Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
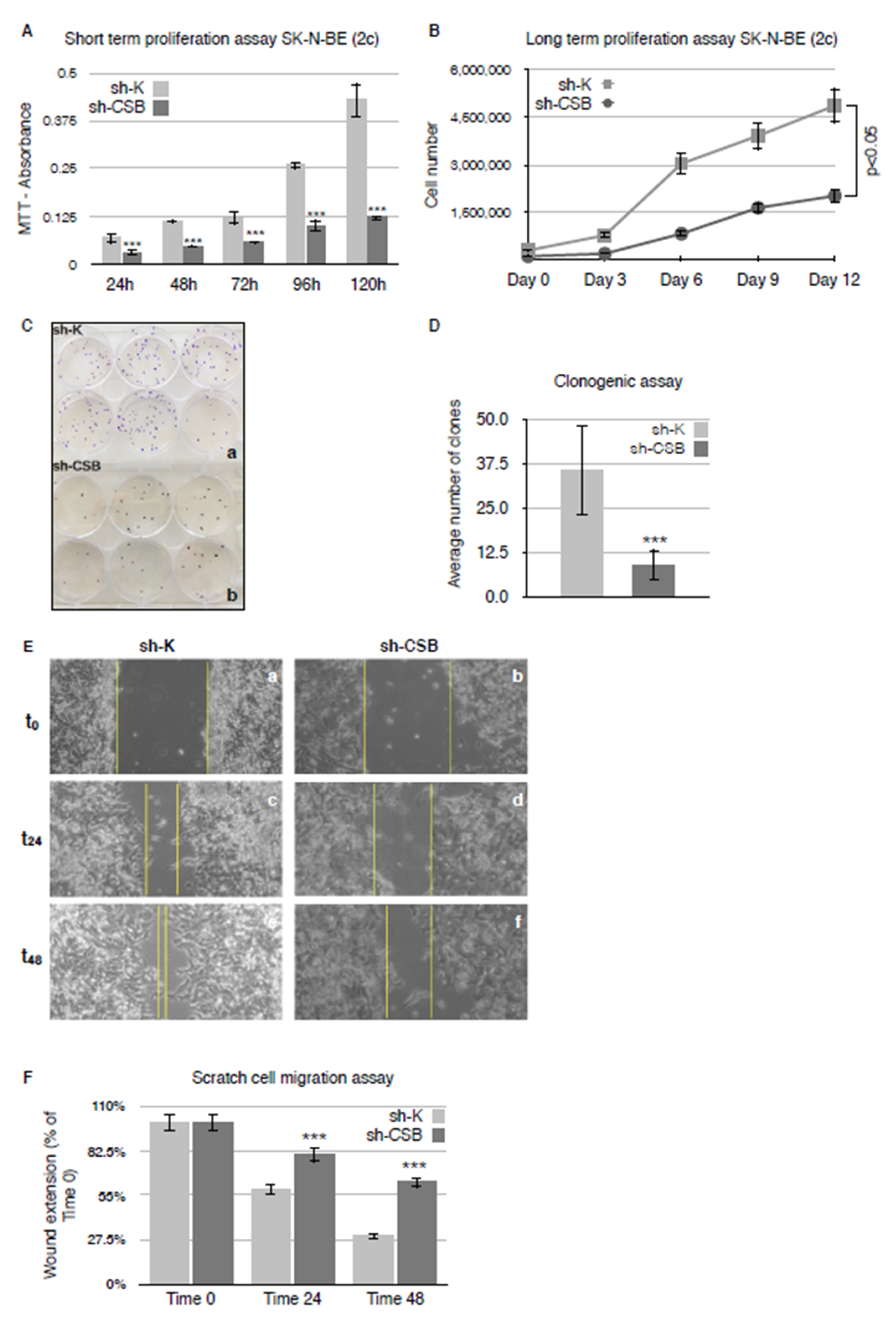
2.1. CSB Suppression Affects Proliferation, Clonogenicity, and Invasivity of Neuroblastoma Cells
2.2. CSB Suppression Determines a Massive Apoptosis Induction in Neuroblastoma Cells
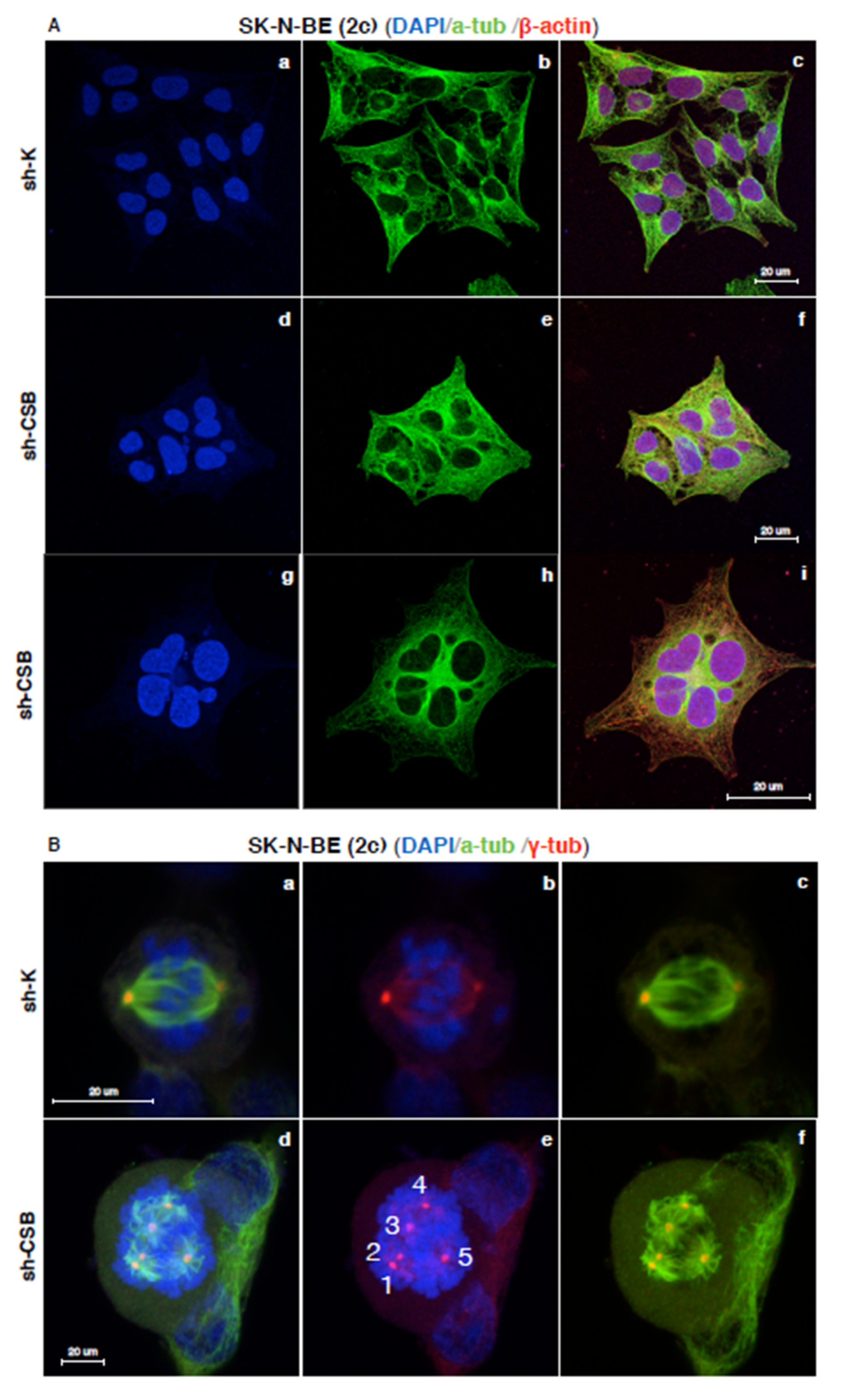
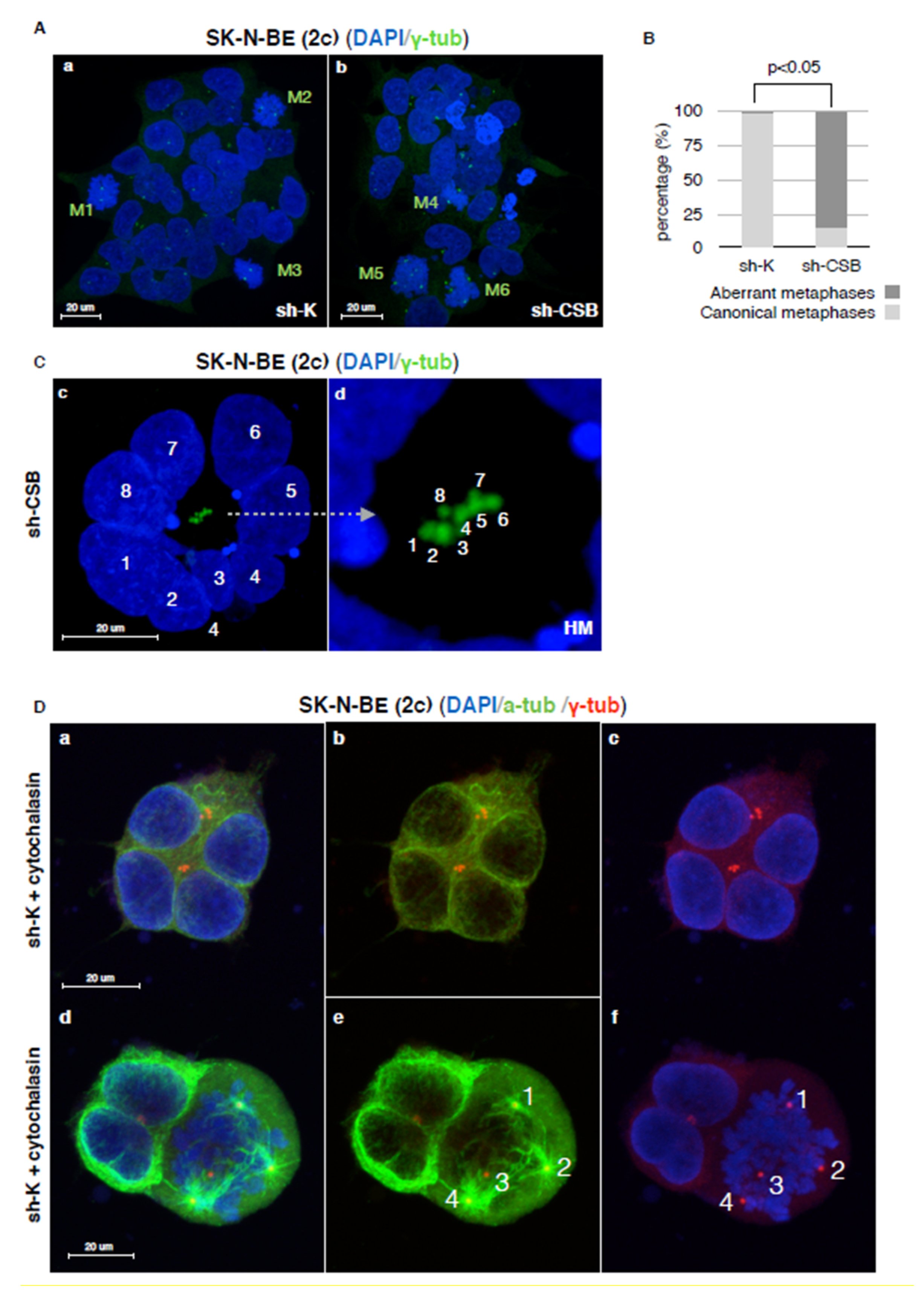
2.3. Centrosomal Abnormalities and Spindle Multipolarity in CSB Suppressed Neuroblastoma Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture and Silencing
4.2. Western Blot Analysis
4.3. Retrotranscription and Real-Time Quantitative PCR
4.4. MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium Bromide] Cell Proliferation Assay
4.5. Long-Term Proliferation Assay
4.6. Clonogenic Assay
4.7. Scratch Cell Migration Assay
4.8. Apoptosis Assay
4.9. Immunofluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomolonis, J.A.; Agarwal, S.; Shohet, J.M. Neuroblastoma pathogenesis: Deregulation of embryonic neural crest development. Cell Tissue Res. 2018, 372, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Dyberg, C.; Wickström, M. Neuroblastoma-A Neural Crest Derived Embryonal Malignancy. Front. Mol. Neurosci. 2019, 12, 9. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Yalçin, B.; Kremer, L.C.; Caron, H.N.; van Dalen, E.C. High-dose chemotherapy and autologoushaematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst. Rev. 2013, 3, CD006301. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef]
- Matson, S.W.; Bean, D.W.; George, J.W. DNA helicases: Enzymes with essential roles in all aspects of DNA metabolism. Bioessays 1994, 16, 13–22. [Google Scholar] [CrossRef]
- Citterio, E.; Van Den Boom, V.; Schnitzler, G.; Kanaar, R.; Bonte, E.; Kingston, R.E.; Hoeijmakers, J.H.; Vermeulen, W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell Biol. 2000, 20, 7643–7653. [Google Scholar] [CrossRef] [PubMed]
