
The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin

 , and
, and
Abstract
1. Background
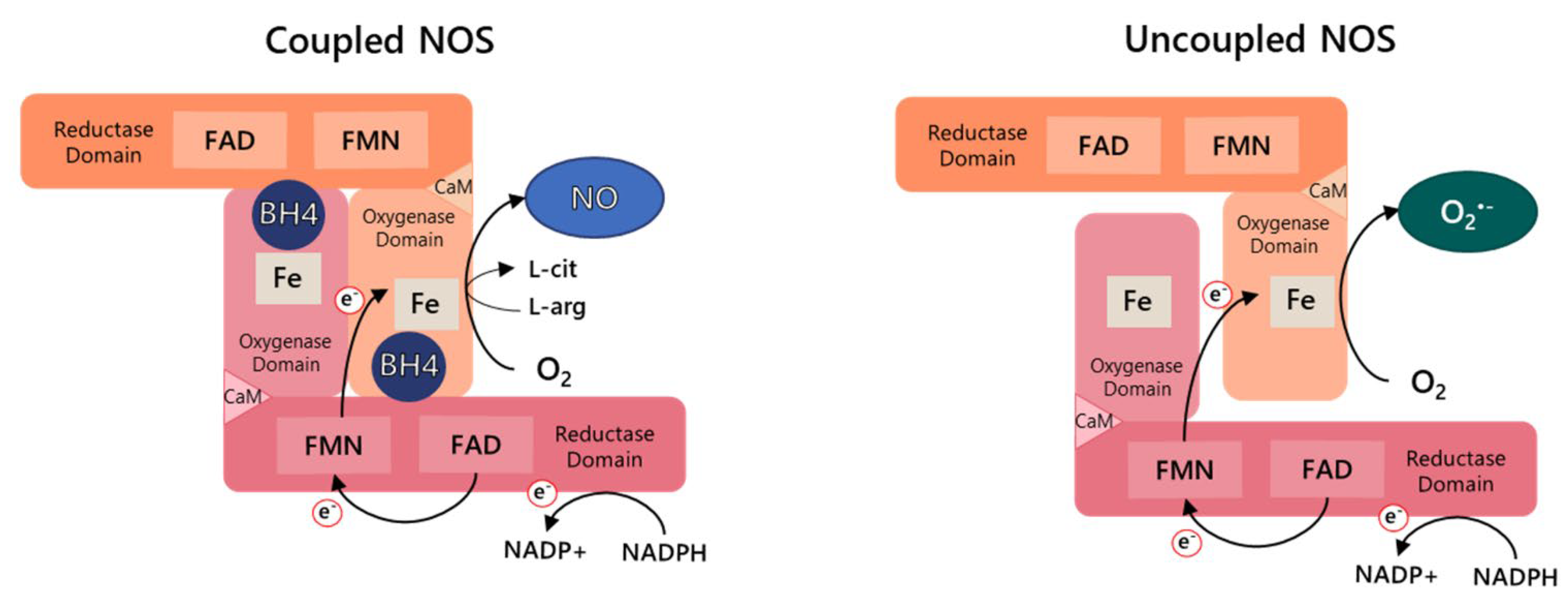
2. Nitric Oxide Synthase
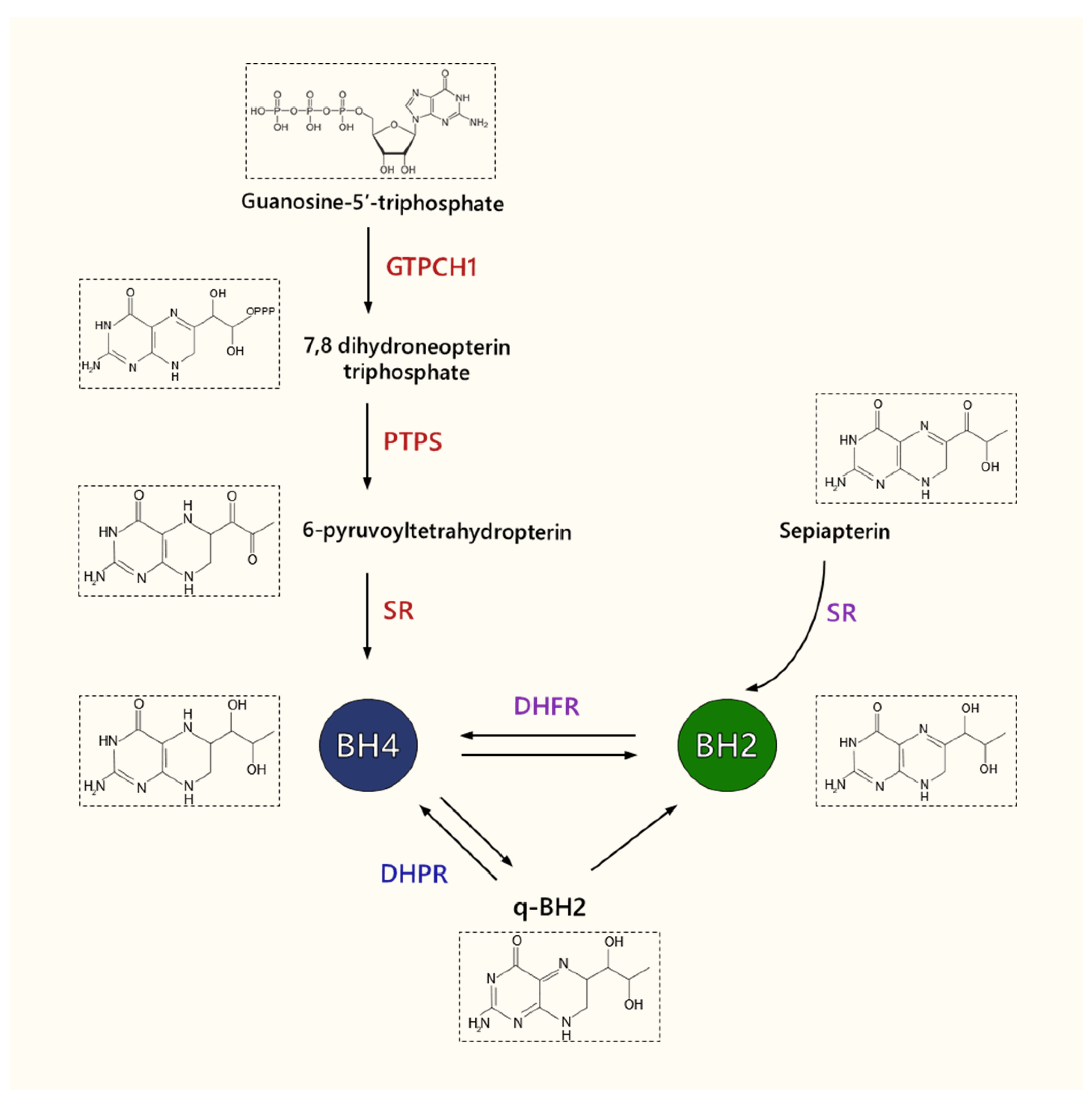
3. Tetrahydrobiopterin
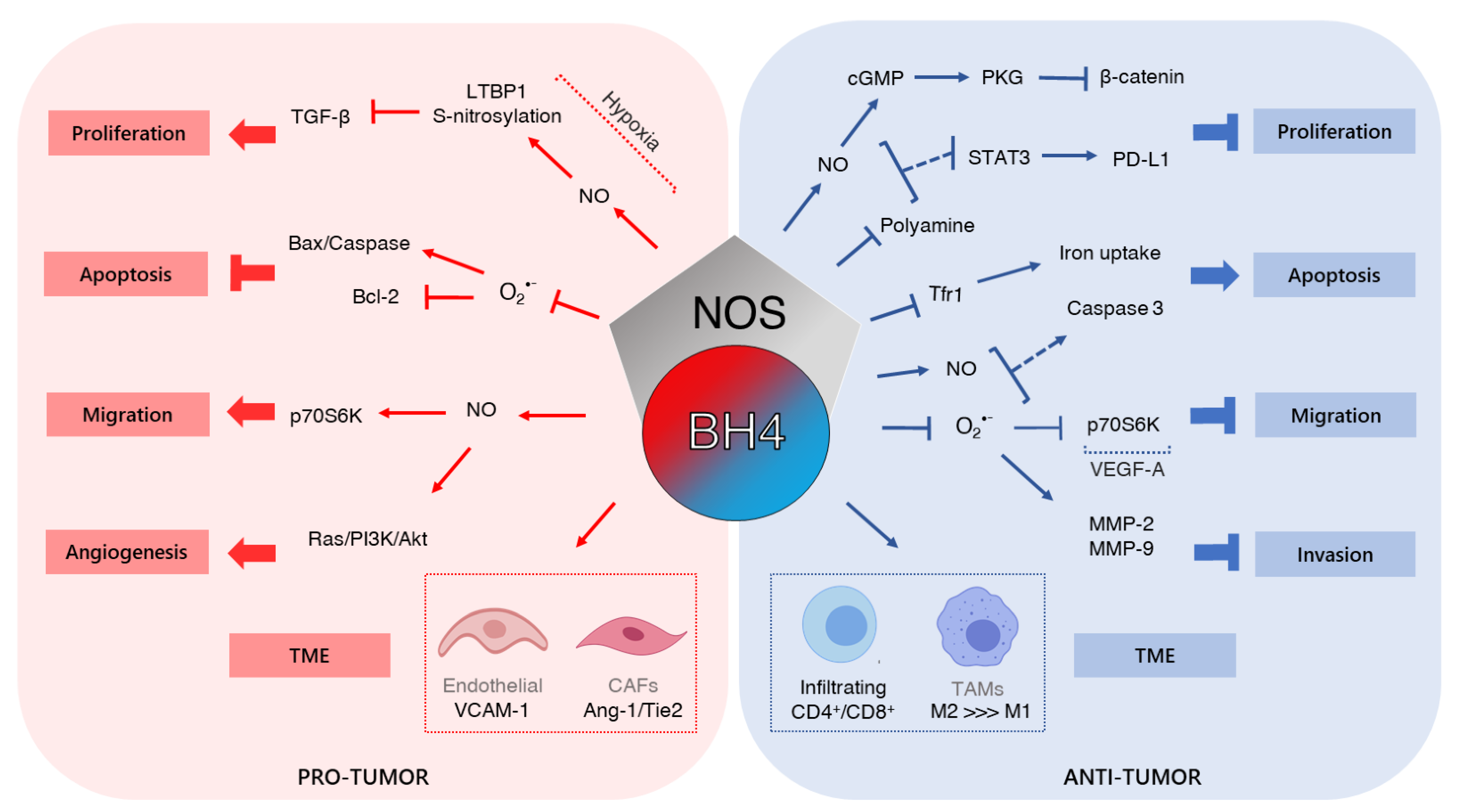
4. Tetrahydrobiopterin and Cancer
4.1. Cell Growth
4.2. Tumor Microenvironment and Angiogenesis
4.3. Migration and Invasion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Montor, W.R.; Salas, A.R.O.S.E.; Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 1–18. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36. [Google Scholar] [CrossRef]
- Arnold, M.; de Vries, E.; Whiteman, D.C.; Jemal, A.; Bray, F.; Parkin, D.M.; Soerjomataram, I. Global burden of cutaneous melanoma attributable to ultraviolet radiation in 2012. Int. J. Cancer 2018, 143, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, in press. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metab. Clin. Exp. 2019, 92, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-boosting, antioxidant and anti-inflammatory food supplements targeting pathogenesis of COVID-19. Front. Immunol. 2020, 11, 2337. [Google Scholar] [CrossRef] [PubMed]
- Bang, E.; Kim, D.H.; Chung, H.Y. Protease-activated receptor 2 induces ROS-mediated inflammation through Akt-mediated NF-κB and FoxO6 modulation during skin photoaging. Redox Biol. 2021, 44, 102022. [Google Scholar] [CrossRef] [PubMed]
- Anavi, S.; Tirosh, O. iNOS as a metabolic enzyme under stress conditions. Free Radic. Biol. Med. 2020, 146, 16–35. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; de Oliveira, É.A.; Faião-Flores, F.; Alicea-Rebecca, G.; Weeraratna, A.T.; Smalley, K.S.M.; Barros, S.B.M.; Maria-Engler, S.S. ER stress promotes antitumor effects in BRAFi/MEKi resistant human melanoma induced by natural compound 4-nerolidylcathecol (4-NC). Pharmacol. Res. 2019, 141, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, D.A.; Xisto, R.; Gonçalves, J.D.; da Silva, D.B.; Moura Soares, J.P.; Icimoto, M.Y.; Sant’Anna, C.; Gimenez, M.; de Angelis, K.; Llesuy, S.; et al. Imbalance between nitric oxide and superoxide anion induced by uncoupled nitric oxide synthase contributes to human melanoma development. Int. J. Biochem. Cell Biol. 2019, 115, 105592. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, Y.; Li, B.; Shen, K.; Li, Q.; Ni, Y.; Huang, L. Mitophagy in carcinogenesis, drug resistance and anticancer therapeutics. Cancer Cell Int. 2021, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L.; Li, H. Nitric oxide synthase and structure-based inhibitor design. Nitric Oxide 2017, 63, 68–77. [Google Scholar] [CrossRef]
- Stuehr, D.J.; Haque, M.M. Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. Br. J. Pharmacol. 2019, 176, 177–188. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J. Tetrahydrobiopterin, superoxide and vascular dysfunction. Free Radic. Biol. Med. 2009, 47, 1108. [Google Scholar] [CrossRef] [PubMed]
- Looft-Wilson, R.C.; Billaud, M.; Johnstone, S.R.; Straub, A.C.; Isakson, B.E. Interaction between nitric oxide signaling and gap junctions: Effects on vascular function. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1895–1902. [Google Scholar] [CrossRef]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Pannirselvam, M.; Simon, V.; Verma, S.; Anderson, T.; Triggle, C.R. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br. J. Pharmacol. 2003, 140, 701. [Google Scholar] [CrossRef]
- İnci, A.; Özaslan, A.; Okur, İ.; Biberoğlu, G.; Güney, E.; Ezgü, F.S.; Tümer, L.; İşeri, E. Autism: Screening of inborn errors of metabolism and unexpected results. Autism Res. 2021, 14, 887–896. [Google Scholar] [CrossRef]
- Infante, T.; Costa, D.; Napoli, C. Novel insights regarding nitric oxide and cardiovascular diseases. Angiology 2021, 72, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Diers, A.R.; Hogg, N. Cancer cell metabolism and the modulating effects of nitric oxide. Free Radic. Biol. Med. 2015, 79, 324. [Google Scholar] [CrossRef]
- Somasundaram, V.; Basudharm, D.; Bharadwaj, G. Molecular mechanisms of nitric oxide in cancer progression, signal transduction, and metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Investig. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, L.-J.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Nitric oxide activated by p38 and NF-κB facilitates apoptosis and cell cycle arrest under oxidative stress in evodiamine-treated human melanoma A375-S2 cells. Free Radic. Res. 2009, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Q.; Pang, B.; Kiziltepe, T.; Trudel, L.J.; Engelward, B.P.; Dedon, P.C.; Wogan, G.N. Threshold effects of nitric oxide-Induced toxicity and cellular responses in wild-type and p53-null human lymphoblastoid cells. Chem. Res. Toxicol. 2006, 19, 399. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, S.; Maksimovic-Ivanic, D.; Mojic, M.; Malaponte, G.; Libra, M.; Cardile, V.; Miljkovic, D.; Harhaji, L.; Dabideen, D.; Cheng, K.F.; et al. Novel nitric oxide-donating compound (S,R)-3-phenyl-4,5-dihydro-5-isoxazole acetic acid–nitric oxide (GIT-27NO) induces p53 mediated apoptosis in human A375 melanoma cells. Nitric Oxide 2008, 19, 177–183. [Google Scholar] [CrossRef]
