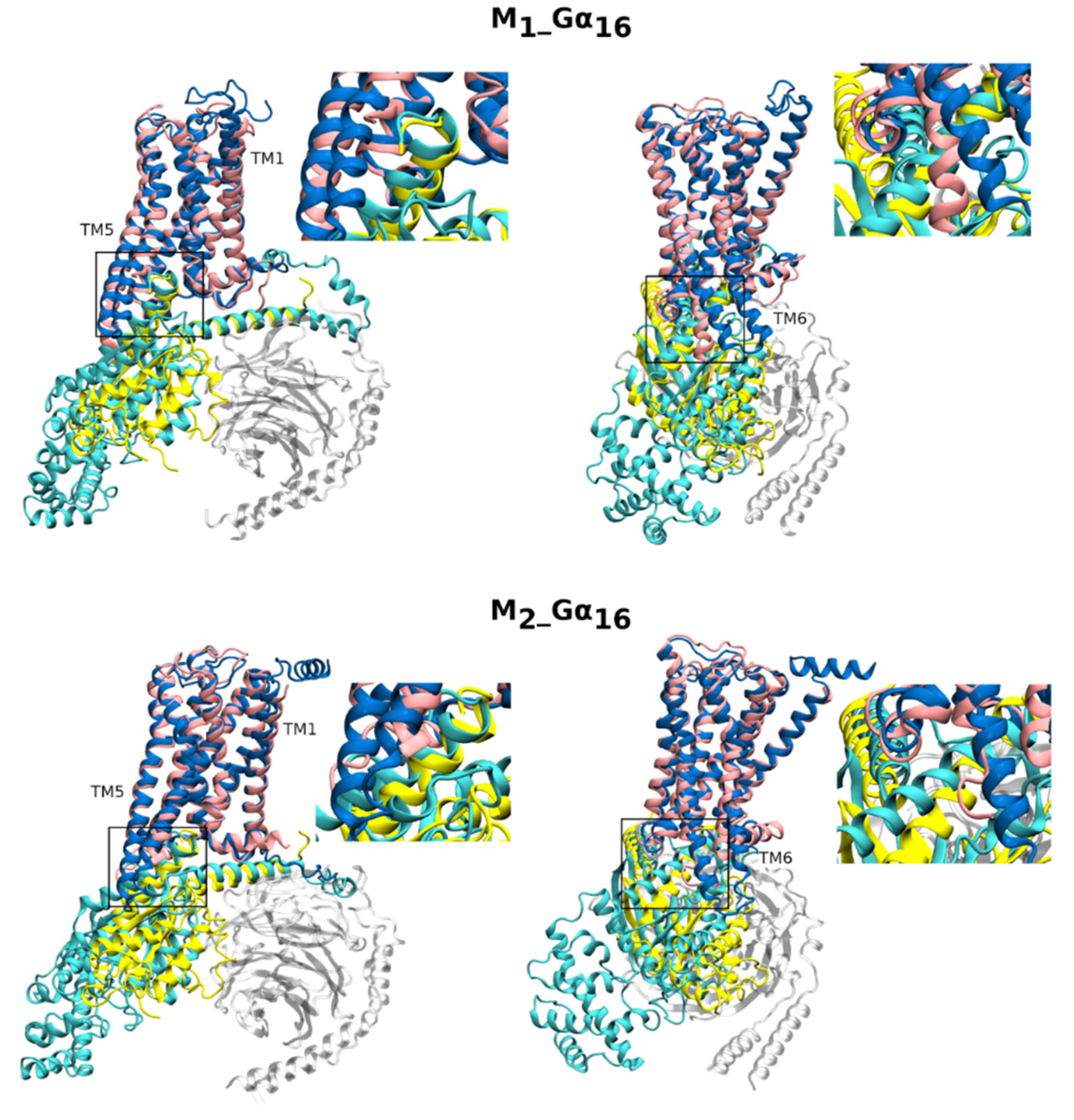
2.1.2. Homology Models of Fusion Proteins
2.1.3. Affinity of [3H]NMS for Gα16 Fused Receptors
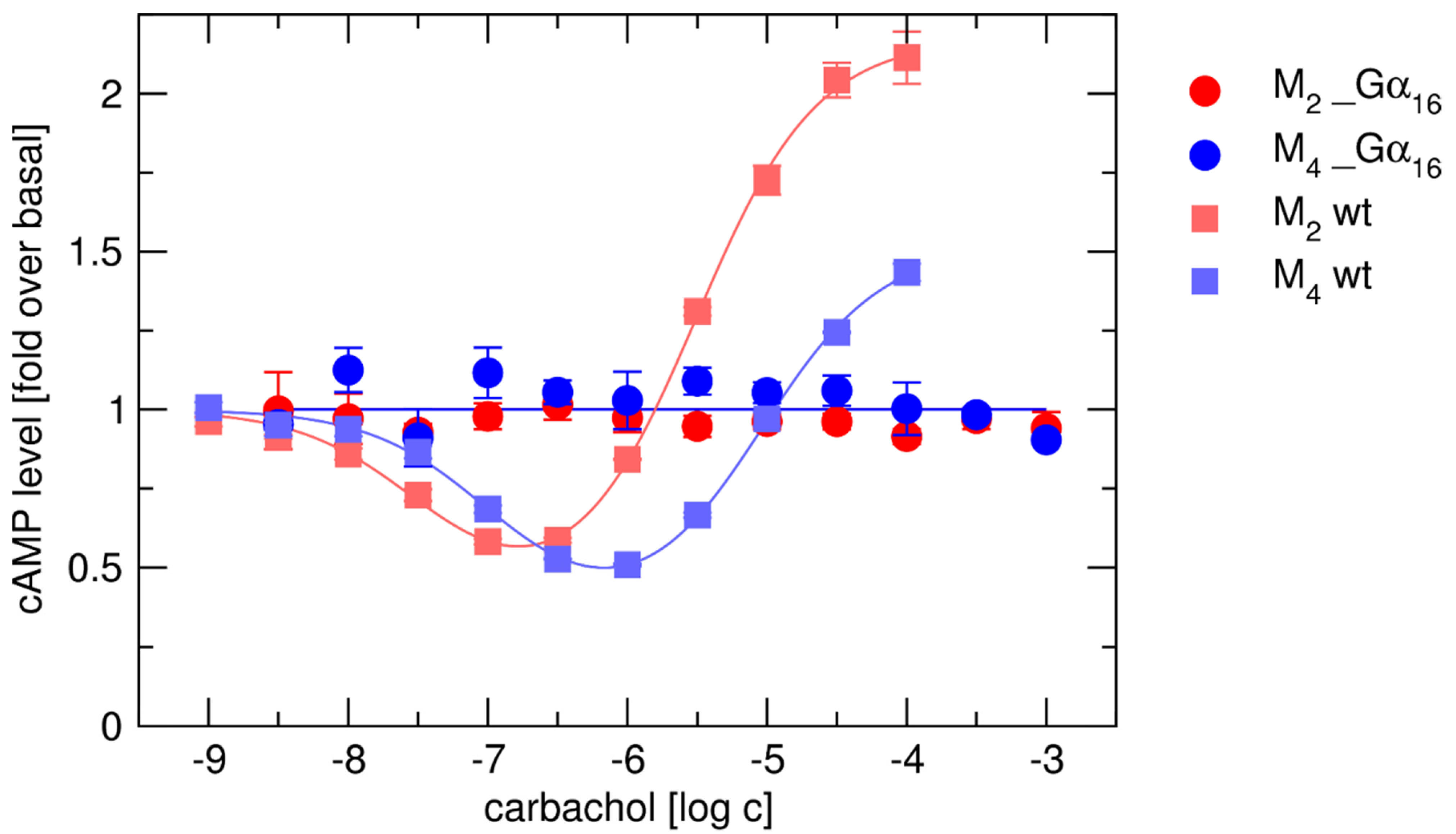
2.2. Lack of Coupling of Gα16_Fused Receptors with Endogenous G-Proteins
2.3. Binding and Functional Analysis of Gα16 Fused Receptors
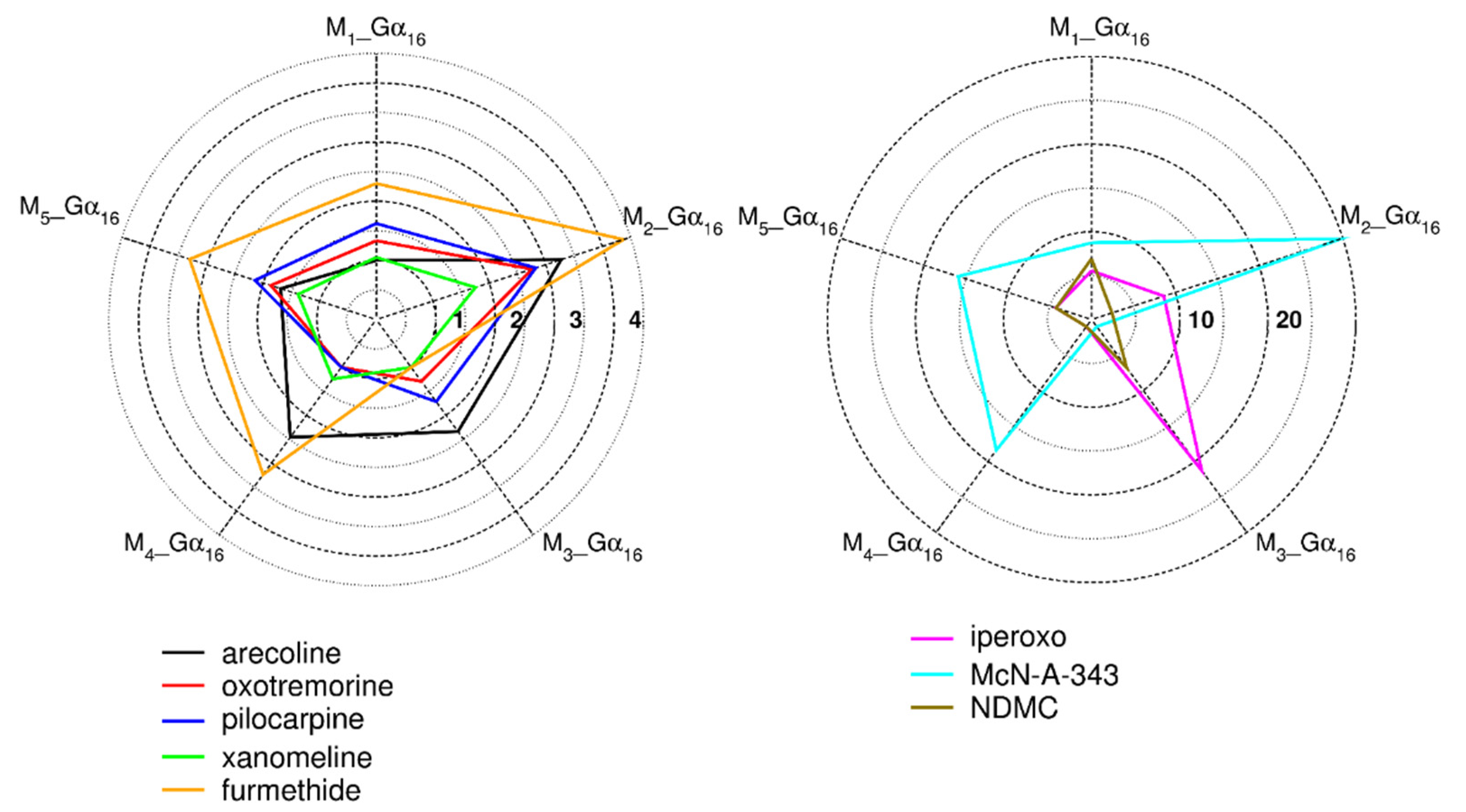
2.3.1. Binding Affinity of Tested Agonists to Gα16 Fused Muscarinic Receptors
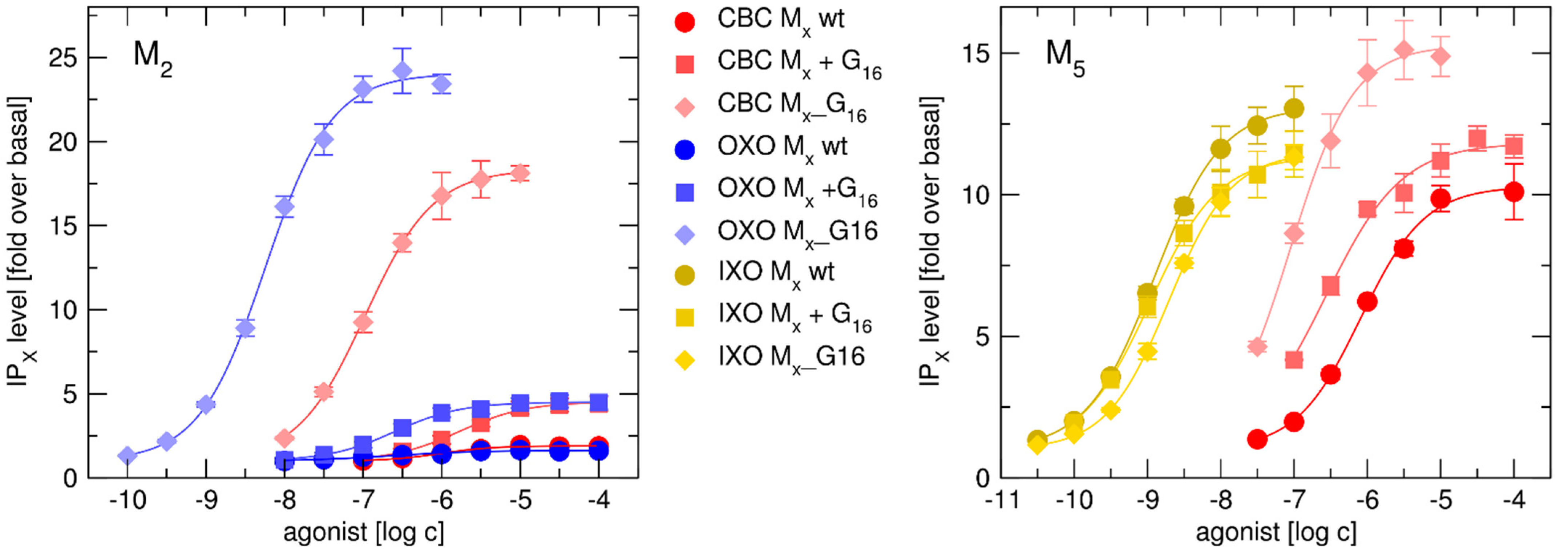
2.3.2. Functional Response of Gα16_Fused Muscarinic Receptors to Agonists
3. Discussion
4. Materials and Methods
4.1. Construct Preparation
4.2. Homology Modeling
4.3. Molecular Dynamics
4.4. Cell Culture and Membrane Preparation
4.5. Radioligand Binding Experiments
4.6. Measurement of Production of cAMP
4.7. Accumulation of Inositol Phosphates
4.8. Used Agonists
4.9. Data and Analysis
4.9.1. [3H]NMS Saturation Binding
4.9.2. Competition Binding
4.9.3. Functional Response
4.9.4. Operational Model of Functional Agonism
4.9.5. Relative Intrinsic Activity
4.9.6. Signaling Bias
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hermans, E. Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol. Ther. 2003, 99, 25–44. [Google Scholar] [CrossRef]
- Jakubík, J.; El-Fakahany, E.E.; Dolezal, V. Differences in kinetics of xanomeline binding and selectivity of activation of G proteins at M(1) and M(2) muscarinic acetylcholine receptors. Mol. Pharmacol. 2006, 70, 656–666. [Google Scholar] [CrossRef]
- Masuho, I.; Ostrovskaya, O.; Kramer, G.M.; Jones, C.D.; Xie, K.; Martemyanov, K.A. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal. 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Jakubík, J.; Bačáková, L.; Lisá, V.; El-Fakahany, E.E.; Tuček, S. Activation of muscarinic acetylcholine receptors via their allosteric binding sites. Proc. Natl. Acad. Sci. USA 1996, 93, 8705–8709. [Google Scholar] [CrossRef]
- Laugwitz, K.L.; Allgeier, A.; Offermanns, S.; Spicher, K.; Van Sande, J.; Dumont, J.E.; Schultz, G. The human thyrotropin receptor: A heptahelical receptor capable of stimulating members of all four G protein families. Proc. Natl. Acad. Sci. USA 1996, 93, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256. [Google Scholar] [CrossRef]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef]
- Lorenzen, E.; Ceraudo, E.; Berchiche, Y.A.; Rico, C.A.; Fürstenberg, A.; Sakmar, T.P.; Huber, T. G protein subtype–specific signaling bias in a series of CCR5 chemokine analogs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Ghahremani, M.H.; Albert, P.R. Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol. Ther. 2019, 200, 148–178. [Google Scholar] [CrossRef] [PubMed]
- Randáková, A.; Jakubík, J. Functionally selective and biased agonists of muscarinic receptors. Pharmacol. Res. 2021, 169, 105641. [Google Scholar] [CrossRef]