- Proietti-De-Santis, L.; Balzerano, A.; Prantera, G. CSB: An Emerging Actionable Target for Cancer Therapy. Trends Cancer 2018, 4, 172–175. [Google Scholar] [CrossRef]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair 2013, 12, 293–299. [Google Scholar] [CrossRef]
- Caputo, M.; Balzerano, A.; Arisi, I.; D’Onofrio, M.; Brandi, R.; Bongiorni, S.; Brancorsini, S.; Frontini, M.; Proietti-De-Santis, L. CSB ablation induced apoptosis is mediated by increased endoplasmic reticulum stress response. PLoS ONE 2017, 12, e0172399. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lian, H.; Sharma, P.; Schreiber-Agus, N.; Russell, R.G.; Chin, L.; van der Horst, G.T.; Bregman, D.B. Disruption of the Cockayne syndrome B gene impairs spontaneous tumorigenesis in cancer-predisposed Ink4a/ARF knockout mice. Mol. Cell Biol. 2001, 21, 1810–1818. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Selby, C.P.; Sancar, A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 1997, 94, 11205–11209. [Google Scholar] [CrossRef]
- Beerens, N.; Hoeijmakers, J.H.; Kanaar, R.; Vermeulen, W.; Wyman, C. The CSB protein actively wraps DNA. J. Biol. Chem. 2005, 280, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Bregman, D.B.; Halaban, R.; van Gool, A.J.; Henning, K.A.; Friedberg, E.C.; Warren, S.L. UV-induced ubiquitination of RNA polymerase II: A novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11586–11590. [Google Scholar] [CrossRef]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci. 2003, 116, 447–451. [Google Scholar] [CrossRef]
- Van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Lainé, J.P.; Egly, J.M. When transcription and repair meet: A complex system. Trends Genet. 2006, 22, 430–436. [Google Scholar] [CrossRef]
- Proietti-De-Santis, L.; Drané, P.; Egly, J.M. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006, 25, 1915–1923. [Google Scholar] [CrossRef]
