
The Emerging Role of Stress Granules in Hepatocellular Carcinoma



Abstract
1. Introduction
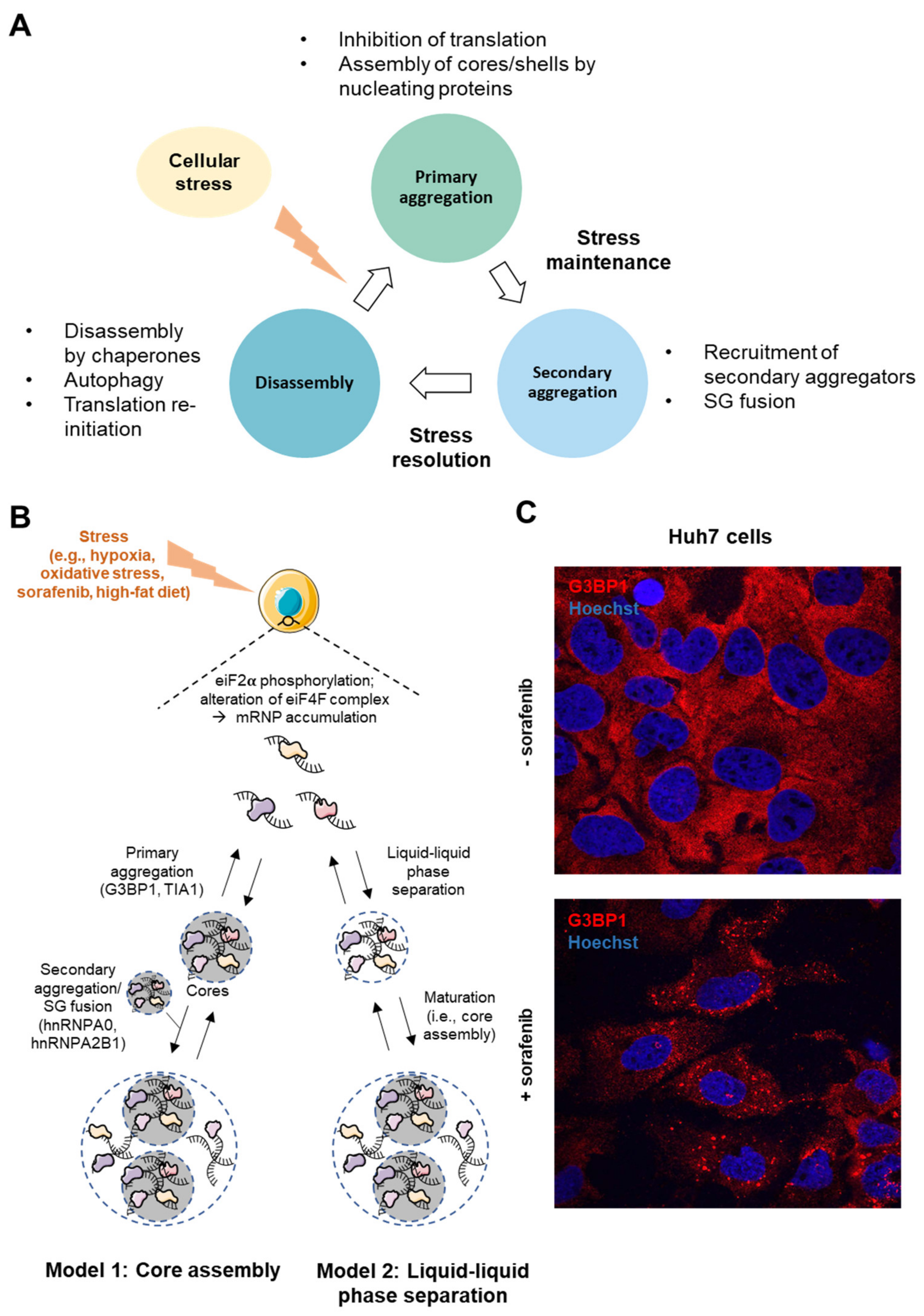
2. Molecular Bases and Complexity of SG Biogenesis
- In the “core first” model, the increased pool of untranslated mRNAs is bound and oligomerized by RBPs (e.g., G3BP1, TIA1, FMRP) bearing either a prion-like domain (PLD) or an intrinsically disordered domain (IDD), necessary for the recruitment of other proteins. This step, forming stable core structures is called “primary aggregation” [20]. The PLD and IDD domains of RBPs are enriched in glycine and uncharged polar residues (i.e., asparagine, glutamine, serine), which promote electrostatic interactions and liquid–liquid phase separation (LLPS)” [28]. Due to these special biophysical properties, SGs behave as hydrogel-like structures and are often considered as “viscous liquid droplets”. Then, the recruitment of additional ribonucleoproteins with weaker interactions (e.g., hnRNPA0, hnRNPA2B1, EWSR1) contributes to the formation of a dynamic shell (secondary aggregation) [20]. During the primary aggregation step, the transport of RBPs within SGs requires functional microtubules and motor proteins (i.e., dyneins and kinesins) [29,30]. Consistent with the role of microtubules in this process, HDAC6, which is a microtubule-associated deacetylase, reduces tubulin-α acetylation (Lys40) and promotes SG formation [31]. Finally, when the stress persists, other SGs components are recruited, allowing the growth and fusion of SGs in a process called “coalescence”, wherein several cores are embedded in a dynamic shell.