- Branco-Price, C.; Evans, C.E.; Johnson, R.S.; Branco-Price, C.; Evans, C.E.; Johnson, R.S. Endothelial hypoxic metabolism in carcinogenesis and dissemination: HIF-A isoforms are a NO metastatic phenomenon. Oncotarget 2013, 4, 2567–2576. [Google Scholar] [CrossRef][Green Version]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef]
- Mijatović, S.; Savić-Radojević, A.; Plješa-Ercegovac, M.; Simić, T.; Nicoletti, F.; Maksimović-Ivanić, D. The double-faced role of nitric oxide and reactive oxygen species in solid tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.M.; Hanoteau, A.; Liu, H.-C.; Gaspero, A.; Parikh, F.; Gartrell-Corrado, R.D.; Hart, T.D.; Laoui, D.; Ginderachter, J.A.; Dharmaraj, N.; et al. Immune microenvironment modulation unmasks therapeutic benefit of radiotherapy and checkpoint inhibition. J. Immunother. Cancer 2019, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Rabender, C.S.; Alam, A.; Sundaresan, G.; Cardnell, R.J.; Yakovlev, V.A.; Mukhopadhyay, N.D.; Graves, P.; Zweit, J.; Mikkelsen, R.B. The role of nitric oxide synthase uncoupling in tumor progression. Mol. Cancer Res. 2015, 13, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Kröller-Schön, S.; Oelze, M.; Hahad, O.; Li, H.; Schulz, R.; Steven, S.; Münzel, T. Oxidative stress and inflammation contribute to traffic noise-induced vascular and cerebral dysfunction via uncoupling of nitric oxide synthases. Redox Biol. 2020, 34, 101506. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative stress and its association with cardiometabolic disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef]
- Campanholo, V.M.L.P.; Silva, R.M.; Silva, T.D.; Neto, R.A.; Ribeiro Paiotti, A.P.; Ribeiro, D.A.; Forones, N.M. Oral concentrated grape juice suppresses expression of NF-kappa B, TNF-α and iNOS in experimentally induced colorectal carcinogenesis in wistar rats. Asian Pac. J. Cancer Prev. 2015, 16, 947–952. [Google Scholar] [CrossRef]
- Peñarando, J.; López-Sánchez, L.M.; Mena, R.; Guil-Luna, S.; Conde, F.; Hernández, V.; Toledano, M.; Gudiño, V.; Raponi, M.; Billard, C.; et al. A role for endothelial nitric oxide synthase in intestinal stem cell proliferation and mesenchymal colorectal cancer. BMC Biol. 2018, 16, 3. [Google Scholar] [CrossRef]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. The potential role of iNOS in ovarian cancer progression and chemoresistance. Int. J. Mol. Sci. 2019, 20, 1751. [Google Scholar] [CrossRef]
- Lawal, B.; Lee, C.-Y.; Mokgautsi, N.; Sumitra, M.R.; Khedkar, H.; Wu, A.T.H.; Huang, H.-S. mTOR/EGFR/iNOS/MAP2K1/FGFR/TGFB1 are druggable candidates for N-(2,4-Difluorophenyl)-2′,4′-Difluoro-4-Hydroxybiphenyl-3-Carboxamide (NSC765598), with consequent anticancer implications. Front. Oncol. 2021, 11, 932. [Google Scholar] [CrossRef]
- Thomas, D.D.; Wink, D.A. NOS2 as an emergent player in progression of cancer. Antioxid. Redox Signal. 2017, 26, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 2019, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; Korth, H.-G.; Stenert, V.; Sustmann, R.; Groot, H. The autoxidation of tetrahydrobiopterin revisited: Proof of superoxide formation from reaction of tetrahydrobiopterin with molecular oxygen. J. Biol. Chem. 2003, 278, 24481–24490. [Google Scholar] [CrossRef]
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Blau, N.; Thöny, B. Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Channon, K.M. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 2011, 25, 81–88. [Google Scholar] [CrossRef]
- Peterson, T.E.; d’Uscio, L.V.; Cao, S.; Wang, X.-L.; Katusic, Z.S. Guanosine triphosphate cyclohydrolase I expression and enzymatic activity are present in caveolae of endothelial cells. Hypertension 2009, 53, 189–195. [Google Scholar] [CrossRef]
- Tatham, A.L.; Crabtree, M.J.; Warrick, N.; Cai, S.; Alp, N.J.; Channon, K.M. GTP cyclohydrolase I expression, protein, and activity determine intracellular tetrahydrobiopterin levels, independent of GTP cyclohydrolase feedback regulatory protein expression. J. Biol. Chem. 2009, 284, 13660–13668. [Google Scholar] [CrossRef]
