
Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
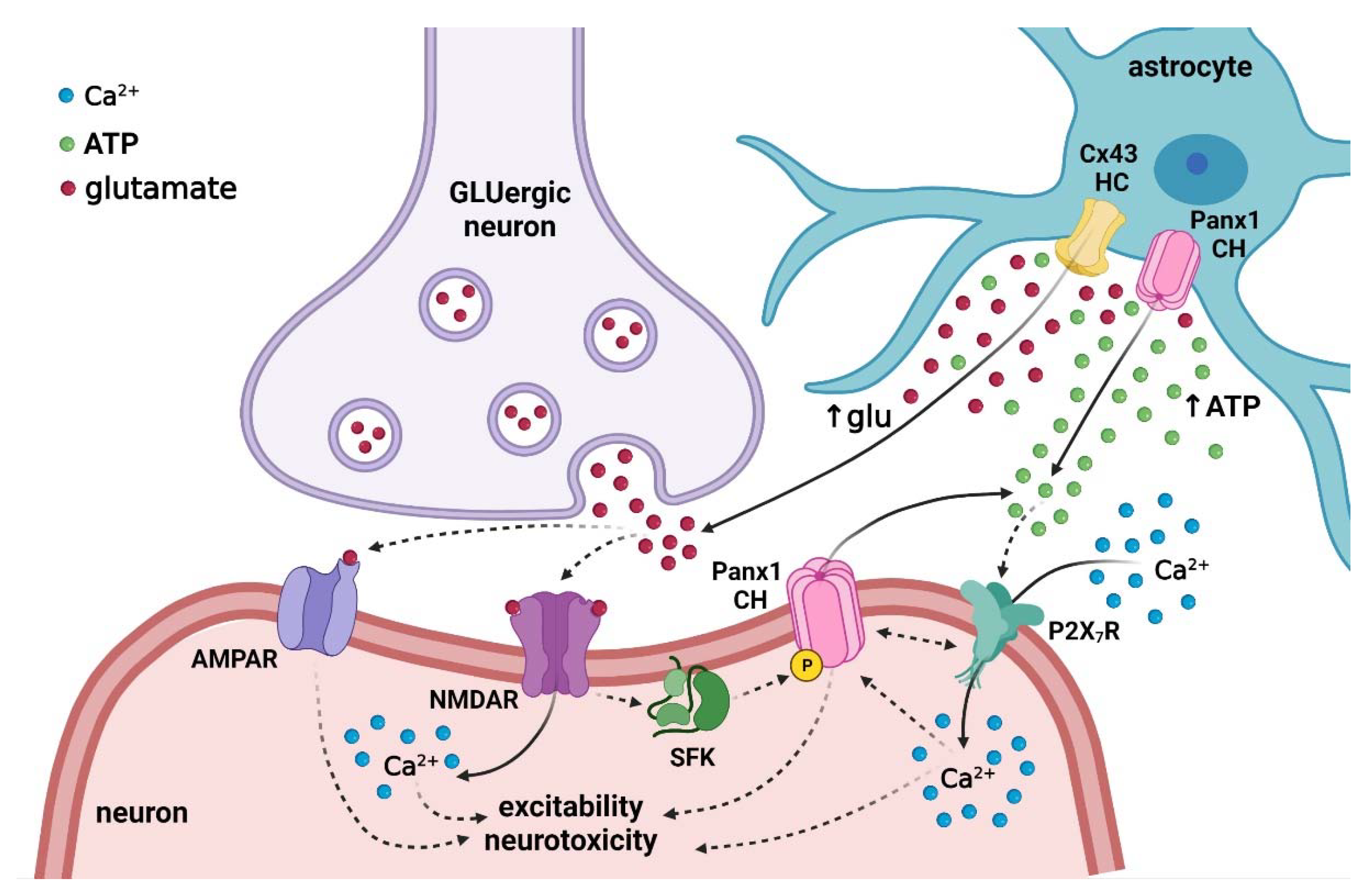{kind=link}
Abstract
1. Introduction

2. Astrocytes: Emerging Stars in the Healthy and Diseased Brain
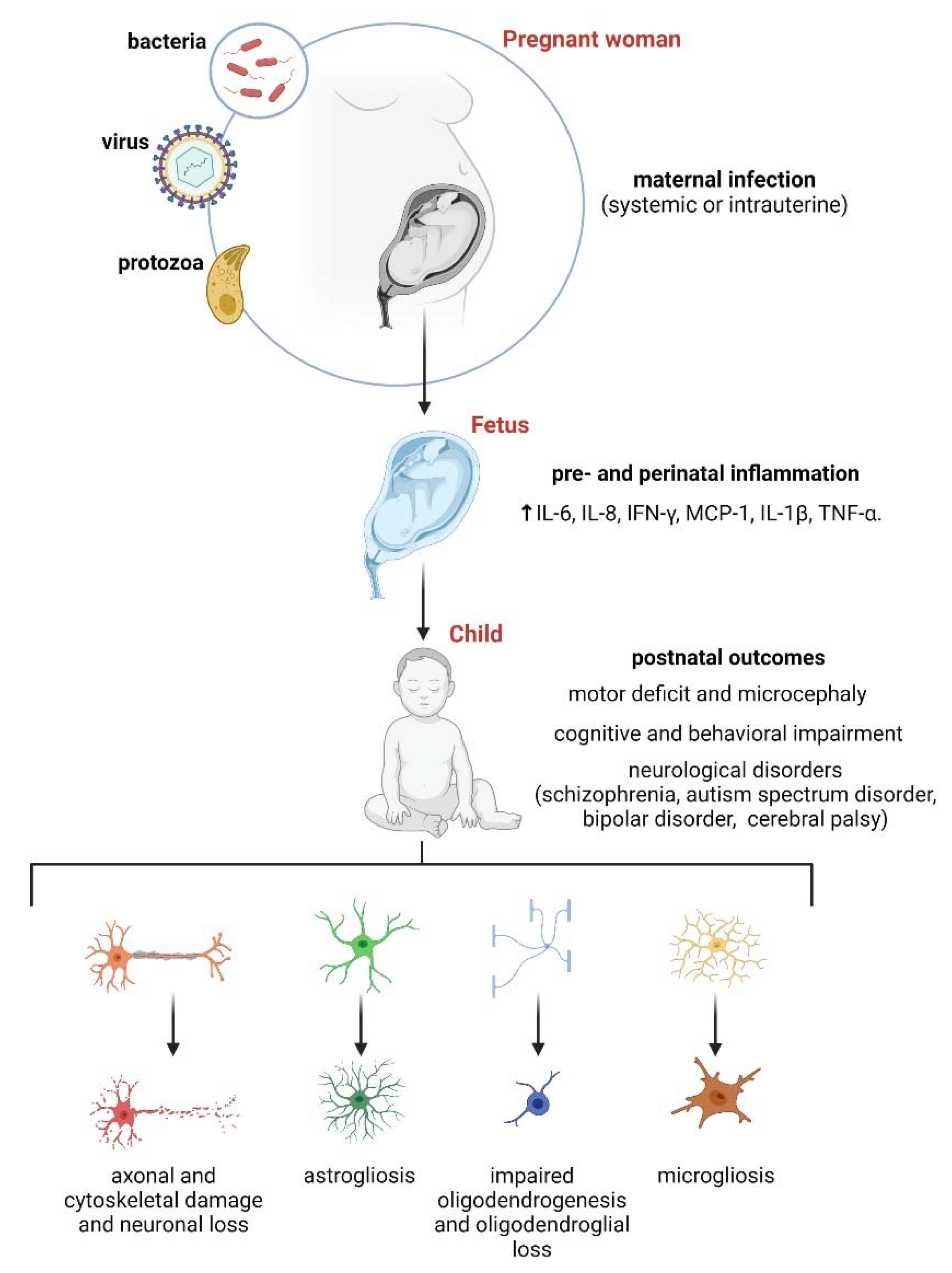
3. Maternal Inflammation and Its Impact on Astrocytes
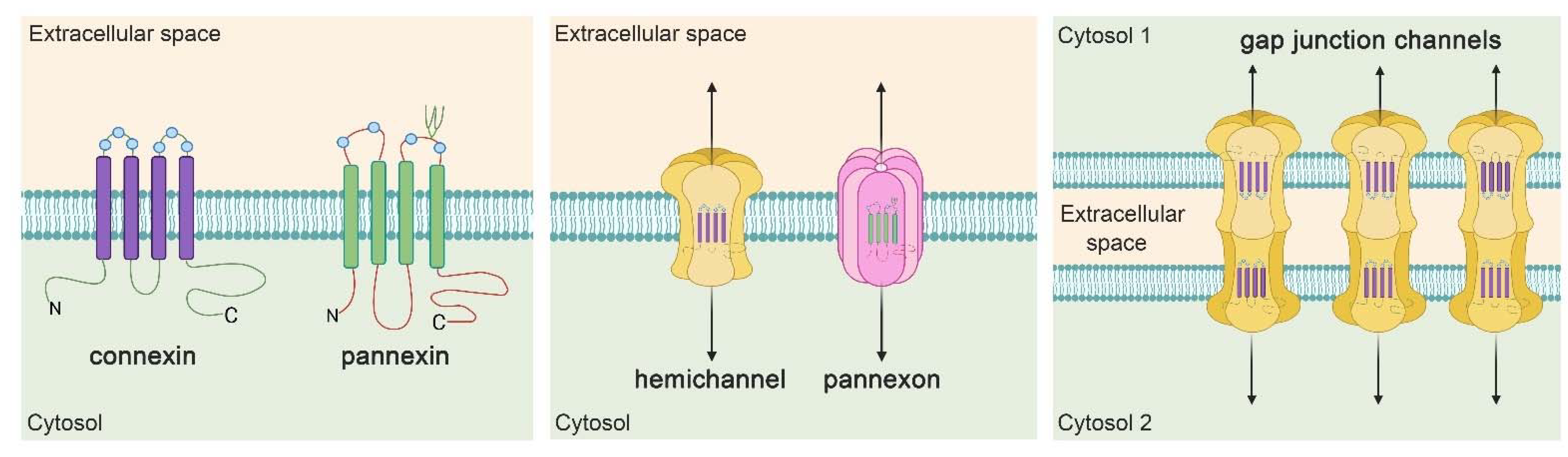
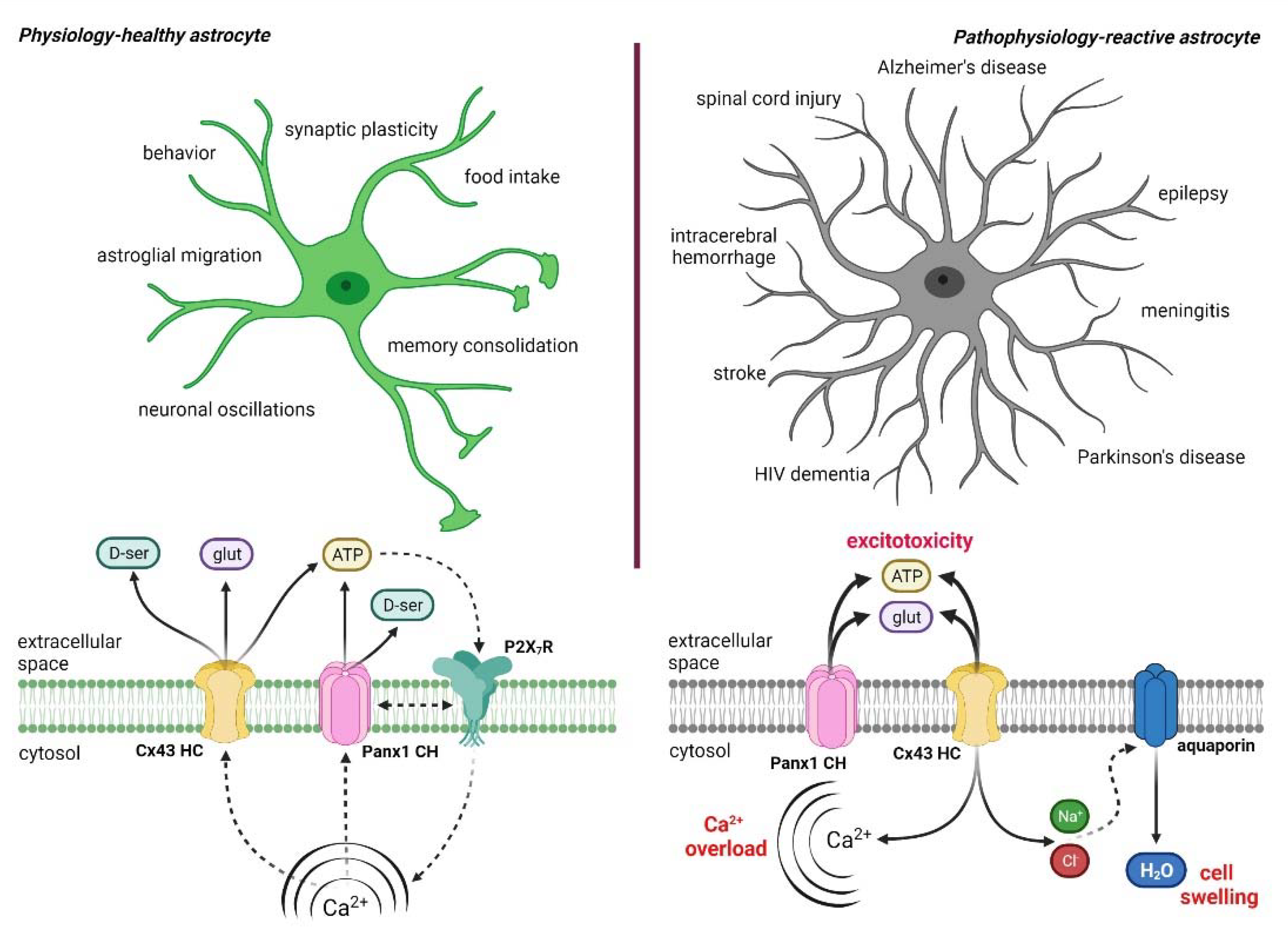
4. Hemichannel and Pannexons: Protagonists on Astroglial Physiology and Pathophysiology
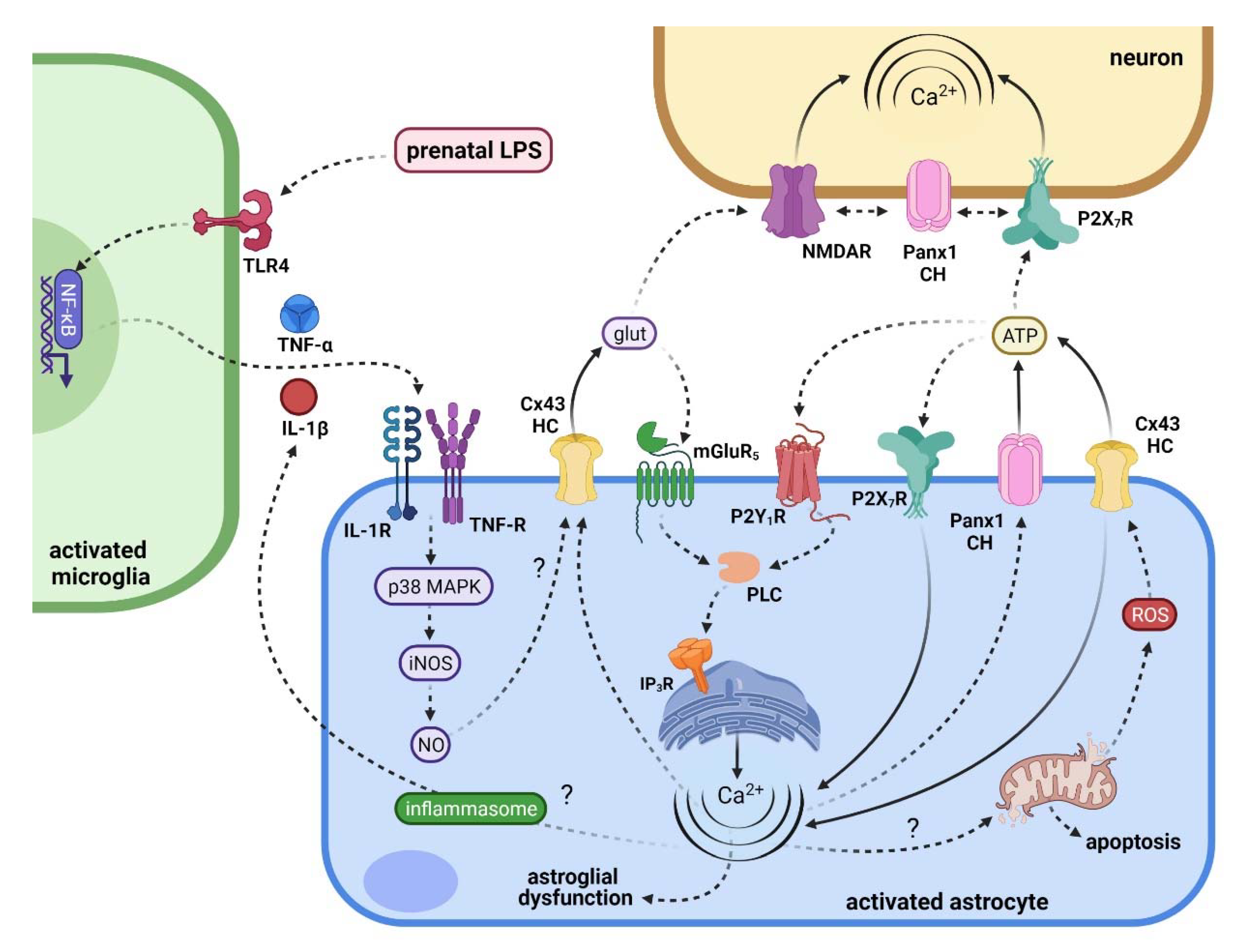
5. Connecting Maternal Inflammation with the Activation of Hemichannels and Pannexons in Offspring Astrocytes
6. Repercussions of Hemichannel and Pannexon Activation in the Offspring Brain following Maternal Inflammation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASD | autism spectrum disorder |
| CNS | Central nervous system |
| COVID-19 | coronavirus disease 2019 |
| Cx43 | Connexin 43 |
| Etd | Ethidium |
| GFAP | glial fibrillary acidic protein |
| IL-1β | Interleukin-1β |
| iNOS | inducible NO synthase |
| [Ca2+]i | Intracellular Ca2+ |
| LPS | lipopolysaccharide |
| NAD+ | Nicotinamide adenine dinucleotide |
| NMDAR | N-methyl-d-aspartate receptor |
| NO | nitric oxide |
| Panx1 PGE2 | Pannexin-1 Prostaglandin E |
| Poly (I:C) | polyriboinosinic-polyribocytidilic acid |
| P2X7Rs | P2X7 receptors |
| p38 MAPK | p38 mitogen-activated protein kinase |
| ROS | reactive oxygen species |
| SFK | Src family kinase |
| IFNs | type I interferons |
| TNF-α | Tumor necrosis factor-α |
References
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- Prudhomme, J.; Morey, C. Epigenesis and plasticity of mouse trophoblast stem cells. Cell Mol. Life Sci. 2016, 73, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, V.; Cardoso, R.C.; Puttabyatappa, M. Developmental Programming, a Pathway to Disease. Endocrinology 2016, 157, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U. Neurodevelopmental Resilience and Susceptibility to Maternal Immune Activation. Trends Neurosci. 2019, 42, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Gumusoglu, S.B.; Stevens, H.E. Maternal Inflammation and Neurodevelopmental Programming: A Review of Preclinical Outcomes and Implications for Translational Psychiatry. Biol. Psychiatry 2019, 85, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Torrey, E.F.; Peterson, M.R. Slow and latent viruses in schizophrenia. Lancet 1973, 2, 22–24. [Google Scholar] [CrossRef]
- Menninger, K.A. Psychoses Associated with Influenza I. General Data: Statistical Analysis. J. Am. Med. Assoc. 1919, 72, 235–241. [Google Scholar] [CrossRef]
- Mednick, S.A.; Machon, R.A.; Huttunen, M.O.; Bonett, D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 1988, 45, 189–192. [Google Scholar] [CrossRef]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef]
- Choudhury, Z.; Lennox, B. Maternal Immune Activation and Schizophrenia-Evidence for an Immune Priming Disorder. Front. Psychiatry 2021, 12, 585742. [Google Scholar] [CrossRef]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child. Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef]
- Libbey, J.E.; Sweeten, T.L.; McMahon, W.M.; Fujinami, R.S. Autistic disorder and viral infections. J. Neurovirol. 2005, 11, 1–10. [Google Scholar] [CrossRef]
- Murphy, D.J.; Sellers, S.; MacKenzie, I.Z.; Yudkin, P.L.; Johnson, A.M. Case-control study of antenatal and intrapartum risk factors for cerebral palsy in very preterm singleton babies. Lancet 1995, 346, 1449–1454. [Google Scholar] [CrossRef]
- Clark, S.M.; Ghulmiyyah, L.M.; Hankins, G.D. Antenatal antecedents and the impact of obstetric care in the etiology of cerebral palsy. Clin. Obstet. Gynecol. 2008, 51, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Camp, B.W.; Broman, S.H.; Nichols, P.L.; Leff, M. Maternal and neonatal risk factors for mental retardation: Defining the ‘at-risk’ child. Early Hum. Dev. 1998, 50, 159–173. [Google Scholar] [CrossRef]
- Hamdani, N.; Daban-Huard, C.; Lajnef, M.; Richard, J.R.; Delavest, M.; Godin, O.; Le Guen, E.; Vederine, F.E.; Lepine, J.P.; Jamain, S.; et al. Relationship between Toxoplasma gondii infection and bipolar disorder in a French sample. J. Affect. Disord. 2013, 148, 444–448. [Google Scholar] [CrossRef]
- Zimmer, A.; Youngblood, A.; Adnane, A.; Miller, B.J.; Goldsmith, D.R. Prenatal exposure to viral infection and neuropsychiatric disorders in offspring: A review of the literature and recommendations for the COVID-19 pandemic. Brain Behav. Immun. 2021, 91, 756–770. [Google Scholar] [CrossRef]
- Boksa, P. Effects of prenatal infection on brain development and behavior: A review of findings from animal models. Brain Behav. Immun. 2010, 24, 881–897. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Zhao, Y.; Shao, F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20, 527–533. [Google Scholar] [CrossRef]
- Kimura, M.; Toth, L.A.; Agostini, H.; Cady, A.B.; Majde, J.A.; Krueger, J.M. Comparison of acute phase responses induced in rabbits by lipopolysaccharide and double-stranded RNA. Am. J. Physiol. 1994, 267, R1596–R1605. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Nyffeler, M.; Engler, A.; Urwyler, A.; Schedlowski, M.; Knuesel, I.; Yee, B.K.; Feldon, J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 4752–4762. [Google Scholar] [CrossRef]