- Nicolai, S.; Filippi, S.; Caputo, M.; Cipak, L.; Gregan, J.; Ammerer, G.; Frontini, M.; Willems, D.; Prantera, G.; Balajee, A.S.; et al. Identification of Novel Proteins Co-Purifying with Cockayne Syndrome Group B (CSB) Reveals Potential Roles for CSB in RNA Metabolism and Chromatin Dynamics. PLoS ONE 2015, 10, e0128558. [Google Scholar] [CrossRef]
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.A.; Caputo, M.; Ye, T.; Donnio, L.M.; Proietti-de-Santis, L.; Coin, F.; Laugel, V.; Egly, J.M. Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol. Cell. 2017, 68, 1054–1066.e6. [Google Scholar] [CrossRef]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.M.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin-proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef]
- Paccosi, E.; Proietti-De-Santis, L. The emerging role of Cockayne group A and B proteins in ubiquitin/proteasome-directed protein degradation. Mech. Ageing Dev. 2021, 195, 111466. [Google Scholar] [CrossRef]
- Latini, P.; Frontini, M.; Caputo, M.; Gregan, J.; Cipak, L.; Filippi, S.; Kumar, V.; Vélez-Cruz, R.; Stefanini, M.; Proietti-De-Santis, L. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle 2011, 10, 3719–3730. [Google Scholar] [CrossRef]
- Filippi, S.; Latini, P.; Frontini, M.; Palitti, F.; Egly, J.M.; Proietti-De-Santis, L. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J. 2008, 27, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; Martin, S.J. Caspase activation cascades in apoptosis. Biochem. Soc. Trans. 2008, 36 Pt 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Urbani, L.; Stearns, T. The centrosome. Curr. Biol. 1999, 9, R315–R317. [Google Scholar] [CrossRef]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef] [PubMed]
- Swift, C.C.; Eklund, M.J.; Kraveka, J.M.; Alazraki, A.L. Updates in Diagnosis, Management, and Treatment of Neuroblastoma. Radiographics 2018, 38, 566–580. [Google Scholar] [CrossRef]