- In a second model called “LLPS First”, oligomerization of mRNAs with proteins containing IDD is believed to promote LLPS. Then, in a further step, the high density of core components stabilizes the core structures, which are assembled inside LLPS.
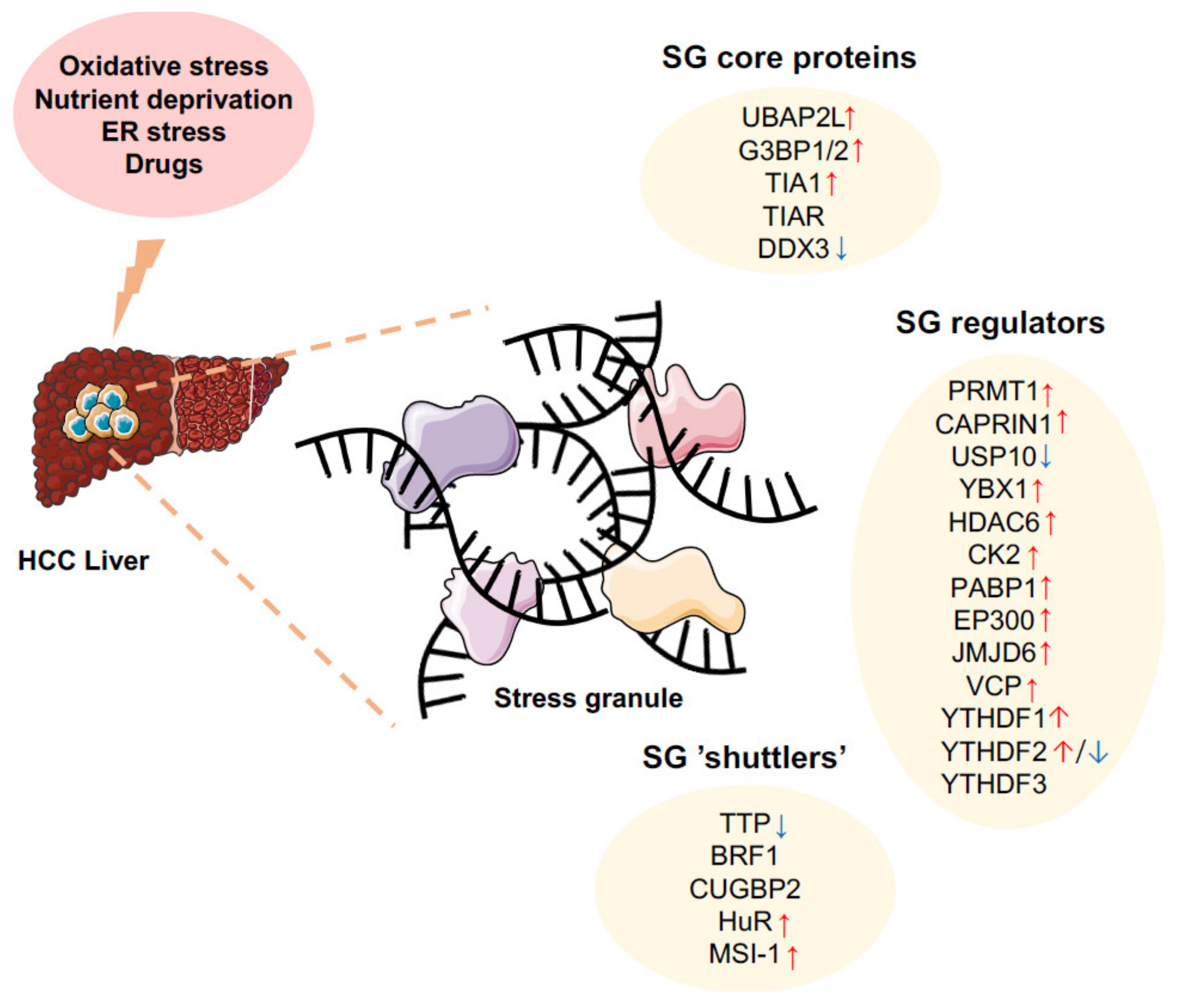
3. SGs in Hepatic Carcinogenesis
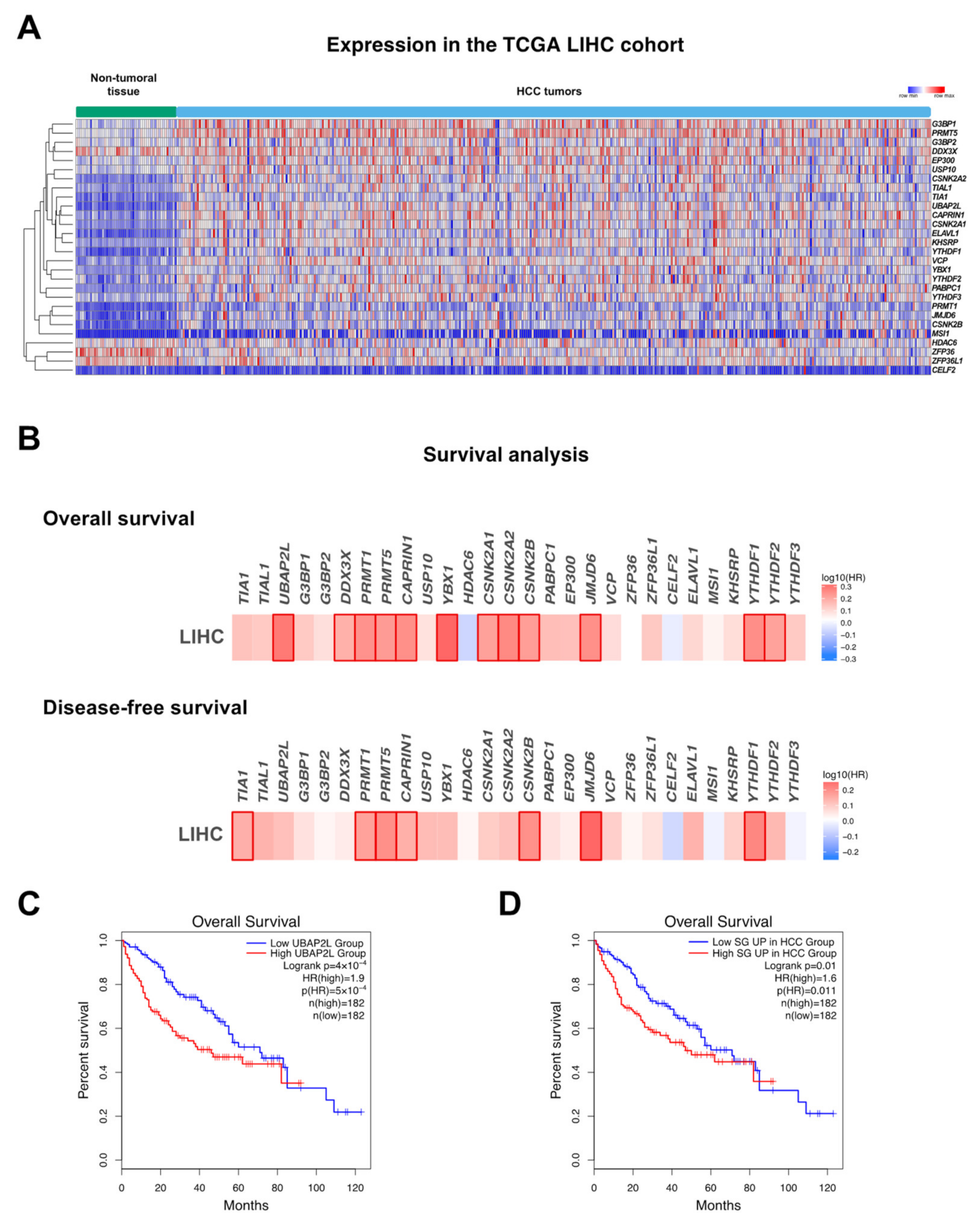
3.1. Nucleic Acids and Proteins Involved in SG Formation
3.1.1. UBAP2L (Ubiquitin-Associated Protein 2-Like)
3.1.2. G3BPs (Ras GTPase-Activating Protein-Binding Proteins)
3.1.3. T-Cell-Restricted Intracellular Antigen-1 (TIA1)
3.1.4. DDX3 (DEAD-Box RNA Helicase 3 or CAP-Rf)
3.1.5. G4DNA (G-Quadruplex DNA Structures)
3.1.6. tRNA-Derived Stress-Induced RNAs (tiRNAs)
3.1.7. m6A RNA-Related Proteins
3.2. Mechanisms Regulating SG Clearance in HCC
3.3. RNA-Binding Proteins Controlling mRNA Stability/Translation
3.3.1. TTP
3.3.2. BRF1 (Butyrate Response Factor 1, ZFP36L1)
3.3.3. HuR
3.3.4. CUGBP2
3.3.5. Musashi-1 (Msi-1)
4. Are SG Potential Therapeutic Targets in HCC?
4.1. Targeting SG Nucleators
4.2. Targeting Regulatory Pathways Involved in SG Assembly
4.3. Targeting Post-Translational Modification of SG Components
4.4. Increasing SG Clearance
4.5. Targeting Microtubules
4.6. Targeting AUBPs Associated with SGs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Kleiner, D.E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 2016, 65, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127 (Suppl. S1), S35–S50. [Google Scholar] [CrossRef]
- Cui, J.; Placzek, W.J. Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. Int. J. Mol. Sci. 2018, 19, 308. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Wong, T.C.; Lo, C.M. Resection strategies for hepatocellular carcinoma. Semin. Liver Dis. 2013, 33, 273–281. [Google Scholar] [CrossRef]
- Santopaolo, F.; Lenci, I.; Milana, M.; Manzia, T.M.; Baiocchi, L. Liver transplantation for hepatocellular carcinoma: Where do we stand? World J. Gastroenterol. 2019, 25, 2591–2602. [Google Scholar] [CrossRef]