- Shimizu, S.; Hiroi, T.; Ishii, M.; Hagiwara, T.; Wajima, T.; Miyazaki, A.; Kiuchi, Y. Hydrogen peroxide stimulates tetrahydrobiopterin synthesis through activation of the Jak2 tyrosine kinase pathway in vascular endothelial cells. Int. J. Biochem. Cell Biol. 2008, 40, 755–765. [Google Scholar] [CrossRef]
- Xue, J.; Yu, C.; Sheng, W.; Zhu, W.; Luo, J.; Zhang, Q.; Yang, H.; Cao, H.; Wang, W.; Zhou, J.; et al. The Nrf2/GCH1/BH4 axis ameliorates radiation-induced skin injury by modulating the ROS cascade. J. Investig. Dermatol. 2017, 137, 2059–2068. [Google Scholar] [CrossRef]
- Cardnell, R.J.G.; Rabender, C.S.; Ross, G.R.; Guo, C.; Howlett, E.L.; Alam, A.; Wang, X.Y.; Akbarali, H.I.; Mikkelsen, R.B. Sepiapterin ameliorates chemically induced murine colitis and azoxymethane-induced colon cancer. J. Pharmacol. Exp. Ther. 2013, 347, 117–125. [Google Scholar] [CrossRef]
- Melo, F.H.M.; Molognoni, F.; Morais, A.S.; Toricelli, M.; Mouro, M.G.; Higa, E.M.S.; Lopes, J.D.; Jasiulionis, M.G. Endothelial nitric oxide synthase uncoupling as a key mediator of melanocyte malignant transformation associated with sustained stress conditions. Free Radic. Biol. Med. 2011, 50, 1263–1273. [Google Scholar] [CrossRef]
- Zhong, G.C.; Zhao, Z.B.; Cheng, Y.; Wang, Y.B.; Qiu, C.; Mao, L.H.; Hu, J.J.; Cai, D.; Liu, Y.; Gong, J.P.; et al. Epigenetic silencing of GCH1promotes hepatocellular carcinoma growth by activating superoxide anion-mediated ASK1/p38 signaling via inhibiting tetrahydrobiopterin de novo biosynthesis. Free Radic. Biol. Med. 2021, 168, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Wang, K.; Tang, J.; Chen, Y.; Jin, G.; Liu, X. The knockdown of the sepiapterin reductase gene suppresses the proliferation of breast cancer by inducing ROS-mediated apoptosis. Int. J. Clin. Exp. Pathol. 2020, 13, 2228–2239. [Google Scholar] [PubMed]
- Tran, A.N.; Walker, K.; Harrison, D.G.; Chen, W.; Mobley, J.; Hocevar, L.; Hackney, J.R.; Sedaka, R.S.; Pollock, J.S.; Goldberg, M.S.; et al. Reactive species balance via GTP cyclohydrolase i regulates glioblastoma growth and tumor initiating cell maintenance. Neuro-Oncology 2018, 20, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, W.; Cao, J.; Wang, F.; Geng, Y.; Cao, J.; Xu, X.; Zhou, J.; Liu, P.; Zhang, S. Upregulation of AUF1 is involved in the proliferation of esophageal squamous cell carcinoma through GCH1. Int. J. Oncol. 2016, 49, 2001–2010. [Google Scholar] [CrossRef]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zheng, K.; Ma, C.; Li, J.; Zhuo, L.; Huang, W.; Chen, T.; Jiang, Y. PTPS Facilitates compartmentalized LTBP1 S-Nitrosylation and promotes tumor growth under hypoxia. Mol. Cell 2020, 77, 95–107.e5. [Google Scholar] [CrossRef]
- Dai, Y.; Cui, J.; Gan, P.; Li, W. Downregulation of tetrahydrobiopterin inhibits tumor angiogenesis in BALB/c-nu mice with hepatocellular carcinoma. Oncol. Rep. 2016, 36, 669–675. [Google Scholar] [CrossRef][Green Version]
- Chen, L.; Zeng, X.; Wang, J.; Briggs, S.S.; O’Neill, E.; Li, J.; Leek, R.; Kerr, D.J.; Harris, A.L.; Cai, S. Roles of tetrahydrobiopterin in promoting tumor angiogenesis. Am. J. Pathol. 2010, 177, 2671–2680. [Google Scholar] [CrossRef]
- Pickert, G.; Lim, H.Y.; Weigert, A.; Häussler, A.; Myrczek, T.; Waldner, M.; Labocha, S.; Ferreirós, N.; Geisslinger, G.; Lötsch, J.; et al. Inhibition of GTP cyclohydrolase attenuates tumor growth by reducing angiogenesis and M2-like polarization of tumor associated macrophages. Int. J. Cancer 2013, 132, 591–604. [Google Scholar] [CrossRef]
- Chen, L.; Zeng, X.; Kleibeuker, E.; Buffa, F.; Barberis, A.; Leek, R.D.; Roxanis, I.; Zhang, W.; Worth, A.; Beech, J.S.; et al. Paracrine effect of GTP cyclohydrolase and angiopoietin-1 interaction in stromal fibroblasts on tumor Tie2 activation and breast cancer growth. Oncotarget 2016, 7, 9353–9367. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Fernando, V.; Sharma, V.; Walia, Y.; Letson, J.; Furuta, S. Correction of arginine metabolism with sepiapterin—The precursor of nitric oxide synthase cofactor BH4—induces immunostimulatory-shift of breast cancer. Biochem. Pharmacol. 2020, 176, 113887. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The metabolite BH4 controls T–cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564. [Google Scholar] [CrossRef]
- Rabender, C.S.; Bruno, N.; Alam, A.; Sundaresan, G.; Zweit, J.; Mikkelsen, R.B. Sepiapterin enhances tumor radio- and chemosensitivities by promoting vascular normalization. J. Pharmacol. Exp. Ther. 2018, 365, 536–543. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Gollavilli, P.N.; Vasamsetti, S.B.; Karnewar, S.; Gopoju, R.; Ummanni, R.; Kotamraju, S. Statin-induced inhibition of breast cancer proliferation and invasion involves attenuation of iron transport: Intermediacy of nitric oxide and antioxidant defence mechanisms. FEBS J. 2014, 281, 3719–3738. [Google Scholar] [CrossRef]