- Meyer, U.; Murray, P.J.; Urwyler, A.; Yee, B.K.; Schedlowski, M.; Feldon, J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol. Psychiatry 2008, 13, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Golan, H.M.; Lev, V.; Hallak, M.; Sorokin, Y.; Huleihel, M. Specific neurodevelopmental damage in mice offspring following maternal inflammation during pregnancy. Neuropharmacology 2005, 48, 903–917. [Google Scholar] [CrossRef]
- Stolp, H.B.; Turnquist, C.; Dziegielewska, K.M.; Saunders, N.R.; Anthony, D.C.; Molnar, Z. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain J. Neurol. 2011, 134, 3236–3248. [Google Scholar] [CrossRef] [PubMed]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise Reverses Behavioral and Synaptic Abnormalities after Maternal Inflammation. Cell Rep. 2019, 27, 2817–2825.e5. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol. Psychiatry 2014, 75, 307–315. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J.; Schedlowski, M.; Yee, B.K. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci. Biobehav. Rev. 2005, 29, 913–947. [Google Scholar] [CrossRef] [PubMed]
- Golan, H.; Stilman, M.; Lev, V.; Huleihel, M. Normal aging of offspring mice of mothers with induced inflammation during pregnancy. Neuroscience 2006, 141, 1909–1918. [Google Scholar] [CrossRef]
- Girard, S.; Kadhim, H.; Beaudet, N.; Sarret, P.; Sebire, G. Developmental motor deficits induced by combined fetal exposure to lipopolysaccharide and early neonatal hypoxia/ischemia: A novel animal model for cerebral palsy in very premature infants. Neuroscience 2009, 158, 673–682. [Google Scholar] [CrossRef]
- Gilmore, J.H.; Jarskog, L.F. Exposure to infection and brain development: Cytokines in the pathogenesis of schizophrenia. Schizophr. Res. 1997, 24, 365–367. [Google Scholar] [CrossRef]
- Saliba, E.; Henrot, A. Inflammatory mediators and neonatal brain damage. Biol. Neonate 2001, 79, 224–227. [Google Scholar] [PubMed]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef]
- Mac Giollabhui, N.; Breen, E.C.; Murphy, S.K.; Maxwell, S.D.; Cohn, B.A.; Krigbaum, N.Y.; Cirillo, P.M.; Perez, C.; Alloy, L.B.; Drabick, D.A.G.; et al. Maternal inflammation during pregnancy and offspring psychiatric symptoms in childhood: Timing and sex matter. J. Psychiatr. Res. 2019, 111, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Meyer, U. Maternal Immune Activation and Neuropsychiatric Illness: A Translational Research Perspective. Am. J. Psychiatry 2018, 175, 1073–1083. [Google Scholar] [CrossRef]
- Girard, S.; Tremblay, L.; Lepage, M.; Sebire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Nilsen, N.; Nonstad, U.; Khan, N.; Knetter, C.F.; Akira, S.; Sundan, A.; Espevik, T.; Lien, E. Lipopolysaccharide and double-stranded RNA up-regulate toll-like receptor 2 independently of myeloid differentiation factor 88. J. Biol. Chem. 2004, 279, 39727–39735. [Google Scholar] [CrossRef] [PubMed]
- Gromkowski, S.H.; Mama, K.; Yagi, J.; Sen, R.; Rath, S. Double-stranded RNA and bacterial lipopolysaccharide enhance sensitivity to TNF-alpha-mediated cell death. Int. Immunol. 1990, 2, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef]
- Reimer, T.; Brcic, M.; Schweizer, M.; Jungi, T.W. poly(I:C) and LPS induce distinct IRF3 and NF-kappaB signaling during type-I IFN and TNF responses in human macrophages. J. Leukoc. Biol. 2008, 83, 1249–1257. [Google Scholar] [CrossRef]
- Hopwood, N.; Maswanganyi, T.; Harden, L.M. Comparison of anorexia, lethargy, and fever induced by bacterial and viral mimetics in rats. Can. J. Physiol. Pharm. 2009, 87, 211–220. [Google Scholar] [CrossRef]
- Goldstein, J.A.; Gallagher, K.; Beck, C.; Kumar, R.; Gernand, A.D. Maternal-Fetal Inflammation in the Placenta and the Developmental Origins of Health and Disease. Front. Immunol. 2020, 11, 531543. [Google Scholar] [CrossRef]
- Rosenfeld, C.S. The placenta-brain-axis. J. Neurosci. Res. 2021, 99, 271–283. [Google Scholar] [CrossRef]
- Wright-Jin, E.C.; Gutmann, D.H. Microglia as Dynamic Cellular Mediators of Brain Function. Trends Mol. Med. 2019, 25, 967–979. [Google Scholar] [CrossRef]
- Zhang, Z.; Bassam, B.; Thomas, A.G.; Williams, M.; Liu, J.; Nance, E.; Rojas, C.; Slusher, B.S.; Kannan, S. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiol. Dis. 2016, 94, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Kato, D.; Ikegami, A.; Hashimoto, A.; Sugio, S.; Guo, Z.; Shibushita, M.; Tatematsu, T.; Haruwaka, K.; Moorhouse, A.J.; et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci. Rep. 2020, 10, 21378. [Google Scholar] [CrossRef]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.; Eggen, B.J.L. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Antonson, A.M.; Lawson, M.A.; Caputo, M.P.; Matt, S.M.; Leyshon, B.J.; Johnson, R.W. Maternal viral infection causes global alterations in porcine fetal microglia. Proc. Natl. Acad. Sci. USA 2019, 116, 20190–20200. [Google Scholar] [CrossRef] [PubMed]
- Delpech, J.C.; Wei, L.; Hao, J.; Yu, X.; Madore, C.; Butovsky, O.; Kaffman, A. Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav. Immun. 2016, 57, 79–93. [Google Scholar] [CrossRef]
- Abbink, M.R.; van Deijk, A.F.; Heine, V.M.; Verheijen, M.H.; Korosi, A. The involvement of astrocytes in early-life adversity induced programming of the brain. Glia 2019, 67, 1637–1653. [Google Scholar] [CrossRef] [PubMed]