- Li, Y.Q.; Shrestha, Y.; Pandey, M.; Chen, M.; Kablan, A.; Gavrilova, O.; Offermanns, S.; Weinstein, L.S. Gq/11 α and Gs α mediate distinct physiological responses to central melanocortins. J. Clin. Investig. 2016, 126, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Kim, K.T. G protein-coupled receptor signalling and cross-talk: Achieving rapidity and specificity. Cell. Signal. 2002, 14, 397–405. [Google Scholar] [CrossRef]
- Denis, C.; Sauliere, A.; Galandrin, S.; Senard, J.-M.; Gales, C. Probing Heterotrimeric G Protein Activation: Applications to Biased Ligands. Curr. Pharm. Des. 2012, 18, 128–144. [Google Scholar] [CrossRef][Green Version]
- Matera, C.; Tata, A. Pharmacological Approaches to Targeting Muscarinic Acetylcholine Receptors. Recent Pat. CNS Drug Discov. 2014, 9, 85–100. [Google Scholar] [CrossRef]
- De Angelis, F.; Maria Tata, A. Analgesic Effects Mediated by Muscarinic Receptors: Mechanisms and Pharmacological Approaches. Cent. Nerv. Syst. Agents Med. Chem. 2016, 16, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Kruse, A.C.A.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef]
- Kruse, A.; Hu, J.; Pan, A.; Arlow, D. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef]
- Thal, D.M.; Sun, B. Crystal Structures of the M1 and M4 Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef]
- Randáková, A.; Nelic, D.; Ungerová, D.; Nwokoye, P.; Su, Q.; Doležal, V.; El-Fakahany, E.E.; Boulos, J.; Jakubík, J. Novel M2-selective, Gi-biased agonists of muscarinic acetylcholine receptors. Br. J. Pharmacol. 2020, 177, 2073–2089. [Google Scholar] [CrossRef]
- Downes, G.B.; Gautam, N. The G protein subunit gene families. Genomics 1999, 62, 544–552. [Google Scholar] [CrossRef]
- Bertin, B.; Freissmuth, M.; Jockers, R.; Strosberg, A.D.; Marullo, S. Cellular signaling by an agonist-activated receptor/G(s)α fusion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 8827–8831. [Google Scholar] [CrossRef]
- Wenzel-Seifert, K.; Seifert, R. Molecular analysis of β2-adrenoceptor coupling to G(s)-, G(i)-, and G(q)-proteins. Mol. Pharmacol. 2000, 58, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Massotte, D.; Brillet, K.; Kieffer, B.L.; Milligan, G. Agonists activate Gi1α or Gi2α fused to the human mu opioid receptor differently. J. Neurochem. 2002, 81, 1372–1382. [Google Scholar] [CrossRef]
- Milligan, G.; Parenty, G.; Stoddart, L.A.; Lane, J.R. Novel pharmacological applications of G-protein-coupled receptor-G protein fusions. Curr. Opin. Pharmacol. 2007, 7, 521–526. [Google Scholar] [CrossRef]
- Lane, J.R.; Powney, B.; Wise, A.; Rees, S.; Milligan, G. Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1, G i2, and Gi3. Mol. Pharmacol. 2007, 71, 1349–1359. [Google Scholar] [CrossRef]
- Suga, H.; Haga, T. Ligand screening system using fusion proteins of G protein-coupled receptors with G protein α subunits. Neurochem. Int. 2007, 51, 140–164. [Google Scholar] [CrossRef]