- Cho, Y.R.; Kim, S.H.; Ko, H.Y.; Kim, M.D.; Choi, S.W.; Seo, D.W. Sepiapterin inhibits cell proliferation and migration of ovarian cancer cells via down-regulation of p70 S6K-dependent VEGFR-2 expression. Oncol. Rep. 2011, 26, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthese (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rozenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xiao, X.; Zhang, C.; Yu, W.; Guo, W.; Zhang, Z.; Li, Z.; Feng, X.; Hao, J.; Zhang, K.; et al. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J. Pineal Res. 2017, 62, 12380. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, S.; Pang, L.; Yang, J.; Li, H.J.; Huo, X.; Qian, S.Y. Celastrol suppresses nitric oxide synthases and the angiogenesis pathway in colorectal cancer. Free Radic. Res. 2019, 53, 324–334. [Google Scholar] [CrossRef]
- Krzystek-Korpacka, M.; Szczęśniak-Sięga, B.; Szczuka, I.; Fortuna, P.; Zawadzki, M.; Kubiak, A.; Mierzchała-Pasierb, M.; Fleszar, M.G.; Lewandowski, Ł.; Serek, P.; et al. L-arginine/nitric oxide pathway is altered in colorectal cancer and can be modulated by novel derivatives from oxicam class of non-steroidal anti-inflammatory drugs. Cancers 2020, 12, 2594. [Google Scholar] [CrossRef]
- Gochman, E.; Mahajna, J.; Shenzer, P.; Dahan, A.; Blatt, A.; Elyakim, R.; Reznick, A.Z. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem. 2012, 114, 827–835. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2018, 14, 618–625. [Google Scholar] [CrossRef]
- Youn, J.; Lee, J.S.; Na, H.K.; Kundu, J.K.; Surh, Y.J. Resveratrol and piceatannol inhibit iNOS expression and NF-kappaB activation in dextran sulfate sodium-induced mouse colitis. Nutr. Cancer 2009, 61, 847–854. [Google Scholar] [CrossRef]
- Burhannudin, M.N.; Widyarini, S.; Purnomosari, D. Chemopreventive effects of Edible Canna (Canna edulis Kerr.) against colorectal carcinogenesis: Effects on expression of adenomatous polyposis coli and inducible nitric oxide synthase in rat inflammatory model. Asian Pac. J. Cancer Prev. 2018, 19, 839–844. [Google Scholar] [CrossRef]
- Lin, S.; Li, Y.; Zamyatnin, A.A.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell. Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef]
- Campos, A.C.E.; Molognoni, F.; Melo, F.H.M.; Galdieri, L.C.; Carneiro, C.R.W.; D’Almeida, V.; Correa, M.; Jasiulionis, M.G. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia 2007, 9, 1111–1121. [Google Scholar] [CrossRef]
- Fu, H.; Tang, B.; Lang, J.; Du, Y.; Cao, B.; Jin, L.; Fang, M.; Hu, Z.; Cheng, C.; Liu, X.; et al. High-fat diet promotes macrophage-mediated hepatic inflammation and aggravates diethylnitrosamine-induced hepatocarcinogenesis in mice. Front. Nutr. 2020, 7, 247. [Google Scholar] [CrossRef]
- González, R.; Rodríguez-Hernández, M.A.; Negrete, M.; Ranguelova, K.; Rossin, A.; Choya-Foces, C.; Cruz-Ojeda, P.; Miranda-Vizuete, A.; Martínez-Ruiz, A.; Rius-Pérez, S.; et al. Downregulation of thioredoxin-1-dependent CD95 S-nitrosation by Sorafenib reduces liver cancer. Redox Biol. 2020, 34, 101528. [Google Scholar] [CrossRef]
- Marisi, G.; Petracci, E.; Raimondi, F.; Faloppi, L.; Foschi, F.G.; Lauletta, G.; Iavarone, M.; Canale, M.; Valgiusti, M.; Neri, L.M.; et al. ANGPT2 and NOS3 polymorphisms and clinical outcome in advanced hepatocellular carcinoma patients receiving sorafenib. Cancers 2019, 11, 1023. [Google Scholar] [CrossRef]
- Casadei-Gardini, A.; Marisi, G.; Dadduzio, V.; Gramantieri, L.; Faloppi, L.; Ulivi, P.; Foschi, F.G.; Tamburini, E.; Vivaldi, C.; Rizzato, M.D.; et al. Association of NOS3 and ANGPT2 gene polymorphisms with survival in patients with hepatocellular carcinoma receiving sorafenib: Results of the multicenter prospective innovate study. Clin. Cancer Res. 2020, 26, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Dhar, D.K.; Yamaguchi, E.; Maruyama, S.; Sato, T.; Hayashi, H.; Ono, T.; Yamanoi, A.; Kohno, H.; Nagasue, N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver. Clin. Cancer Res. 2001, 7, 1325–1332. [Google Scholar]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Geerts, D.; Feith, D.J.; Mocz, G.; Koster, J.; Bachmann, A.S. Novel interaction of ornithine decarboxylase with sepiapterin reductase regulates neuroblastoma cell proliferation. J. Mol. Biol. 2014, 426, 332–346. [Google Scholar] [CrossRef]