- Snyder-Keller, A.; Stark, P.F. Prenatal inflammatory effects on nigrostriatal development in organotypic cultures. Brain Res. 2008, 1233, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Jennische, E.; Hansson, H.A.; Holmang, A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1345–R1356. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; Heijnen, C.J.; Benders, M.J.; van Bel, F.; Fleiss, B.; Gressens, P.; Nijboer, C.H. Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Prog. Neurobiol. 2016, 136, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Hoshiai, K.; Tanabe, K.; Nakamura, A.; Funamoto, K.; Aoyagi, A.; Chisaka, H.; Okamura, K.; Yaegashi, N.; Kimura, Y. Maternal undernutrition with vaginal inflammation impairs the neonatal oligodendrogenesis in mice. Tohoku J. Exp. Med. 2011, 223, 215–222. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 1994, 263, 1768–1771. [Google Scholar] [CrossRef]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef]
- Rusakov, D.A. Disentangling calcium-driven astrocyte physiology. Nat. Rev. Neurosci. 2015, 16, 226–233. [Google Scholar] [CrossRef]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Dallerac, G.; Zapata, J.; Rouach, N. Versatile control of synaptic circuits by astrocytes: Where, when and how? Nat. Rev. Neurosci. 2018, 19, 729–743. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Mayorquin, L.C.; Rodriguez, A.V.; Sutachan, J.J.; Albarracin, S.L. Connexin-Mediated Functional and Metabolic Coupling Between Astrocytes and Neurons. Front. Mol. Neurosci. 2018, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109 (Suppl. S1), 87–93. [Google Scholar] [CrossRef]
- Weber, B.; Barros, L.F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb. Perspect. Biol. 2015, 7, a020396. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yu, S.; Zheng, Y.; Yang, H.; Zhang, J. Oxidative Modification and Its Implications for the Neurodegeneration of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. S2), 115–125. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef]
- Jackson, J.G.; Robinson, M.B. Reciprocal Regulation of Mitochondrial Dynamics and Calcium Signaling in Astrocyte Processes. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 15199–15213. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Werth, J.L.; Thayer, S.A. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. Off. J. Soc. Neurosci. 1994, 14, 348–356. [Google Scholar] [CrossRef]
- Bernardi, P.; Petronilli, V. The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenerg. Biomembr. 1996, 28, 131–138. [Google Scholar] [CrossRef]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605. [Google Scholar] [CrossRef]
- O’Donnell, J.C.; Jackson, J.G.; Robinson, M.B. Transient Oxygen/Glucose Deprivation Causes a Delayed Loss of Mitochondria and Increases Spontaneous Calcium Signaling in Astrocytic Processes. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7109–7127. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhauser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef]
- Folkerth, R.D.; Keefe, R.J.; Haynes, R.L.; Trachtenberg, F.L.; Volpe, J.J.; Kinney, H.C. Interferon-gamma expression in periventricular leukomalacia in the human brain. Brain Pathol. 2004, 14, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.L.; Folkerth, R.D.; Keefe, R.J.; Sung, I.; Swzeda, L.I.; Rosenberg, P.A.; Volpe, J.J.; Kinney, H.C. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J. Neuropathol. Exp. Neurol. 2003, 62, 441–450. [Google Scholar] [CrossRef]
- Cai, Z.; Pan, Z.L.; Pang, Y.; Evans, O.B.; Rhodes, P.G. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr. Res. 2000, 47, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Rousset, C.I.; Chalon, S.; Cantagrel, S.; Bodard, S.; Andres, C.; Gressens, P.; Saliba, E. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr. Res. 2006, 59, 428–433. [Google Scholar] [CrossRef]
- Rousset, C.I.; Kassem, J.; Olivier, P.; Chalon, S.; Gressens, P.; Saliba, E. Antenatal bacterial endotoxin sensitizes the immature rat brain to postnatal excitotoxic injury. J. Neuropathol. Exp. Neurol. 2008, 67, 994–1000. [Google Scholar] [CrossRef]
- Hao, L.Y.; Hao, X.Q.; Li, S.H.; Li, X.H. Prenatal exposure to lipopolysaccharide results in cognitive deficits in age-increasing offspring rats. Neuroscience 2010, 166, 763–770. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflamm. 2017, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Emamian, E.S.; Sidwell, R.W.; Kist, D.A.; Stary, J.M.; Earle, J.A.; Thuras, P. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol. Psychiatry 2002, 7, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, U.; Quinn, T.A.; Castillo-Melendez, M.; Dickinson, H.; Walker, D.W. Behaviour and hippocampus-specific changes in spiny mouse neonates after treatment of the mother with the viral-mimetic Poly I:C at mid-pregnancy. Brain Behav. Immun. 2012, 26, 1288–1299. [Google Scholar] [CrossRef]
- Esshili, A.; Manitz, M.P.; Freund, N.; Juckel, G. Induction of inducible nitric oxide synthase expression in activated microglia and astrocytes following pre- and postnatal immune challenge in an animal model of schizophrenia. Eur. Neuropsychopharmacol. 2020, 35, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Duchatel, R.J.; Meehan, C.L.; Harms, L.R.; Michie, P.T.; Bigland, M.J.; Smith, D.W.; Walker, F.R.; Jobling, P.; Hodgson, D.M.; Tooney, P.A. Late gestation immune activation increases IBA1-positive immunoreactivity levels in the corpus callosum of adult rat offspring. Psychiatry Res. 2018, 266, 175–185. [Google Scholar] [CrossRef]