- Giannone, F.; Malpeli, G.; Lisi, V.; Grasso, S.; Shukla, P.; Ramarli, D.; Sartoris, S.; Monsurró, V.; Krampera, M.; Amato, E.; et al. The puzzling uniqueness of the heterotrimeric G15 protein and its potential beyond hematopoiesis. J. Mol. Endocrinol. 2010, 44, 259–269. [Google Scholar] [CrossRef]
- Grant, K.R.; Harnett, W.; Milligant, G. Differential G-protein expression during B- and T-cell development. Immunology 1997, 90, 564–571. [Google Scholar] [CrossRef]
- Giovinazzo, F.; Malpeli, G.; Zanini, S.; Parenti, M.; Piemonti, L.; Colombatti, M.; Valenti, M.T.; Dalle Carbonare, L.; Scarpa, A.; Sinnett-Smith, J.; et al. Ectopic expression of the heterotrimeric G15 protein in pancreatic carcinoma and its potential in cancer signal transduction. Cell. Signal. 2013, 25, 651–659. [Google Scholar] [CrossRef]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef]
- Jakubík, J.; Randáková, A.; Rudajev, V.; Zimčík, P.; El-Fakahany, E.E.; Doležal, V. Applications and limitations of fitting of the operational model to determine relative efficacies of agonists. Sci. Rep. 2019, 9, 4637. [Google Scholar] [CrossRef]
- Schrage, R.; Seemann, W.K.; Klöckner, J.; Dallanoce, C.; Racké, K.; Kostenis, E.; De Amici, M.; Holzgrabe, U.; Mohr, K. Agonists with supraphysiological efficacy at the muscarinic M2 ACh receptor. Br. J. Pharmacol. 2013, 169, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Schrage, R.; Holze, J.; Klöckner, J.; Balkow, A.; Klause, A.S.; Schmitz, A.L.; De Amici, M.; Kostenis, E.; Tränkle, C.; Holzgrabe, U.; et al. New insight into active muscarinic receptors with the novel radioagonist [3H]iperoxo. Biochem. Pharmacol. 2014, 90, 307–319. [Google Scholar] [CrossRef]
- Sur, C.; Mallorga, P.J.; Wittmann, M.; Jacobson, M.A.; Pascarella, D.; Williams, J.B.; Brandish, P.E.; Pettibone, D.J.; Scolnick, E.M.; Conn, P.J. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc. Natl. Acad. Sci. USA 2003, 100, 13674–13679. [Google Scholar] [CrossRef]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Antoni, F.A. Calcium regulation of adenylyl cyclase: Relevance for endocrine control. Trends Endocrinol. Metab. 1997, 8, 7–14. [Google Scholar] [CrossRef]
- Delmas, P.; Abogadie, F.C.; Milligan, G.; Buckley, N.J.; Brown, D.A. βγ dimers derived from G0 and G1 proteins contribute different components of adrenergic inhibition of Ca2+ channels in rat sympathetic neurones. J. Physiol. 1999, 518, 23–36. [Google Scholar] [CrossRef]
- Li, S.; Huang, S.; Peng, S. Bin Overexpression of G protein-coupled receptors in cancer cells: Involvement in tumor progression. Int. J. Oncol. 2005, 27, 1329–1339. [Google Scholar] [CrossRef]
- Corvol, J.C.; Muriel, M.P.; Valjent, E.; Féger, J.; Hanoun, N.; Girault, J.A.; Hirsch, E.C.; Hervé, D. Persistent increase in olfactory type G-protein α subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J. Neurosci. 2004, 24, 7007–7014. [Google Scholar] [CrossRef]
- Campbell, A.P.; Smrcka, A.V.; Arbor, A. Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. 2019, 17, 789–803. [Google Scholar] [CrossRef]