- Gamble, L.D.; Purgato, S.; Murray, J.; Xiao, L.; Yu, D.M.T.; Hanssen, K.M.; Giorgi, F.M.; Carter, D.R.; Gifford, A.J.; Valli, E.; et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 2019, 11, 1099. [Google Scholar] [CrossRef]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081. [Google Scholar] [CrossRef]
- Palumbo, P.; Miconi, G.; Cinque, B.; Lombardi, F.; La Torre, C.; Dehcordi, S.R.; Galzio, R.; Cimini, A.; Giordano, A.; Cifone, M.G. NOS2 expression in glioma cell lines and glioma primary cell cultures: Correlation with neurosphere generation and SOX-2 expression. Oncotarget 2017, 8, 25582. [Google Scholar] [CrossRef] [PubMed]
- Zucconi, B.E.; Wilson, G.M. Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Front. Biosci. 2011, 16, 2307. [Google Scholar] [CrossRef]
- Al-Khalaf, H.H.; Aboussekhra, A. AUF1 positively controls angiogenesis through mRNA stabilizationdependent up-regulation of HIF-1α and VEGF-A in human osteosarcoma. Oncotarget 2019, 10, 4868–4879. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Li, J.; Liu, T.H.; Li, D.N.; Wang, J.J.; Zhang, H.; Deng, Z.L.; Chen, F.J.; Cai, J.P. The overexpression of AUF1 in colorectal cancer predicts a poor prognosis and promotes cancer progression by activating ERK and AKT pathways. Cancer Med. 2020, 9, 8612–8623. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Zhang, Y.; Liu, Y.; Yan, S.; Hou, Z.; Cao, M.; Liu, G.; Dong, C.; Gao, J.; Liu, G. Neferine induces apoptosis by modulating the ROS-mediated JNK pathway in esophageal squamous cell carcinoma. Oncol. Rep. 2020, 44, 1116–1126. [Google Scholar] [CrossRef]
- Liu, Q.; He, J.; Zhou, X.; Han, M.; Li, J.; Liu, C.; Yuan, H. ACP-5862 suppresses esophageal squamous cell carcinoma growth through inducing apoptosis via activation of endoplasmic reticulum stress and ROS production. Biochem. Biophys. Res. Commun. 2021, 534, 995–1002. [Google Scholar] [CrossRef]
- Giovanelli, J.; Campos, K.L.; Kaufman, S. Tetrahydrobiopterin, a cofactor for rat cerebellar nitric oxide synthase, does not function as a reactant in the oxygenation of arginine. Proc. Natl. Acad. Sci. USA 1991, 88, 7091–7095. [Google Scholar] [CrossRef]
- Chung, J.Y.-F.; Chan, M.K.-K.; Li, J.S.-F.; Chan, A.S.-W.; Tang, P.C.-T.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. TGF-β Signaling: From tissue fibrosis to tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef]
- Chambers, A.; Kundranda, M.; Rao, S.; Mahmoud, F.; Niu, J. Anti-angiogenesis revisited: Combination with immunotherapy in solid tumors. Curr. Oncol. Reports 2021, 23, 1–9. [Google Scholar] [CrossRef]
- Ribatti, D.; Solimando, A.G.; Pezzella, F. The anti-VEGF(R) drug discovery legacy: Improving attrition rates by breaking the vicious cycle of angiogenesis in cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef]
- Brouet, A.; Dewever, J.; Martinive, P.; Havaux, X.; Bouzin, C.; Sonveaux, P.; Feron, O. Antitumor effects of in vivo caveolin gene delivery are associated with the inhibition of the proangiogenic and vasodilatory effects of nitric oxide. FASEB J. 2005, 19, 1–15. [Google Scholar] [CrossRef]
- Lin, M.I.; Yu, J.; Murata, T.; Sessa, W.C. Caveolin-1—Deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res. 2007, 67, 2849–2856. [Google Scholar] [CrossRef]
- Wei, X.; Guan, L.; Fan, P.; Liu, X.; Liu, R.; Liu, Y.; Bai, H. Direct current electric field stimulates nitric oxide production and promotes NO-dependent angiogenesis: Involvement of the PI3K/Akt signaling pathway. J. Vasc. Res. 2020, 57, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Michaelis, R.; Suhan, T.; Schmidt, H.; Mohamed, A.; Doerr, H.W.; Cinatl, J. Ribavirin inhibits angiogenesis by tetrahydrobiopterin depletion. FASEB J. 2007, 21, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.H.; Zhang, F.; Yao, S.; Tang, L.; Zeng, H.T.; Zhu, L.P.; Yang, Z. Shear stress triggers angiogenesis of late endothelial progenitor cells via the PTEN/Akt/GTPCH/BH4 pathway. Stem Cells Int. 2020, 2020, 5939530. [Google Scholar] [CrossRef] [PubMed]
- Marinos, R.S.; Zhang, W.; Wu, G.; Kelly, K.A.; Meininger, C.J. Tetrahydrobiopterin levels regulate endothelial cell proliferation. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H482–H489. [Google Scholar] [CrossRef]
- Marshall, H.E.; Foster, M.W. S-nitrosylation of Ras in breast cancer. Breast Cancer Res. 2012, 14, 113. [Google Scholar] [CrossRef]
- Santos, A.I.; Carreira, B.P.; Izquierdo-Álvarez, A.; Ramos, E.; Lourenço, A.S.; Filipa Santos, D.; Morte, M.I.; Ribeiro, L.F.; Marreiros, A.; Sánchez-López, N.; et al. S-Nitrosylation of ras mediates nitric oxide-dependent post-injury neurogenesis in a seizure model. Antioxid. Redox Signal. 2018, 28, 15–30. [Google Scholar] [CrossRef]