- Ding, S.; Hu, Y.; Luo, B.; Cai, Y.; Hao, K.; Yang, Y.; Zhang, Y.; Wang, X.; Ding, M.; Zhang, H.; et al. Age-related changes in neuroinflammation and prepulse inhibition in offspring of rats treated with Poly I:C in early gestation. Behav. Brain Funct. 2019, 15, 3. [Google Scholar] [CrossRef]
- Arsenault, D.; St-Amour, I.; Cisbani, G.; Rousseau, L.S.; Cicchetti, F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav. Immun. 2014, 38, 77–90. [Google Scholar] [CrossRef]
- Giovanoli, S.; Weber-Stadlbauer, U.; Schedlowski, M.; Meyer, U.; Engler, H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav. Immun. 2016, 55, 25–38. [Google Scholar] [CrossRef]
- Giovanoli, S.; Notter, T.; Richetto, J.; Labouesse, M.A.; Vuillermot, S.; Riva, M.A.; Meyer, U. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflamm. 2015, 12, 221. [Google Scholar] [CrossRef]
- Paylor, J.W.; Lins, B.R.; Greba, Q.; Moen, N.; de Moraes, R.S.; Howland, J.G.; Winship, I.R. Developmental disruption of perineuronal nets in the medial prefrontal cortex after maternal immune activation. Sci. Rep. 2016, 6, 37580. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Zeng, H.C.; Zhang, L.; Li, Y.Y.; Wang, Y.J.; Xia, W.; Lin, Y.; Wei, J.; Xu, S.Q. Inflammation-like glial response in rat brain induced by prenatal PFOS exposure. Neurotoxicology 2011, 32, 130–139. [Google Scholar] [CrossRef]
- Bennett, G.A.; Palliser, H.K.; Shaw, J.C.; Walker, D.; Hirst, J.J. Prenatal Stress Alters Hippocampal Neuroglia and Increases Anxiety in Childhood. Dev. Neurosci. 2015, 37, 533–545. [Google Scholar] [CrossRef]
- Sowa, J.E.; Slusarczyk, J.; Trojan, E.; Chamera, K.; Leskiewicz, M.; Regulska, M.; Kotarska, K.; Basta-Kaim, A. Prenatal stress affects viability, activation, and chemokine signaling in astroglial cultures. J. Neuroimmunol. 2017, 311, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Duran-Carabali, L.E.; Arcego, D.M.; Odorcyk, F.K.; Reichert, L.; Cordeiro, J.L.; Sanches, E.F.; Freitas, L.D.; Dalmaz, C.; Pagnussat, A.; Netto, C.A. Prenatal and Early Postnatal Environmental Enrichment Reduce Acute Cell Death and Prevent Neurodevelopment and Memory Impairments in Rats Submitted to Neonatal Hypoxia Ischemia. Mol. Neurobiol. 2018, 55, 3627–3641. [Google Scholar] [CrossRef] [PubMed]
- Frahm, K.A.; Handa, R.J.; Tobet, S.A. Embryonic Exposure to Dexamethasone Affects Nonneuronal Cells in the Adult Paraventricular Nucleus of the Hypothalamus. J. Endocr. Soc. 2018, 2, 140–153. [Google Scholar] [CrossRef]
- Mayordomo, F.; Renau-Piqueras, J.; Megias, L.; Guerri, C.; Iborra, F.J.; Azorin, I.; Ledig, M. Cytochemical and stereological analysis of rat cortical astrocytes during development in primary culture. Effect of prenatal exposure to ethanol. Int. J. Dev. Biol. 1992, 36, 311–321. [Google Scholar]
- Onoda, A.; Takeda, K.; Umezawa, M. Pretreatment with N-acetyl cysteine suppresses chronic reactive astrogliosis following maternal nanoparticle exposure during gestational period. Nanotoxicology 2017, 11, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Pistell, P.J.; Purpera, M.N.; Gupta, S.; Fernandez-Kim, S.O.; Hise, T.L.; Keller, J.N.; Ingram, D.K.; Morrison, C.D.; Bruce-Keller, A.J. Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: Contributions of maternal diet. Neurobiol. Dis. 2009, 35, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Glendining, K.A.; Grattan, D.R.; Jasoni, C.L. Maternal obesity leads to increased proliferation and numbers of astrocytes in the developing fetal and neonatal mouse hypothalamus. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2016, 53, 18–25. [Google Scholar] [CrossRef]
- Orellana, J.A.; Shoji, K.F.; Abudara, V.; Ezan, P.; Amigou, E.; Saez, P.J.; Jiang, J.X.; Naus, C.C.; Saez, J.C.; Giaume, C. Amyloid beta-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef]
- Orellana, J.A.; Froger, N.; Ezan, P.; Jiang, J.X.; Bennett, M.V.; Naus, C.C.; Giaume, C.; Saez, J.C. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J. Neurochem. 2011, 118, 826–840. [Google Scholar] [CrossRef]
- Hu, J.; Ferreira, A.; Van Eldik, L.J. S100beta induces neuronal cell death through nitric oxide release from astrocytes. J. Neurochem. 1997, 69, 2294–2301. [Google Scholar] [CrossRef]
- Abudara, V.; Roux, L.; Dallerac, G.; Matias, I.; Dulong, J.; Mothet, J.P.; Rouach, N.; Giaume, C. Activated microglia impairs neuroglial interaction by opening Cx43 hemichannels in hippocampal astrocytes. Glia 2015, 63, 795–811. [Google Scholar] [CrossRef]
- Gajardo-Gomez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibanez, C.A.; Saez, J.C.; Giaume, C.; Orellana, J.A. Cannabinoids prevent the amyloid beta-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Karpuk, N.; Burkovetskaya, M.; Fritz, T.; Angle, A.; Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Garre, J.M.; Yang, G.; Bukauskas, F.F.; Bennett, M.V. FGF-1 Triggers Pannexin-1 Hemichannel Opening in Spinal Astrocytes of Rodents and Promotes Inflammatory Responses in Acute Spinal Cord Slices. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4785–4801. [Google Scholar] [CrossRef]