- Sobolewski, C.; Calo, N.; Portius, D.; Foti, M. MicroRNAs in fatty liver disease. Semin. Liver Dis. 2015, 35, 12–25. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N.; Ivanov, P. Stress granules, P-bodies and cancer. Biochim. Biophys. Acta 2015, 1849, 861–870. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jiang, L.; Gong, Y.; Chen, X.; Ying, M.; Zhu, H.; He, Q.; Yang, B.; Cao, J. Stress granule: A promising target for cancer treatment. Br. J. Pharm. 2019, 176, 4421–4433. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157.e5. [Google Scholar] [CrossRef] [PubMed]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian stress granules and P bodies at a glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef] [PubMed]
- Omer, A.; Patel, D.; Lian, X.J.; Sadek, J.; Di Marco, S.; Pause, A.; Gorospe, M.; Gallouzi, I.E. Stress granules counteract senescence by sequestration of PAI-1. EMBO Rep. 2018, 19, e44722. [Google Scholar] [CrossRef]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef]
- Adjibade, P.; St-Sauveur, V.G.; Quevillon Huberdeau, M.; Fournier, M.J.; Savard, A.; Coudert, L.; Khandjian, E.W.; Mazroui, R. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget 2015, 6, 43927–43943. [Google Scholar] [CrossRef]
- Marcelo, A.; Koppenol, R.; de Almeida, L.P.; Matos, C.A.; Nobrega, C. Stress granules, RNA-binding proteins and polyglutamine diseases: Too much aggregation? Cell Death Dis. 2021, 12, 592. [Google Scholar] [CrossRef]
- Hofmann, S.; Cherkasova, V.; Bankhead, P.; Bukau, B.; Stoecklin, G. Translation suppression promotes stress granule formation and cell survival in response to cold shock. Mol. Biol. Cell 2012, 23, 3786–3800. [Google Scholar] [CrossRef]
- Bai, Y.; Dong, Z.; Shang, Q.; Zhao, H.; Wang, L.; Guo, C.; Gao, F.; Zhang, L.; Wang, Q. Pdcd4 Is Involved in the Formation of Stress Granule in Response to Oxidized Low-Density Lipoprotein or High-Fat Diet. PLoS ONE 2016, 11, e0159568. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J.M. CancerMine: A literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Chernov, K.G.; Barbet, A.; Hamon, L.; Ovchinnikov, L.P.; Curmi, P.A.; Pastre, D. Role of microtubules in stress granule assembly: Microtubule dynamical instability favors the formation of micrometric stress granules in cells. J. Biol. Chem. 2009, 284, 36569–36580. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.A.; Chudinova, E.M.; Nadezhdina, E.S. Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp. Cell Res. 2003, 290, 227–233. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Heberle, A.M.; Razquin Navas, P.; Langelaar-Makkinje, M.; Kasack, K.; Sadik, A.; Faessler, E.; Hahn, U.; Marx-Stoelting, P.; Opitz, C.A.; Sers, C.; et al. The PI3K and MAPK/p38 pathways control stress granule assembly in a hierarchical manner. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Sfakianos, A.P.; Mellor, L.E.; Pang, Y.F.; Kritsiligkou, P.; Needs, H.; Abou-Hamdan, H.; Desaubry, L.; Poulin, G.B.; Ashe, M.P.; Whitmarsh, A.J. The mTOR-S6 kinase pathway promotes stress granule assembly. Cell Death Differ. 2018, 25, 1766–1780. [Google Scholar] [CrossRef]
- Fournier, M.J.; Coudert, L.; Mellaoui, S.; Adjibade, P.; Gareau, C.; Cote, M.F.; Sonenberg, N.; Gaudreault, R.C.; Mazroui, R. Inactivation of the mTORC1-eukaryotic translation initiation factor 4E pathway alters stress granule formation. Mol. Cell. Biol. 2013, 33, 2285–2301. [Google Scholar] [CrossRef]
- Gal, J.; Chen, J.; Na, D.Y.; Tichacek, L.; Barnett, K.R.; Zhu, H. The Acetylation of Lysine-376 of G3BP1 Regulates RNA Binding and Stress Granule Dynamics. Mol. Cell. Biol. 2019, 39, e00052-19. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Gayatri, S.; Reineke, L.C.; Sbardella, G.; Bedford, M.T.; Lloyd, R.E. Arginine Demethylation of G3BP1 Promotes Stress Granule Assembly. J. Biol. Chem. 2016, 291, 22671–22685. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Reineke, L.C.; Jain, A.; Jung, S.Y.; Lloyd, R.E. Histone arginine demethylase JMJD6 is linked to stress granule assembly through demethylation of the stress granule-nucleating protein G3BP1. J. Biol. Chem. 2017, 292, 18886–18896. [Google Scholar] [CrossRef]
- Pene, V.; Li, Q.; Sodroski, C.; Hsu, C.S.; Liang, T.J. Dynamic Interaction of Stress Granules, DDX3X, and IKK-alpha Mediates Multiple Functions in Hepatitis C Virus Infection. J. Virol. 2015, 89, 5462–5477. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Silva Afonso, M.; Kelemen, S.E.; Ray, M.; Vrakas, C.N.; Burke, A.C.; Scalia, R.G.; Moore, K.; Autieri, M.V. Regulation of Stress Granule Formation by Inflammation, Vascular Injury, and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 2014–2027. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Y.; Dai, H.; Zhang, H.; Xie, M.; Zhang, H.; Chen, F.; Kang, X.; Bai, X.; Chen, Z. UBAP2L arginine methylation by PRMT1 modulates stress granule assembly. Cell Death Differ. 