- Rasenick, M.M.; Watanabe, M.; Lazarevic, M.B.; Hatta, S.; Hamm, H.E. Synthetic peptides as probes for G protein function: Carboxyl-terminal Gαs peptides mimic Gs and evoke high affinity agonist binding to β-adrenergic receptors. J. Biol. Chem. 1994, 269, 21519–21525. [Google Scholar] [CrossRef]
- Krumins, A.M.; Gilman, A.G. Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J. Biol. Chem. 2006, 281, 10250–10262. [Google Scholar] [CrossRef]
- Jakubík, J.; Janíčková, H.; Randáková, A.; El-Fakahany, E.E.; Doležal, V. Subtype differences in pre-coupling of muscarinic acetylcholine receptors. PLoS ONE 2011, 6, e27732. [Google Scholar] [CrossRef]
- Barr, A.J.; Brass, L.F.; Manning, D.R. Reconstitution of receptors and GTP-binding regulatory proteins (G proteins) in Sf9 cells. A direct evaluation of selectivity in receptor-G protein coupling. J. Biol. Chem. 1997, 272, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Houston, C.; Wenzel-Seifert, K.; Bürckstümmer, T.; Seifert, R. The human histamine H2-receptor couples more efficiently to Sf9 insect cell Gs-proteins than to insect cell Gq-proteins: Limitations of Sf9 cells for the analysis of receptor/Gq-protein coupling. J. Neurochem. 2002, 80, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Uustare, A.; Näsman, J.; Åkerman, K.E.O.; Rinken, A. Characterization of M2 muscarinic receptor activation of different G protein subtypes. Neurochem. Int. 2004, 44, 119–124. [Google Scholar] [CrossRef]
- Parker, E.M.; Kameyama, K.; Higashijima, T.; Ross, E.M. Reconstitutively active G protein-coupled receptors purified from baculovirus-infected insect cells. J. Biol. Chem. 1991, 266, 519–527. [Google Scholar] [CrossRef]
- Heitz, F.; McClue, S.J.; Harris, B.A.; Guenet, C. Expression of human M2 muscarinic receptors in Sf9 cells: Characterisation and reconstitution with G-proteins. J. Recept. Signal Transduct. Res. 1995, 15, 55–70. [Google Scholar] [CrossRef] [PubMed]
- DeLapp, N.W. The antibody-capture [35S]GTPγS scintillation proximity assay: A powerful emerging technique for analysis of GPCR pharmacology. Trends Pharmacol. Sci. 2004, 25, 400–401. [Google Scholar] [CrossRef]
- Galés, C.; Rebois, R.V.; Hogue, M.; Trieu, P.; Breit, A.; Hébert, T.E.; Bouvier, M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2005, 2, 177–184. [Google Scholar] [CrossRef]
- Hein, P.; Frank, M.; Hoffmann, C.; Lohse, M.J.; Bünemann, M. Dynamics of receptor/G protein coupling in living cells. EMBO J. 2005, 24, 4106–4114. [Google Scholar] [CrossRef]
- Wessler, I.; Kilbinger, H.; Bittinger, F.; Unger, R.; Kirkpatrick, C.J. The non-neuronal cholinergic system in humans: Expression, function and pathophysiology. Life Sci. 2003, 72, 2055–2061. [Google Scholar] [CrossRef]
- Pereira, A.; McLaren, A.; Bell, W.R.; Copolov, D.; Dean, B. Potential clozapine target sites on peripheral hematopoietic cells and stromal cells of the bone marrow. Pharmacogenom. J. 2003, 3, 227–234. [Google Scholar] [CrossRef][Green Version]