- Santiago, M.F.; Veliskova, J.; Patel, N.K.; Lutz, S.E.; Caille, D.; Charollais, A.; Meda, P.; Scemes, E. Targeting pannexin1 improves seizure outcome. PLoS ONE 2011, 6, e25178. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef]
- Gajardo-Gomez, R.; Santibanez, C.A.; Labra, V.C.; Gomez, G.I.; Eugenin, E.A.; Orellana, J.A. HIV gp120 Protein Increases the Function of Connexin 43 Hemichannels and Pannexin-1 Channels in Astrocytes: Repercussions on Astroglial Function. Int. J. Mol. Sci. 2020, 21, 2503. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar]
- Gomez, G.I.; Falcon, R.V.; Maturana, C.J.; Labra, V.C.; Salgado, N.; Rojas, C.A.; Oyarzun, J.E.; Cerpa, W.; Quintanilla, R.A.; Orellana, J.A. Heavy Alcohol Exposure Activates Astroglial Hemichannels and Pannexons in the Hippocampus of Adolescent Rats: Effects on Neuroinflammation and Astrocyte Arborization. Front. Cell Neurosci. 2018, 12, 472. [Google Scholar] [CrossRef]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharm. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J. Cell Biol. 1991, 115, 1077–1089. [Google Scholar] [CrossRef]
- Dahl, G. The Pannexin1 membrane channel: Distinct conformations and functions. FEBS Lett. 2018, 592, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, J.; Michalski, K.; Kawate, T.; Furukawa, H. On the molecular nature of large-pore channels. J. Mol. Biol. 2021, 433, 166994. [Google Scholar] [CrossRef]
- Patel, D.; Zhang, X.; Veenstra, R.D. Connexin hemichannel and pannexin channel electrophysiology: How do they differ? FEBS Lett. 2014, 588, 1372–1378. [Google Scholar] [CrossRef]
- Giaume, C.; Naus, C.C.; Saez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiol. Rev. 2021, 101, 93–145. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed]
- Chever, O.; Lee, C.Y.; Rouach, N. Astroglial connexin43 hemichannels tune basal excitatory synaptic transmission. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 11228–11232. [Google Scholar] [CrossRef]
- Ma, W.; Compan, V.; Zheng, W.; Martin, E.; North, R.A.; Verkhratsky, A.; Surprenant, A. Pannexin 1 forms an anion-selective channel. Pflug. Arch. Eur. J. Physiol. 2012, 463, 585–592. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef]
- Contreras, J.E.; Saez, J.C.; Bukauskas, F.F.; Bennett, M.V. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 11388–11393. [Google Scholar] [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef]
- Ebihara, L.; Liu, X.; Pal, J.D. Effect of external magnesium and calcium on human connexin46 hemichannels. Biophys. J. 2003, 84, 277–286. [Google Scholar] [CrossRef]
- Bruzzone, R.; Barbe, M.T.; Jakob, N.J.; Monyer, H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 2005, 92, 1033–1043. [Google Scholar] [CrossRef]
- Giaume, C.; Fromaget, C.; el Aoumari, A.; Cordier, J.; Glowinski, J.; Gros, D. Gap junctions in cultured astrocytes: Single-channel currents and characterization of channel-forming protein. Neuron 1991, 6, 133–143. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, R.; Dahl, G.; Qiu, F.; Spray, D.C.; Scemes, E. Pannexin 1: The molecular substrate of astrocyte “hemichannels”. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 7092–7097. [Google Scholar] [CrossRef]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef]
- Pan, H.C.; Chou, Y.C.; Sun, S.H. P2X7 R-mediated Ca(2+) -independent d-serine release via pannexin-1 of the P2X7 R-pannexin-1 complex in astrocytes. Glia 2015, 63, 877–893. [Google Scholar] [CrossRef]
- Roux, L.; Madar, A.; Lacroix, M.M.; Yi, C.; Benchenane, K.; Giaume, C. Astroglial Connexin 43 Hemichannels Modulate Olfactory Bulb Slow Oscillations. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 15339–15352. [Google Scholar] [CrossRef]
- Alvarez, A.; Lagos-Cabre, R.; Kong, M.; Cardenas, A.; Burgos-Bravo, F.; Schneider, P.; Quest, A.F.; Leyton, L. Integrin-mediated transactivation of P2X7R via hemichannel-dependent ATP release stimulates astrocyte migration. Biochim. Biophys. Acta 2016, 1863, 2175–2188. [Google Scholar] [CrossRef]
- Guillebaud, F.; Barbot, M.; Barbouche, R.; Brezun, J.M.; Poirot, K.; Vasile, F.; Lebrun, B.; Rouach, N.; Dallaporta, M.; Gaige, S.; et al. Blockade of Glial Connexin 43 Hemichannels Reduces Food Intake. Cells 2020, 9, 2387. [Google Scholar] [CrossRef] [PubMed]
- Meunier, C.; Wang, N.; Yi, C.; Dallerac, G.; Ezan, P.; Koulakoff, A.; Leybaert, L.; Giaume, C. Contribution of Astroglial Cx43 Hemichannels to the Modulation of Glutamatergic Currents by D-Serine in the Mouse Prefrontal Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 9064–9075. [Google Scholar] [CrossRef] [PubMed]
- Ardiles, A.O.; Flores-Munoz, C.; Toro-Ayala, G.; Cardenas, A.M.; Palacios, A.G.; Munoz, P.; Fuenzalida, M.; Saez, J.C.; Martinez, A.D. Pannexin 1 regulates bidirectional hippocampal synaptic plasticity in adult mice. Front. Cell Neurosci. 2014, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Prochnow, N.; Abdulazim, A.; Kurtenbach, S.; Wildforster, V.; Dvoriantchikova, G.; Hanske, J.; Petrasch-Parwez, E.; Shestopalov, V.I.; Dermietzel, R.; Manahan-Vaughan, D.; et al. Pannexin1 stabilizes synaptic plasticity and is needed for learning. PLoS ONE 2012, 7, e51767. [Google Scholar] [CrossRef]