2020, 27, 227–241. [Google Scholar] [CrossRef]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Cirillo, L.; Cieren, A.; Barbieri, S.; Khong, A.; Schwager, F.; Parker, R.; Gotta, M. UBAP2L Forms Distinct Cores that Act in Nucleating Stress Granules Upstream of G3BP1. Curr. Biol. 2020, 30, 698–707.e6. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Cai, L.; Li, Z.; Guo, X. UBAP2L silencing inhibits cell proliferation and G2/M phase transition in breast cancer. Breast Cancer 2018, 25, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Huang, Y. Knockdown of ubiquitin associated protein 2-like inhibits the growth and migration of prostate cancer cells. Oncol. Rep. 2014, 32, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, W.; Hu, Y.C.; Yin, T.T.; He, J. Knockdown of Ubiquitin Associated Protein 2-Like (UBAP2L) Inhibits Growth and Metastasis of Hepatocellular Carcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 7109–7118. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, M.; Peng, Y.; He, J. Ubiquitin Associated Protein 2-Like (UBAP2L) Overexpression in Patients with Hepatocellular Carcinoma and its Clinical Significance. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 4779–4788. [Google Scholar] [CrossRef][Green Version]
- Ye, T.; Xu, J.; Du, L.; Mo, W.; Liang, Y.; Xia, J. Downregulation of UBAP2L Inhibits the Epithelial-Mesenchymal Transition via SNAIL1 Regulation in Hepatocellular Carcinoma Cells. Cell Physiol. Biochem. 2017, 41, 1584–1595. [Google Scholar] [CrossRef]
- Zhang, X.P.; Jiang, Y.B.; Zhong, C.Q.; Ma, N.; Zhang, E.B.; Zhang, F.; Li, J.J.; Deng, Y.Z.; Wang, K.; Xie, D.; et al. PRMT1 Promoted HCC Growth and Metastasis In Vitro and In Vivo via Activating the STAT3 Signalling Pathway. Cell Physiol. Biochem. 2018, 47, 1643–1654. [Google Scholar] [CrossRef]
- Wei, H.; Liu, Y.; Min, J.; Zhang, Y.; Wang, J.; Zhou, M.; Xiong, E.; Yu, G.; Zhou, H.; He, J.; et al. Protein arginine methyltransferase 1 promotes epithelial-mesenchymal transition via TGF-beta1/Smad pathway in hepatic carcinoma cells. Neoplasma 2019, 66, 918–929. [Google Scholar] [CrossRef]