- Onfroy, L.; Galandrin, S.; Pontier, S.M.; Seguelas, M.H.; N’Guyen, D.; Sénard, J.M.; Galés, C. G protein stoichiometry dictates biased agonism through distinct receptor-G protein partitioning. Sci. Rep. 2017, 7, 7885. [Google Scholar] [CrossRef]
- Randáková, A.; Dolejší, E.; Rudajev, V.; Zimčík, P.; Doležal, V.; El-Fakahany, E.E.; Jakubík, J. Classical and atypical agonists activate M1 muscarinic acetylcholine receptors through common mechanisms. Pharmacol. Res. 2015, 97, 27–39. [Google Scholar] [CrossRef]
- Jakubík, J.; Tuek, S.; El-Fakahany, E.E. Allosteric modulation by persistent binding of xanomeline of the interaction of competitive ligands with the M1 muscarinic acetylcholine receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1033–1041. [Google Scholar] [CrossRef]
- Valant, C.; Gregory, K.J.; Hall, N.E.; Scammells, P.J.; Lew, M.J.; Sexton, P.M.; Christopoulos, A. A novel mechanism of G protein-coupled receptor functional selectivity: Muscarinic partial agonist McN-A-343 as a bitopic orthosteric/allosteric ligand. J. Biol. Chem. 2008, 283, 29312–29321. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Haga, T. Palmitoylation of muscarinic acetylcholine receptor m2 subtypes: Reduction in their ability to activate G proteins by mutation of a putative palmitoylation site, cysteine 457, in the carboxyl-terminal tail. Arch. Biochem. Biophys. 1997, 340, 376–382. [Google Scholar] [CrossRef]
- Chen, C.A.; Manning, D.R. Regulation of G proteins by covalent modification. Oncogene 2001, 20, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–565. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 2019, 488, 1–13. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Gurwitz, D.; Haring, R.; Heldman, E.; Fraser, C.M.; Manor, D.; Fisher, A. Discrete activation of transduction pathways associated with acetylcholine m1 receptor by several muscarinic ligands. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 267, 21–31. [Google Scholar] [CrossRef]
- Burford, N.T.; Tobin, A.B.; Nahorski, S.R. Differential coupling of m1, m2 and m3 muscarinic receptor subtypes to inositol 1,4,5-trisphosphate and adenosine 3′,5′-cyclic monophosphate accumulation in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1995, 274, 134–142. [Google Scholar] [PubMed]
- Michal, P.; El-Fakahany, E.E.; Dolezal, V. Muscarinic M2 receptors directly activate Gq/11 and Gs G-proteins. J. Pharmacol. Exp. Ther. 2007, 320, 607–614. [Google Scholar] [CrossRef]