- Walrave, L.; Vinken, M.; Albertini, G.; De Bundel, D.; Leybaert, L.; Smolders, I.J. Inhibition of Connexin43 Hemichannels Impairs Spatial Short-Term Memory without Affecting Spatial Working Memory. Front. Cell Neurosci. 2016, 10, 288. [Google Scholar] [CrossRef]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverria, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Deng, F.; Chen, Y.; Qin, Y.; Hao, Y.; Guo, X. Ultrafine carbon black induces glutamate and ATP release by activating connexin and pannexin hemichannels in cultured astrocytes. Toxicology 2014, 323, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Cortes, C.J.; Reuss, L.; Bennett, M.V.; Saez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef]
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.E.; Saez, J.C.; Reuss, L.; Altenberg, G.A. Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 2012, 287, 40826–40834. [Google Scholar] [CrossRef]
- Schalper, K.A.; Sanchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Saez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef]
- Locovei, S.; Wang, J.; Dahl, G. Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 2006, 580, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernandez, P.; Schmitt, J.; Saez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef] [PubMed]
- Walrave, L.; Pierre, A.; Albertini, G.; Aourz, N.; De Bundel, D.; Van Eeckhaut, A.; Vinken, M.; Giaume, C.; Leybaert, L.; Smolders, I. Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 2018, 66, 1788–1804. [Google Scholar] [CrossRef]
- Wellmann, M.; Alvarez-Ferradas, C.; Maturana, C.J.; Saez, J.C.; Bonansco, C. Astroglial Ca(2+)-Dependent Hyperexcitability Requires P2Y1 Purinergic Receptors and Pannexin-1 Channel Activation in a Chronic Model of Epilepsy. Front. Cell Neurosci. 2018, 12, 446. [Google Scholar] [CrossRef]
- Orellana, J.A.; Saez, J.C.; Bennett, M.V.; Berman, J.W.; Morgello, S.; Eugenin, E.A. HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 2014, 128, 752–763. [Google Scholar] [CrossRef]
- Berman, J.W.; Carvallo, L.; Buckner, C.M.; Luers, A.; Prevedel, L.; Bennett, M.V.; Eugenin, E.A. HIV-tat alters Connexin43 expression and trafficking in human astrocytes: Role in NeuroAIDS. J. Neuroinflamm. 2016, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yang, L.; Chen, J.; Chen, Y.; Zhang, L.; Wang, L.; Li, X.; Li, Y.; Yu, H. Inhibition of Connexin43 hemichannels with Gap19 protects cerebral ischemia/reperfusion injury via the JAK2/STAT3 pathway in mice. Brain Res. Bull. 2019, 146, 124–135. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Wang, N.; Bechberger, J.F.; De Bock, M.; Lampe, P.D.; Leybaert, L.; Naus, C.C. Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 2019, 216, 916–935. [Google Scholar] [CrossRef]
- Yu, H.; Cao, X.; Li, W.; Liu, P.; Zhao, Y.; Song, L.; Chen, J.; Chen, B.; Yu, W.; Xu, Y. Targeting connexin 43 provides anti-inflammatory effects after intracerebral hemorrhage injury by regulating YAP signaling. J. Neuroinflamm. 2020, 17, 322. [Google Scholar] [CrossRef]
- Mao, Y.; Tonkin, R.S.; Nguyen, T.; O’Carroll, S.J.; Nicholson, L.F.; Green, C.R.; Moalem-Taylor, G.; Gorrie, C.A. Systemic Administration of Connexin43 Mimetic Peptide Improves Functional Recovery after Traumatic Spinal Cord Injury in Adult Rats. J. Neurotrauma 2017, 34, 707–719. [Google Scholar] [CrossRef]
- Avendano, B.C.; Montero, T.D.; Chavez, C.E.; von Bernhardi, R.; Orellana, J.A. Prenatal exposure to inflammatory conditions increases Cx43 and Panx1 unopposed channel opening and activation of astrocytes in the offspring effect on neuronal survival. Glia 2015, 63, 2058–2072. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, E.; Decrock, E.; De Bock, M.; Yamasaki, H.; Naus, C.C.; Evans, W.H.; Leybaert, L. Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell 2007, 18, 34–46. [Google Scholar] [CrossRef]
- Saez, J.C.; Vargas, A.A.; Hernandez, D.E.; Ortiz, F.C.; Giaume, C.; Orellana, J.A. Permeation of Molecules through Astroglial Connexin 43 Hemichannels Is Modulated by Cytokines with Parameters Depending on the Permeant Species. Int. J. Mol. Sci. 2020, 21, 3970. [Google Scholar]
- Vinken, M. Regulation of connexin signaling by the epigenetic machinery. Biochim. Biophys. Acta 2016, 1859, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Chavez, C.E.; Oyarzun, J.E.; Avendano, B.C.; Mellado, L.A.; Inostroza, C.A.; Alvear, T.F.; Orellana, J.A. The Opening of Connexin 43 Hemichannels Alters Hippocampal Astrocyte Function and Neuronal Survival in Prenatally LPS-Exposed Adult Offspring. Front. Cell Neurosci. 2019, 13, 460. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.; Pierrat, B.; Mary, J.L.; Lesslauer, W. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 1997, 272, 28373–28380. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; St Pierre, B.A.; Parkinson, J.F.; Medberry, P.; Wong, J.L.; Rogers, N.E.; Ignarro, L.J.; Merrill, J.E. Inducible nitric-oxide synthase and nitric oxide production in human fetal astrocytes and microglia. A kinetic analysis. J. Biol. Chem. 1997, 272, 11327–11335. [Google Scholar] [CrossRef] [PubMed]
- Chamera, K.; Kotarska, K.; Szuster-Gluszczak, M.; Trojan, E.; Skorkowska, A.; Pomierny, B.; Krzyzanowska, W.; Bryniarska, N.; Basta-Kaim, A. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: A link to schizophrenia-like behaviours. J. Neuroinflamm. 2020, 17, 247. [Google Scholar] [CrossRef]