- Batista, W.L.; Ogata, F.T.; Curcio, M.F.; Miguel, R.B.; Arai, R.J.; Matsuo, A.L.; Moraes, M.S.; Stern, A.; Monteiro, H.P. S-nitrosoglutathione and endothelial nitric oxide synthase-derived nitric oxide regulate compartmentalized ras S-nitrosylation and stimulate cell proliferation. Antioxid. Redox Signal. 2013, 18, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Levy, B.D.; Michel, T. Tetrahydrobiopterin recycling, a key determinant of endothelial nitric-oxide synthase-dependent signaling pathways in cultured vascular endothelial cells. J. Biol. Chem. 2009, 284, 12691–12700. [Google Scholar] [CrossRef]
- Fukushi, J.; Ono, M.; Morikawa, W.; Iwamoto, Y.; Kuwano, M. The activity of soluble VCAM-1 in angiogenesis stimulated by IL-4 and IL-13. J. Immunol. 2000, 165, 2818–2823. [Google Scholar] [CrossRef]
- Nakao, S.; Kuwano, T.; Ishibashi, T.; Kuwano, M.; Ono, M. Synergistic effect of TNF-α in soluble VCAM-1-induced angiogenesis through α 4 integrins. J. Immunol. 2003, 170, 5704–5711. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.-X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Chen, C.; Xu, Z.; Zhao, J.; Ou, B.; Sun, J.; Zheng, M.; Zong, Y.; Lu, A. CCR6 promotes tumor angiogenesis via the AKT/NF-κB/VEGF pathway in colorectal cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 387–397. [Google Scholar] [CrossRef]
- Ikemoto, K.; Matsumoto, T.; Ohtsuki, M.; Itoh, M.; Tada, S.; Udagawa, Y.; Sumi-Ichinose, C.; Kondo, K.; Nomura, T. 2,4-Diamino-6-hydroxypyrimidine (DAHP) suppresses cytokine-induced VCAM-1 expression on the cell surface of human umbilical vein endothelial cells in a BH4-independent manner. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef]
- Abubakar, M.; Zhang, J.; Ahearn, T.U.; Koka, H.; Guo, C.; Lawrence, S.M.; Mutreja, K.; Figueroa, J.D.; Ying, J.; Lissowska, J.; et al. Tumor-associated stromal cellular density as a predictor of recurrence and mortality in breast cancer: Results from ethnically diverse study populations. Cancer Epidemiol. Prev. Biomark. 2021, 30, 1397–1407. [Google Scholar] [CrossRef]
- Ji, Z.; Tian, W.; Gao, W.; Zang, R.; Wang, H.; Yang, G. Cancer-associated fibroblast-derived interleukin-8 promotes ovarian cancer cell stemness and malignancy through the Notch3-mediated signaling. Front. Cell Dev. Biol. 2021, 9, 1655. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Xia, C.; Ding, L.; Pu, Y.; Hu, X.; Cai, H.; Hu, Q. Integrated analysis of single-cell RNA-seq and bulk RNA-seq reveals distinct cancer-associated fibroblasts in head and neck squamous cell carcinoma. Ann. Transl. Med. 2021, 9, 1017. [Google Scholar] [CrossRef]
- Cui, X.; Chen, J.; Zacharek, A.; Roberts, C.; Yang, Y.; Chopp, M. Nitric oxide donor up-regulation of SDF1/CXCR4 and Ang1/Tie2 promotes neuroblast cell migration after stroke. J. Neurosci. Res. 2009, 87, 86–95. [Google Scholar] [CrossRef]
- Zacharek, A.; Chen, J.; Zhang, C.; Cui, X.; Roberts, C.; Jiang, H.; Teng, H.; Chopp, M. Nitric oxide regulates Angiopoietin1/Tie2 expression after stroke. Neurosci. Lett. 2006, 404, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, M.B.; Kuschman, H.P.; Bovee, R.; Hickok, J.R.; Thomas, D.D. Vorinostat exhibits anticancer effects in triple-negative breast cancer cells by preventing nitric oxide-driven histone deacetylation. Biol. Chem. 2021, 402, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Q.; Zheng, Z.; Liu, S.; Meng, L.; Dong, L.; Jiang, X. Vascular normalization in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed. Pharmacother. 2021, 139, 111607. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, S.H.; Kim, J.S.; Moon, J.H.; Jeung, I.C.; Lee, N.G.; Park, J.; Hong, H.J.; Cho, Y.L.; Jung, H.; et al. Stimulation of angiogenesis and survival of endothelial cells by human monoclonal Tie2 receptor antibody. Biomaterials 2015, 51, 119–128. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-Oncol. 2017, 19, 1435–1446. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-associated macrophages in tumor immunity. Front. Immunol. 2020, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Mansouri, S.; Krager, A.; Grimminger, F.; Seeger, W.; Pullamsetti, S.S.; Wheelock, C.E.; Savai, R. Metabolism in tumour-associated macrophages: A quid pro quo with the tumour microenvironment. Eur. Respir. Rev. 2020, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Crezee, T.; Rabold, K.; de Jong, L.; Jaeger, M.; Netea-Maier, R.T. Metabolic programming of tumor associated macrophages in the context of cancer treatment. Ann. Transl. Med. 2020, 8, 1028. [Google Scholar] [CrossRef]
- Zou, S.; Wang, X.; Liu, P.; Ke, C.; Xu, S. Arginine metabolism and deprivation in cancer therapy. Biomed. Pharmacother. 2019, 118, 109210. [Google Scholar] [CrossRef] [PubMed]