- Burt, A.R.; Sautel, M.; Wilson, M.A.; Rees, S.; Wise, A.; Milligan, G. Agonist occupation of an alpha2A-adrenoreceptor-Gi1alpha fusion protein results in activation of both receptor-linked and endogenous Gi proteins. Comparisons of their contributions to GTPase activity and signal transduction and analysis of receptor-G prot. J. Biol. Chem. 1998, 273, 10367–10375. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; Janíčková, H.; El-Fakahany, E.E.; Doležal, V. Negative cooperativity in binding of muscarinic receptor agonists and GDP as a measure of agonist efficacy. Br. J. Pharmacol. 2011, 162, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Devree, B.T.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.J.; Edwald, E.; Fung, J.J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef]
- Griffin, M.T.; Figueroa, K.W.; Liller, S.; Ehlert, F.J. Estimation of agonist activity at g protein-coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o, G s, and G15. J. Pharmacol. Exp. Ther. 2007, 321, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Bock, A.; Krebs, F.; Holzgrabe, U.; Mohr, K.; Lohse, M.J.; Wolber, G. Ligand-Specific Restriction of Extracellular Conformational Dynamics Constrains Signaling of the M2 Muscarinic Receptor. ACS Chem. Biol. 2017, 12, 1743–1748. [Google Scholar] [CrossRef]
- Holze, J.; Bermudez, M.; Pfeil, E.M.; Kauk, M.; Bödefeld, T.; Irmen, M.; Matera, C.; Dallanoce, C.; De Amici, M.; Holzgrabe, U.; et al. Ligand-Specific Allosteric Coupling Controls G-Protein-Coupled Receptor Signaling. ACS Pharmacol. Transl. Sci. 2020, 3, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Amatruda, T.T.; Steele, D.A.; Slepak, V.Z.; Simon, M.I. Gα16, a G protein α subunit specifically expressed in hematopoietic cells. Proc. Natl. Acad. Sci. USA 1991, 88, 5587–5591. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinforma. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Konagurthu, A.S.; Whisstock, J.; Stuckey, P.J. Progressive multiple alignment using sequence triplet optimizations and three-residue exchange costs. J. Bioinform. Comput. Biol. 2004, 2, 719–745. [Google Scholar] [CrossRef]
- El-Fakahany, E.E.; Jakubik, J. Radioligand Binding at Muscarinic Receptors. In Neuromethods; Myslivecek, J., Jakubík, J., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 107, pp. 37–68. [Google Scholar]
- Black, J.W.; Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond.-Biol. Sci. 1983, 220, 141–162. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M1_Gα16 | M2_Gα16 | M3_Gα16 | M4_Gα16 | M5_Gα16 | |
|---|---|---|---|---|---|
| Arecoline | 5.19 ± 0.06 | 4.68 ± 0.03 | 5.17 ± 0.08 | 4.68 ± 0.01 | 5.16 ± 0.04 |
| Carbachol | 4.87 ± 0.01 | 4.62 ± 0.01 | 4.77 ± 0.02 | 4.61 ± 0.02 | 4.72 ± 0.01 |
| Furmethide | 5.79 ± 0.01 | 4.69 ± 0.04 | 5.27 ± 0.03 | 4.71 ± 0.01 | 5.25 ± 0.02 |
| Iperoxo (high) | 8.35 ± 0.12 | n.d. | n.d. | n.d. | n.d. |
| Iperoxo (low) | 6.20 ± 0.08 | 5.83 ± 0.03 | 6.06 ± 0.04 | 5.96 ± 0.03 | 6.99 ± 0.02 |
| McN-A-343 | 4.24 ± 0.04 | 6.54 ± 0.04 | 5.14 ± 0.02 | 6.41 ± 0.02 | 5.34 ± 0.06 |
| NDMC | 7.06 ± 0.01 | 6.51 ± 0.04 | 6.75 ± 0.02 | 6.40 ± 0.01 | 6.77 ± 0.03 |
| Oxotremorine | 6.61 ± 0.01 | 5.70 ± 0.04 | 6.24 ± 0.03 | 5.86 ± 0.02 | 6.16 ± 0.03 |
| Pilocarpine | 5.26 ± 0.02 | 4.52 ± 0.01 | 4.92 ± 0.02 | 4.54 ± 0.03 | 4.88 ± 0.04 |