- Valter, K.; Zhivotovsky, B.; Gogvadze, V. Cell death-based treatment of neuroblastoma. Cell Death Dis. 2018, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Braithwaite, A.W.; Robinson, P.J.; Chircop, M. Dynamin inhibitors induce caspase-mediated apoptosis following cytokinesis failure in human cancer cells and this is blocked by Bcl-2 overexpression. Mol. Cancer 2011, 10, 78. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Lartigue, L.; Perkins, G. Targeting Mcl-1 and other Bcl-2 family member proteins in cancer therapy. Pharmacol. Ther. 2019, 195, 13–20. [Google Scholar] [CrossRef]
- Gizatullin, F.; Yao, Y.; Kung, V.; Harding, M.W.; Loda, M.; Shapiro, G.I. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006, 66, 7668–7677. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Tsao, T.; Nakakuma, H.; Andreeff, M. Concomitant inhibition of Mdm2-p53 interaction and Aurora kinases activates the p53-dependent postmitotic checkpoints and synergistically induces p53-mediated mitochondrial apoptosis along with reduced endoreduplication in acute myelogenous leukemia. Blood 2008, 112, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Roschke, A.V.; Kirsch, I.R. Targeting karyotypic complexity and chromosomal instability of cancer cells. Curr. Drug Targets 2010, 11, 1341–1350. [Google Scholar] [CrossRef]
- Burrell, R.A.; Juul, N.; Johnston, S.R.; Reis-Filho, J.S.; Szallasi, Z.; Swanton, C. Targeting chromosomal instability and tumour heterogeneity in HER2-positive breast cancer. J. Cell Biochem. 2010, 111, 782–790. [Google Scholar] [CrossRef]
- McKenzie, C.; D’Avino, P.P. Investigating cytokinesis failure as a strategy in cancer therapy. Oncotarget 2016, 7, 87323–87341. [Google Scholar] [CrossRef]