- Tellez-Merlo, G.; Morales-Medina, J.C.; Camacho-Abrego, I.; Juarez-Diaz, I.; Aguilar-Alonso, P.; de la Cruz, F.; Iannitti, T.; Flores, G. Prenatal immune challenge induces behavioral deficits, neuronal remodeling, and increases brain nitric oxide and zinc levels in the male rat offspring. Neuroscience 2019, 406, 594–605. [Google Scholar] [CrossRef]
- Lohman, A.W.; Weaver, J.L.; Billaud, M.; Sandilos, J.K.; Griffiths, R.; Straub, A.C.; Penuela, S.; Leitinger, N.; Laird, D.W.; Bayliss, D.A.; et al. S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 2012, 287, 39602–39612. [Google Scholar] [CrossRef] [PubMed]
- Poornima, V.; Vallabhaneni, S.; Mukhopadhyay, M.; Bera, A.K. Nitric oxide inhibits the pannexin 1 channel through a cGMP-PKG dependent pathway. Nitric Oxide Biol. Chem. Off. J. Nitric Oxide Soc. 2015, 47, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Barbera-Cremades, M.; Pelegrin, P. The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 2013, 1828, 79–93. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef]
- Poornima, V.; Madhupriya, M.; Kootar, S.; Sujatha, G.; Kumar, A.; Bera, A.K. P2X7 receptor-pannexin 1 hemichannel association: Effect of extracellular calcium on membrane permeabilization. J. Mol. Neurosci. MN 2012, 46, 585–594. [Google Scholar] [CrossRef]
- Iglesias, R.; Locovei, S.; Roque, A.; Alberto, A.P.; Dahl, G.; Spray, D.C.; Scemes, E. P2X7 receptor-Pannexin1 complex: Pharmacology and signaling. Am. J. Physiol. Cell Physiol. 2008, 295, C752–C760. [Google Scholar] [CrossRef] [PubMed]
- Sorge, R.E.; Trang, T.; Dorfman, R.; Smith, S.B.; Beggs, S.; Ritchie, J.; Austin, J.S.; Zaykin, D.V.; Vander Meulen, H.; Costigan, M.; et al. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat. Med. 2012, 18, 595–599. [Google Scholar] [CrossRef]
- Qiu, F.; Dahl, G. A permeant regulating its permeation pore: Inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 2009, 296, C250–C255. [Google Scholar] [CrossRef] [PubMed]
- Ventura, L.; Freiberger, V.; Thiesen, V.B.; Dias, P.; Dutra, M.L.; Silva, B.B.; Schlindwein, A.D.; Comim, C.M. Involvement of NLRP3 inflammasome in schizophrenia-like behaviour in young animals after maternal immune activation. Acta Neuropsychiatr. 2020, 32, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Kim, Y.G.; Chen, G.; Park, J.H.; Franchi, L.; Vandenabeele, P.; Nunez, G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 2007, 26, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Murphy, N.; Cowley, T.R.; Richardson, J.C.; Virley, D.; Upton, N.; Walter, D.; Lynch, M.A. The neuroprotective effect of a specific P2X(7) receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathol. 2012, 22, 295–306. [Google Scholar] [CrossRef]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Bol, M.; Decrock, E.; Ponsaerts, R.; Bultynck, G.; Dupont, G.; Leybaert, L. Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 2012, 287, 12250–12266. [Google Scholar] [CrossRef]
- Lopez, X.; Palacios-Prado, N.; Guiza, J.; Escamilla, R.; Fernandez, P.; Vega, J.L.; Rojas, M.; Marquez-Miranda, V.; Chamorro, E.; Cardenas, A.M.; et al. A physiologic rise in cytoplasmic calcium ion signal increases pannexin1 channel activity via a C-terminus phosphorylation by CaMKII. Proc. Natl. Acad. Sci. USA 2021, 118, e2108967118. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.H.; Wu, W.W.; Lei, J.; McLane, M.; Xie, H.; Hart, K.D.; Pereira, L.; Burd, I.; Maylie, J. Functional changes in hippocampal synaptic signaling in offspring survivors of a mouse model of intrauterine inflammation. J. Neuroinflamm. 2017, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef]
- Rovegno, M.; Soto, P.A.; Saez, P.J.; Naus, C.C.; Saez, J.C.; von Bernhardi, R. Connexin43 hemichannels mediate secondary cellular damage spread from the trauma zone to distal zones in astrocyte monolayers. Glia 2015, 63, 1185–1199. [Google Scholar] [CrossRef]
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 Receptor Primes IL-1beta and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front. Cell Neurosci. 2017, 11, 227. [Google Scholar] [CrossRef]
- Wei, L.; Sheng, H.; Chen, L.; Hao, B.; Shi, X.; Chen, Y. Effect of pannexin-1 on the release of glutamate and cytokines in astrocytes. J. Clin. Neurosci. 2016, 23, 135–141. [Google Scholar] [CrossRef]
- Agulhon, C.; Sun, M.Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front. Pharm. 2012, 3, 139. [Google Scholar] [CrossRef]
- Ishikawa, M.; Iwamoto, T.; Nakamura, T.; Doyle, A.; Fukumoto, S.; Yamada, Y. Pannexin 3 functions as an ER Ca(2+) channel, hemichannel, and gap junction to promote osteoblast differentiation. J. Cell Biol. 2011, 193, 1257–1274. [Google Scholar] [CrossRef]