| Xanomeline | 7.29 ± 0.01 | 6.82 ± 0.02 | 7.19 ± 0.04 | 7.04 ± 0.03 | 7.06 ± 0.02 |
| JR-6 (high) | n.d. | n.d. | n.d. | 6.73 ± 0.28 | n.d. |
| JR-6 (low) | 4.97 ± 0.07 | 5.74 ± 0.10 | 5.07 ± 0.04 | 5.29 ± 0.21 | 5.44 ± 0.05 |
| JR-7 | 4.34 ± 0.05 | 5.17 ± 0.07 | 4.22 ± 0.06 | 4.82 ± 0.03 | 4.46 ± 0.04 |
| Arecoline | Carbachol | Furmethide | Iperoxo | McN-A343 | NDMC | Oxotremorine | Pilocarpine | Xanomeline | JR6 | JR7 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1_G16 | τ | 0.594 | ± | 0.057 | 0.887 | ± | 0.017 | 0.655 | ± | 0.005 | 1.577 | ± | 0.055 | 0.472 | ± | 0.02 | 0.5 | ± | 0.009 | 0.869 | ± | 0.027 | 0.668 | ± | 0.04 | 0.795 | ± | 0.007 | 0 | 0 |
| pKA | 6.68 | ± | 0.01 | 6.8 | ± | 0.06 | 6.3 | ± | 0.03 | 8.24 | ± | 0.03 | 6.53 | ± | 0.1 | 7.43 | ± | 0.05 | 7.82 | ± | 0.05 | 6.29 | ± | 0.02 | 8.25 | ± | 0.02 | n.c. | n.c. | |
| RAi | 0.45 | ± | 0.025 | 1 | ± | 0.01 | 0.234 | ± | 0.001 | 45.9 | ± | 0.9 | 0.248 | ± | 0.006 | 2.4 | ± | 0.02 * | 10.3 | ± | 0.2 | 0.234 | ± | 0.008 | 25 | ± | 0.1 | 0 | 0 | |
| M2_G16 | τ | 1.566 | ± | 0.038 | 1.41 | ± | 0.028 | 1.669 | ± | 0.034 | 9.61 | ± | 0.227 | 1.033 | ± | 0.021 | 0.951 | ± | 0.016 | 2.799 | ± | 0.081 | 0.946 | ± | 0.02 | 1.669 | ± | 0.034 | 0 | 0 |
| pKA | 6.73 | ± | 0.06 | 6.6 | ± | 0.06 | 6.18 | ± | 0.03 | 7.62 | ± | 0.08 | 6.68 | ± | 0.04 | 6.7 | ± | 0.02 | 7.64 | ± | 0.08 | 6.39 | ± | 0.02 | 8.15 | ± | 0.03 | n.c. | n.c. | |
| RAi | 1.47 | ± | 0.02 * | 1 | ± | 0.01 | 0.446 | ± | 0.005 * | 71.3 | ± | 1 | 0.875 | ± | 0.010 * | 0.846 | ± | 0.008 | 21.4 | ± | 0.4 * | 0.407 | ± | 0.005 * | 41.9 | ± | 0.5 * | 0 | 0 | |
| M3_G16 | τ | 0.541 | ± | 0.004 | 0.918 | ± | 0.02 | 0.656 | ± | 0.009 | 2.926 | ± | 0.876 | 0.307 | ± | 0.046 | 0.497 | ± | 0.003 | 0.834 | ± | 0.027 | 0.697 | ± | 0.039 | 0.777 | ± | 0.022 | 0 | 0 |
| pKA | 7.1 | ± | 0.01 | 6.8 | ± | 0.08 | 5.95 | ± | 0.31 | 8.56 | ± | 0.1 | 5.81 | ± | 0.13 | 7.43 | ± | 0.06 | 7.84 | ± | 0.05 | 6.31 | ± | 0.01 | 8.25 | ± | 0.03 | n.c. | n.c. | |
| RAi | 1.05 | ± | 0 | 1 | ± | 0.01 | 0.102 | ± | 0.001 | 176 | ± | 30 * | 0.028 | ± | 0.002 | 2.35 | ± | 0.01 | 10 | ± | 0.2 | 0.247 | ± | 0.008 | 23.8 | ± | 0.4 | 0 | 0 | |
| M4_G16 | τ | 0.804 | ± | 0.071 | 0.897 | ± | 0.04 | 0.865 | ± | 0.084 | 1.273 | ± | 0.055 | 0.704 | ± | 0.07 | 0.623 | ± | 0.05 | 0.994 | ± | 0.049 | 0.557 | ± | 0.054 | 0.866 | ± | 0.084 | 0 | 0 |
| pKA | 7.19 | ± | 0.04 | 7.1 | ± | 0.01 | 6.64 | ± | 0.01 | 7.87 | ± | 0.03 | 6.93 | ± | 0.02 | 6.8 | ± | 0.02 | 7.95 | ± | 0.01 | 6.47 | ± | 0.01 | 8.59 | ± | 0.01 | n.c. | n.c. | |
| RAi | 1.11 | ± | 0.06 | 1 | ± | 0.03 | 0.33 | ± | 0.018 | 8.26 | ± | 0.21 | 0.523 | ± | 0.03 | 0.346 | ± | 0.016 | 7.76 | ± | 0.22 | 0.144 | ± | 0.008 | 29.6 | ± | 1.7 | 0 | 0 | |
| M5_G16 | τ | 0.51 | ± | 0.005 | 1.126 | ± | 0.015 | 1.072 | ± | 0.01 | 0.825 | ± | 0.031 | 0.358 | ± | 0.009 | 0.709 | ± | 0.009 | 1.555 | ± | 0.016 | 0.8 | ± | 0.023 | 1.172 | ± | 0.015 | 0 | 0 |