- Ryu, J.W.; Kim, S.K.; Son, M.Y.; Jeon, S.J.; Oh, J.H.; Lim, J.H.; Cho, S.; Jung, C.R.; Hamamoto, R.; Kim, D.S.; et al. Novel prognostic marker PRMT1 regulates cell growth via downregulation of CDKN1A in HCC. Oncotarget 2017, 8, 115444–115455. [Google Scholar] [CrossRef] [PubMed]
- French, J.; Stirling, R.; Walsh, M.; Kennedy, H.D. The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem. J. 2002, 34, 223–231. [Google Scholar] [CrossRef]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Dai, L.; Liu, X.; Pan, G.; Chen, H.; Huang, J.; Xu, Q. Upregulation of caprin1 expression is associated with poor prognosis in hepatocellular carcinoma. Pathol. Res. Pract. 2017, 213, 1563–1567. [Google Scholar] [CrossRef]
- Li, X.Q.; Song, J.Y.; Lv, W.; Zhang, D.; Wu, J.Z. Circular circ_0000885 promotes hepatocellular carcinoma proliferation by epigenetically upregulating Caprin1. Eur. Rev. Med. Pharm. Sci. 2019, 23, 7848–7854. [Google Scholar]
- Zhang, Y.; You, W.; Zhou, H.; Chen, Z.; Han, G.; Zuo, X.; Zhang, L.; Wu, J.; Wang, X. Downregulated miR-621 promotes cell proliferation via targeting CAPRIN1 in hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 2116–2129. [Google Scholar]
- Lu, C.; Ning, Z.; Wang, A.; Chen, D.; Liu, X.; Xia, T.; Tekcham, D.S.; Wang, W.; Li, T.; Liu, X.; et al. USP10 suppresses tumor progression by inhibiting mTOR activation in hepatocellular carcinoma. Cancer Lett. 2018, 436, 139–148. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; He, H.; Zhao, W.; Chen, J.; Shao, R.G. GAP161 targets and downregulates G3BP to suppress cell growth and potentiate cisplaitin-mediated cytotoxicity to colon carcinoma HCT116 cells. Cancer Sci. 2012, 103, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.M.; Huang, H.X.; Chang, P.H.; Tseng, K.C.; Miyajima, A.; Chern, E. Y-box binding protein-1 promotes hepatocellular carcinoma-initiating cell progression and tumorigenesis via Wnt/beta-catenin pathway. Oncotarget 2017, 8, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.Z.; Chen, C.T.; Li, N.C.; Lin, L.C.; Huang, B.S.; Chang, Y.H.; Chow, L.P. Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2020, 22, 224. [Google Scholar] [CrossRef]