- Clemente, G.S.; van Waarde, A.; Antunes, I.F.; Dömling, A.; Elsinga, P.H. Arginase as a potential biomarker of disease progression: A molecular imaging perspective. Int. J. Mol. Sci. 2020, 21, 5291. [Google Scholar] [CrossRef]
- Akinyele, O.; Wallace, H.M. Characterising the response of human breast cancer cells to polyamine modulation. Biomolecules 2021, 11, 743. [Google Scholar] [CrossRef]
- Cervelli, M.; Pietropaoli, S.; Signore, F.; Amendola, R.; Mariottini, P. Polyamines metabolism and breast cancer: State of the art and perspectives. Breast Cancer Res. Treat. 2014, 148, 233–248. [Google Scholar] [CrossRef]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.-S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences are Emerging—A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ran, R.; Shao, B.; Li, H. Prognostic and clinicopathological value of PD-L1 expression in primary breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2019, 178, 17–33. [Google Scholar] [CrossRef]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of tumor-associated macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef] [PubMed]
- García-Ortiz, A.; Serrador, J.M. Nitric oxide signaling in T cell-mediated immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Williams, M.S.; Noguchi, S.; Henkart, P.A.; Osawa, Y. Nitric oxide synthase plays a signaling role in TCR-triggered apoptotic death. J. Immunol. 1998, 161, 6526. [Google Scholar] [PubMed]
- Pall, M.L. Nitric oxide synthase partial uncoupling as a key switching mechanism for the NO/ONOO—Cycle. Med. Hypotheses 2007, 69, 821–825. [Google Scholar] [CrossRef]
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenes 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Paulo, M.; Costa, D.E.F.R.; Bonaventura, D.; Lunardi, C.N.; Bendhack, L.M. Nitric oxide donors as potential drugs for the treatment of vascular diseases due to endothelium dysfunction. Curr. Pharm. Des. 2020, 26, 3748–3759. [Google Scholar] [CrossRef]
- Channon, K.M. Tetrahydrobiopterin: A vascular redox target to improve endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 705. [Google Scholar] [CrossRef]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R. Kshitiz systems biology of cancer metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Zeeshan, R.; Mutahir, Z. Cancer metastasis—Tricks of the trade. Bosn. J. Basic Med. Sci. 2017, 17, 172. [Google Scholar] [CrossRef][Green Version]
- Hope, H.F.; Binkley, G.M.; Fenton, S.; Kitas, G.D.; Verstappen, S.M.M.; Symmons, D.P.M. Systematic review of the predictors of statin adherence for the primary prevention of cardiovascular disease. PLoS ONE 2019, 14, e0201196. [Google Scholar] [CrossRef]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Ma, C.C.H. Recent developments in the effects of nitric oxide-donating statins on cardiovascular disease through regulation of tetrahydrobiopterin and nitric oxide. Vascul. Pharmacol. 2014, 63, 63–70. [Google Scholar] [CrossRef]
- Aoki, C.; Nakano, A.; Tanaka, S.; Yanagi, K.; Ohta, S.; Jojima, T.; Kasai, K.; Takekawa, H.; Hirata, K.; Hattori, Y. Fluvastatin upregulates endothelial nitric oxide synthase activity via enhancement of its phosphorylation and expression and via an increase in tetrahydrobiopterin in vascular endothelial cells. Int. J. Cardiol. 2012, 156, 55–61. [Google Scholar] [CrossRef]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.-H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, A.; et al. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation 2011, 124, 335. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, U.A. Augmentation of Fluvastatin cytotoxicity against prostate carcinoma PC3 Cell line utilizing alpha lipoic—Ellagic acid nanostructured lipid carrier formula. AAPS Pharm. Sci. Tech. 2018, 19, 3454–3461. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.; Alhakamy, N.A.; Fahmy, U.A.; Ahmed, O.A.A.; Asfour, H.Z.; Althagafi, A.A.; Aldawsari, H.M.; Rizg, W.Y.; Mahdi, W.A.; Alghaith, A.F.; et al. Cytotoxic and pro-apoptotic effects of a sub-toxic concentration of fluvastatin on OVCAR3 ovarian cancer cells after its optimized formulation to melittin nano-conjugates. Front. Pharmacol. 2021, 11, 2500. [Google Scholar] [CrossRef]
- Salis, O.; Okuyucu, A.; Bedir, A.; Gör, U.; Kulcu, C.; Yenen, E.; Kılıç, N. Antimetastatic effect of fluvastatin on breast and hepatocellular carcinoma cells in relation to SGK1 and NDRG1 genes. Tumor Biol. 2015, 37, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jiang, T.; Lu, D.; Luo, Y.; Zheng, C.; Feng, J.; Yang, D.; Chen, C.; Yan, X. NADPH oxidase 4 mediates reactive oxygen species induction of CD146 dimerization in VEGF signal transduction. Free Radic. Biol. Med. 2010, 49, 227–236. [Google Scholar] [CrossRef]