- Jackson, J.R.; Patrick, D.R.; Dar, M.M.; Huang, P.S. Targeted anti-mitotic therapies: Can we improve on tubulin agents? Nat. Rev. Cancer 2007, 7, 107–117. [Google Scholar] [CrossRef]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krssák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, G.J.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Gamble, L.D.; Kees, U.R.; Tweddle, D.A.; Lunec, J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene 2012, 31, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Bate-Eya, L.T.; den Hartog, I.J.; van der Ploeg, I.; Schild, L.; Koster, J.; Santo, E.E.; Westerhout, E.M.; Versteeg, R.; Caron, H.N.; Molenaar, J.J.; et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 2016, 7, 27946–27958. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yan, S.; Attayan, N.; Ramalingam, S.; Thiele, C.J. Combination of an allosteric Akt Inhibitor MK-2206 with etoposide or rapamycin enhances the antitumor growth effect in neuroblastoma. Clin. Cancer Res. 2012, 18, 3603–3615. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paccosi, E.; Costantino, M.; Balzerano, A.; Filippi, S.; Brancorsini, S.; Proietti-De-Santis, L. Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival. Int. J. Mol. Sci. 2021, 22, 10070. https://doi.org/10.3390/ijms221810070
Paccosi E, Costantino M, Balzerano A, Filippi S, Brancorsini S, Proietti-De-Santis L. Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival. International Journal of Molecular Sciences. 2021; 22(18):10070. https://doi.org/10.3390/ijms221810070
Chicago/Turabian StylePaccosi, Elena, Michele Costantino, Alessio Balzerano, Silvia Filippi, Stefano Brancorsini, and Luca Proietti-De-Santis. 2021. "Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival" International Journal of Molecular Sciences 22, no. 18: 10070. https://doi.org/10.3390/ijms221810070
APA StylePaccosi, E., Costantino, M., Balzerano, A., Filippi, S., Brancorsini, S., & Proietti-De-Santis, L. (2021). Neuroblastoma Cells Depend on CSB for Faithful Execution of Cytokinesis and Survival. International Journal of Molecular Sciences, 22(18), 10070. https://doi.org/10.3390/ijms221810070