- Vanden Abeele, F.; Bidaux, G.; Gordienko, D.; Beck, B.; Panchin, Y.V.; Baranova, A.V.; Ivanov, D.V.; Skryma, R.; Prevarskaya, N. Functional implications of calcium permeability of the channel formed by pannexin 1. J. Cell Biol. 2006, 174, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Delalio, L.J.; Best, A.K.; Macal, E.; Milstein, J.; Donnelly, I.; Miller, A.M.; McBride, M.; Shu, X.; Koval, M.; et al. Endothelial Pannexin 1 Channels Control Inflammation by Regulating Intracellular Calcium. J. Immunol. 2020, 204, 2995–3007. [Google Scholar] [CrossRef]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Maatouk, L.; Yi, C.; Carrillo-de Sauvage, M.A.; Compagnion, A.C.; Hunot, S.; Ezan, P.; Hirsch, E.C.; Koulakoff, A.; Pfrieger, F.W.; Tronche, F.; et al. Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 2019, 26, 580–596. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Matute, C. Calcium and glial cell death. Cell Calcium 2005, 38, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Sub-Cell. Biochem. 2007, 45, 481–506. [Google Scholar]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Islam, M.R.; Uramoto, H.; Okada, T.; Sabirov, R.Z.; Okada, Y. Maxi-anion channel and pannexin 1 hemichannel constitute separate pathways for swelling-induced ATP release in murine L929 fibrosarcoma cells. Am. J. Physiol. Cell Physiol. 2012, 303, C924–C935. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Mellstrom, B.; Savignac, M.; Gomez-Villafuertes, R.; Naranjo, J.R. Ca2+-operated transcriptional networks: Molecular mechanisms and in vivo models. Physiol. Rev. 2008, 88, 421–449. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef]
- Lante, F.; Meunier, J.; Guiramand, J.; De Jesus Ferreira, M.C.; Cambonie, G.; Aimar, R.; Cohen-Solal, C.; Maurice, T.; Vignes, M.; Barbanel, G. Late N-acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus 2008, 18, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; O’Leary, D.D. Axon retraction and degeneration in development and disease. Annu Rev. Neurosci. 2005, 28, 127–156. [Google Scholar] [CrossRef] [PubMed]
- Riccomagno, M.M.; Kolodkin, A.L. Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol. 2015, 31, 779–805. [Google Scholar] [CrossRef]
- Fernandez de Cossio, L.; Guzman, A.; van der Veldt, S.; Luheshi, G.N. Prenatal infection leads to ASD-like behavior and altered synaptic pruning in the mouse offspring. Brain Behav. Immun. 2017, 63, 88–98. [Google Scholar] [CrossRef]
- Monte, A.S.; Mello, B.S.F.; Borella, V.C.M.; da Silva Araujo, T.; da Silva, F.E.R.; Sousa, F.C.F.; de Oliveira, A.C.P.; Gama, C.S.; Seeman, M.V.; Vasconcelos, S.M.M.; et al. Two-hit model of schizophrenia induced by neonatal immune activation and peripubertal stress in rats: Study of sex differences and brain oxidative alterations. Behav. Brain Res. 2017, 331, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.J.; Jackson, M.F.; Olah, M.E.; Rungta, R.L.; Hines, D.J.; Beazely, M.A.; MacDonald, J.F.; MacVicar, B.A. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science 2008, 322, 1555–1559. [Google Scholar] [CrossRef]
- Thompson, R.J.; Zhou, N.; MacVicar, B.A. Ischemia opens neuronal gap junction hemichannels. Science 2006, 312, 924–927. [Google Scholar] [CrossRef]
- Schock, S.C.; Leblanc, D.; Hakim, A.M.; Thompson, C.S. ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 2008, 368, 138–144. [Google Scholar] [CrossRef]
- Hansen, D.B.; Ye, Z.C.; Calloe, K.; Braunstein, T.H.; Hofgaard, J.P.; Ransom, B.R.; Nielsen, M.S.; MacAulay, N. Activation, permeability, and inhibition of astrocytic and neuronal large pore (hemi)channels. J. Biol. Chem. 2014, 289, 26058–26073. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Lohman, A.W.; Rakai, B.D.; Ma, E.M.; Bialecki, J.; Maslieieva, V.; Rilea, T.; Bandet, M.V.; Ikuta, N.T.; Scott, L.; et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat. Neurosci. 2016, 19, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 12579–12588. [Google Scholar] [CrossRef] [PubMed]
- Bialecki, J.; Werner, A.; Weilinger, N.L.; Tucker, C.M.; Vecchiarelli, H.A.; Egana, J.; Mendizabal-Zubiaga, J.; Grandes, P.; Hill, M.N.; Thompson, R.J. Suppression of Presynaptic Glutamate Release by Postsynaptic Metabotropic NMDA Receptor Signalling to Pannexin-1. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Orellana, J.A.; Busso, D.; Ramirez, G.; Campos, M.; Rigotti, A.; Eugenin, J.; von Bernhardi, R. Prenatal nicotine exposure enhances Cx43 and Panx1 unopposed channel activity in brain cells of adult offspring mice fed a high-fat/cholesterol diet. Front. Cell Neurosci. 2014, 8, 403. [Google Scholar] [CrossRef]
- Maturana, C.J.; Aguirre, A.; Saez, J.C. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol. 2017, 77, 625–642. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Villalobos, J.; Alvear, T.F.; Liberona, A.; Lucero, C.M.; Martínez-Araya, C.J.; Balmazabal, J.; Inostroza, C.A.; Ramírez, G.; Gómez, G.I.; Orellana, J.A. Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 9503. https://doi.org/10.3390/ijms22179503
Prieto-Villalobos J, Alvear TF, Liberona A, Lucero CM, Martínez-Araya CJ, Balmazabal J, Inostroza CA, Ramírez G, Gómez GI, Orellana JA. Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders. International Journal of Molecular Sciences. 2021; 22(17):9503. https://doi.org/10.3390/ijms22179503
Chicago/Turabian StylePrieto-Villalobos, Juan, Tanhia F. Alvear, Andrés Liberona, Claudia M. Lucero, Claudio J. Martínez-Araya, Javiera Balmazabal, Carla A. Inostroza, Gigliola Ramírez, Gonzalo I. Gómez, and Juan A. Orellana. 2021. "Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders" International Journal of Molecular Sciences 22, no. 17: 9503. https://doi.org/10.3390/ijms22179503
APA StylePrieto-Villalobos, J., Alvear, T. F., Liberona, A., Lucero, C. M., Martínez-Araya, C. J., Balmazabal, J., Inostroza, C. A., Ramírez, G., Gómez, G. I., & Orellana, J. A. (2021). Astroglial Hemichannels and Pannexons: The Hidden Link between Maternal Inflammation and Neurological Disorders. International Journal of Molecular Sciences, 22(17), 9503. https://doi.org/10.3390/ijms22179503