- Liu, W.-J.; Huang, Y.-X.; Wang, W.; Zhang, Y.; Liu, B.-J.; Qiu, J.-G.; Jiang, B.-H.; Liu, L.-Z. NOX4 signaling mediates cancer development and therapeutic resistance through HER3 in ovarian cancer cells. Cells 2021, 10, 1647. [Google Scholar] [CrossRef]
- You, Y.; Fan, Q.; Huang, J.; Wu, Y.; Lin, H.; Zhang, Q. Ferroptosis-related gene signature promotes ovarian cancer by influencing immune infiltration and invasion. J. Oncol. 2021, 2021, 1–16. [Google Scholar] [CrossRef]
- Hefler, L.A.; Grimm, C.; Lantzsch, T.; Lampe, D.; Koelbl, H.; Lebrecht, A.; Heinze, G.; Tempfer, C.; Reinthaller, A.; Zeillinger, R. Polymorphisms of the endothelial nitric oxide synthase gene in breast cancer. Breast Cancer Res. Treat. 2006, 98, 151–155. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Ellerhorst, J.; Smid, C.M.; Prieto, V.G.; Munsell, M.; Buzaid, A.C.; Grimm, E.A. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin. Cancer Res. 2000, 6, 4768–4775. [Google Scholar]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during cancer progression. Crit. Rev. Oncog. 2016, 21, 155. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rivandi, M.; Abedini, S.; Pasdar, A.; Sahebkar, A. Regulators and mechanisms of anoikis in triple-negative breast cancer (TNBC): A review. Crit. Rev. Oncol. Hematol. 2019, 140, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, H.P.; Rodrigues, E.G.; Amorim Reis, A.K.C.; Longo, L.S.; Ogata, F.T.; Moretti, A.I.S.; da Costa, P.E.; Teodoro, A.C.S.; Toledo, M.S.; Stern, A. Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 2019, 89, 1–13. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Altered Tumor Capability | NOS Isoforms | Management Methods of BH4 Levels | BH4/NOS Role | Reference |
|---|---|---|---|---|---|
| Melanoma | Growth/Apoptosis in vitro | eNOS, iNOS | BH4/L-sep supplementation | Anti-tumor | [13] |
| Breast | Growth in vitro/in vivo | eNOS, iNOS | L-sep supplementation | Anti-tumor | [35] |
| Colorectal | Growth in vivo | NOS | L-sep supplementation | Anti-tumor | [52] |
| Melanoma | Anoikis in vitro, Growth in vivo | eNOS | L-sep/DAHP supplementation | Anti-tumor | [53] |
| HCC | Growth in vitro/in vivo | NOS | GCH1 silencing BH4 supplementation | Anti-tumor | [54] |
| Breast | Growth/Apoptosis in vitro | - | SPR silencing | Pro-tumor | [55] |
| Glioblastoma | Growth in vitro/in vivo | - | GCH1 overexpression GCH1 silencing | Pro-tumor | [56] |
| ESCC | Growth/Apoptosis in vitro | - | GCH1 silencing GCH1 regulation | Pro-tumor | [57] |
| Leukemia, lymphoma | Growth in vitro | eNOS | GCH1/SPR/PTS knockout BH2 supplementation | Pro-tumor | [58] |
| Colorectal | Growth in vitro/in vivo | iNOS | BH4 supplementation PTPS silencing | Pro-tumor | [59] |
| HCC | Angiogenesis/Growth in vivo | eNOS | DAHP supplementation | Pro-tumor | [60] |
| CAFs | Growth/Angiogenesis in vivo | eNOS | DAHP supplementation GCH1 silencing | Pro-tumor | [61] |
| Colon, Breast, melanoma, TAMs | Angiogenesis/Growth in vivo/in vivo | eNOS | DAHP supplementation, GCH1 silencing | Pro-tumor | [62] |
| Breast, CAFs | Growth in vitro/in vivo Angiogenesis in vivo | - | DAHP supplementation GCH1 silencing | Pro-tumor | [63] |
| Breast, TAMs | Growth ex vivo | eNOS, iNOS | L-sep supplementation | Anti-tumor | [64] |
| Breast, T cells | Growth in vivo | iNOS | BH4/L-sep supplementation GCH1 overexpression/silencing | Anti-tumor | [65] |
| Breast | Angiogenesis/Apoptosis in vivo | NOS | L-sep supplementation | Anti-tumor | [66] |
| Breast | Invasion/Apoptosis in vitro | iNOS | L-sep supplementation | Anti-tumor | [67] |
| Ovarian | Growth/migration in vitro | NOS | L-sep supplementation | Anti/pro- tumor | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, D.A.; Jasiulionis, M.G.; Melo, F.H.M.d. The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin. Int. J. Mol. Sci. 2021, 22, 9546. https://doi.org/10.3390/ijms22179546
Gonçalves DA, Jasiulionis MG, Melo FHMd. The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin. International Journal of Molecular Sciences. 2021; 22(17):9546. https://doi.org/10.3390/ijms22179546
Chicago/Turabian StyleGonçalves, Diego Assis, Miriam Galvonas Jasiulionis, and Fabiana Henriques Machado de Melo. 2021. "The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin" International Journal of Molecular Sciences 22, no. 17: 9546. https://doi.org/10.3390/ijms22179546
APA StyleGonçalves, D. A., Jasiulionis, M. G., & Melo, F. H. M. d. (2021). The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin. International Journal of Molecular Sciences, 22(17), 9546. https://doi.org/10.3390/ijms22179546