| pKA | 7.02 | ± | 0.14 | 6.7 | ± | 0.02 | 6.25 | ± | 0.03 | 8.46 | ± | 0.14 | 6.96 | ± | 0.11 | 7.07 | ± | 0.03 | 7.72 | ± | 0.03 | 6.34 | ± | 0.01 | 8.2 | ± | 0.01 | n.c. | n.c. | |
| RAi | 0.76 | ± | 0.005 | 1 | ± | 0.01 | 0.336 | ± | 0.002 | 35 | ± | 1 | 0.452 | ± | 0.006 | 1.47 | ± | 0.01 | 14.5 | ± | 0.1 | 0.309 | ± | 0.005 | 33 | ± | 0.2 | 0 | 0 | |
| pEC50 | E′MAX | τ | pKA | RAi | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M2+Gα16 | carbachol | 5.59 | ± | 0.12 | 4.62 | ± | 0.37 | 0.88 | ± | 0.17 | 5.52 | ± | 0.17 † | 1 | ± | 0.03 |
| oxotremorine | 6.53 | ± | 0.13 | 4.46 | ± | 0.33 | 0.85 | ± | 0.15 | 6.26 | ± | 0.10 † | 5.41 | ± | 0.05 † | |
| M2_Gα16 | carbachol | 6.99 | ± | 0.06 | 18 | ± | 0.4 | 1.41 | ± | 0.03 | 6.6 | ± | 0.06 | 1 | ± | 0.01 |
| oxotremorine | 8.22 | ± | 0.08 | 22.4 | ± | 0.6 * | 2.8 | ± | 0.08 * | 7.64 | ± | 0.08 | 21.4 | ± | 0.4 | |
| M2 wt | carbachol | 6.01 | ± | 0.04 | 1.91 | ± | 0.07 | 0.2 | ± | 0.1 | 5.9 | ± | 0.1 | 1 | ± | 0.01 |
| oxotremorine | 6.68 | ± | 0.05 | 1.60 | ± | 0.05 | 0.2 | ± | 0.1 | 6.6 | ± | 0.1 | 3.08 | ± | 0.28 | |
| M5+Gα16 | carbachol | 6.61 | ± | 0.08 | 11.7 | ± | 0.4 | 0.814 | ± | 0.03 | 6.35 | ± | 0.08 † | 1 | ± | 0.02 |
| iperoxo | 8.95 | ± | 0.14 | 11.4 | ± | 0.8 | 0.785 | ± | 0.057 | 8.7 | ± | 0.14 † | 213 | ± | 9 † | |
| M5_Gα16 | carbachol | 7.03 | ± | 0.02 | 15.3 | ± | 0.2 | 1.126 | ± | 0.015 | 6.7 | ± | 0.02 | 1 | ± | 0.01 |
| iperoxo | 8.72 | ± | 0.14 | 11.3 | ± | 0.4 * | 0.825 | ± | 0.031 * | 8.46 | ± | 0.14 | 35 | ± | 1 | |
| M5 wt | carbachol | 6.09 | ± | 0.16 | 10.1 | ± | 1.1 | 0.68 | ± | 0.077 | 5.86 | ± | 0.16 | 1 | ± | 0.06 |
| iperoxo | 8.93 | ± | 0.16 | 13 | ± | 1.1 * | 1.08 | ± | 0.09 * | 8.61 | ± | 0.16 | 912 | ± | 45 † | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Randáková, A.; Nelic, D.; Hochmalová, M.; Zimčík, P.; Mulenga, M.J.; Boulos, J.; Jakubík, J. Fusion with Promiscuous Gα16 Subunit Reveals Signaling Bias at Muscarinic Receptors. Int. J. Mol. Sci. 2021, 22, 10089. https://doi.org/10.3390/ijms221810089
Randáková A, Nelic D, Hochmalová M, Zimčík P, Mulenga MJ, Boulos J, Jakubík J. Fusion with Promiscuous Gα16 Subunit Reveals Signaling Bias at Muscarinic Receptors. International Journal of Molecular Sciences. 2021; 22(18):10089. https://doi.org/10.3390/ijms221810089
Chicago/Turabian StyleRandáková, Alena, Dominik Nelic, Martina Hochmalová, Pavel Zimčík, Mutale Jane Mulenga, John Boulos, and Jan Jakubík. 2021. "Fusion with Promiscuous Gα16 Subunit Reveals Signaling Bias at Muscarinic Receptors" International Journal of Molecular Sciences 22, no. 18: 10089. https://doi.org/10.3390/ijms221810089
APA StyleRandáková, A., Nelic, D., Hochmalová, M., Zimčík, P., Mulenga, M. J., Boulos, J., & Jakubík, J. (2021). Fusion with Promiscuous Gα16 Subunit Reveals Signaling Bias at Muscarinic Receptors. International Journal of Molecular Sciences, 22(18), 10089. https://doi.org/10.3390/ijms221810089