- Somasekharan, S.P.; El-Naggar, A.; Leprivier, G.; Cheng, H.; Hajee, S.; Grunewald, T.G.; Zhang, F.; Ng, T.; Delattre, O.; Evdokimova, V.; et al. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J. Cell Biol. 2015, 208, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Dou, N.; Chen, J.; Yu, S.; Gao, Y.; Li, Y. G3BP1 contributes to tumor metastasis via upregulation of Slug expression in hepatocellular carcinoma. Am. J. Cancer Res. 2016, 6, 2641–2650. [Google Scholar] [PubMed]
- Reineke, L.C.; Tsai, W.C.; Jain, A.; Kaelber, J.T.; Jung, S.Y.; Lloyd, R.E. Casein Kinase 2 Is Linked to Stress Granule Dynamics through Phosphorylation of the Stress Granule Nucleating Protein G3BP1. Mol. Cell. Biol. 2017, 37, e00596-16. [Google Scholar] [CrossRef]
- Zhang, H.X.; Jiang, S.S.; Zhang, X.F.; Zhou, Z.Q.; Pan, Q.Z.; Chen, C.L.; Zhao, J.J.; Tang, Y.; Xia, J.C.; Weng, D.S. Protein kinase CK2alpha catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015, 6, 34800–34817. [Google Scholar] [CrossRef]
- Yu, W.; Ding, X.; Chen, F.; Liu, M.; Shen, S.; Gu, X.; Yu, L. The phosphorylation of SEPT2 on Ser218 by casein kinase 2 is important to hepatoma carcinoma cell proliferation. Mol. Cell. Biochem. 2009, 325, 61–67. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, K.; Lee, K.H.; Kim, S.J.; Kim, J. Inhibition of casein kinase 2 enhances the death ligand- and natural kiler cell-induced hepatocellular carcinoma cell death. Clin. Exp. Immunol. 2008, 152, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sui, C.; Meng, F.; Tian, X.; Fu, L.; Li, Y.; Qi, X.; Cui, H.; Liu, Y.; Jiang, Y. Stable knockdown of protein kinase CK2-alpha (CK2alpha) inhibits migration and invasion and induces inactivation of hedgehog signaling pathway in hepatocellular carcinoma Hep G2 cells. Acta Histochem. 2014, 116, 1501–1508. [Google Scholar] [CrossRef]
- Yeh, T.S.; Lo, S.J.; Chen, P.J.; Lee, Y.H. Casein kinase II and protein kinase C modulate hepatitis delta virus RNA replication but not empty viral particle assembly. J. Virol. 1996, 70, 6190–6198. [Google Scholar] [CrossRef]
- Choi, S.E.; Kwon, S.; Seok, S.; Xiao, Z.; Lee, K.W.; Kang, Y.; Li, X.; Shinoda, K.; Kajimura, S.; Kemper, B.; et al. Obesity-Linked Phosphorylation of SIRT1 by Casein Kinase 2 Inhibits Its Nuclear Localization and Promotes Fatty Liver. Mol. Cell. Biol. 2017, 37, e00006-17. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Kanno, S.; Nitta, H.; Uesugi, N.; Sugai, T.; Masuda, T.; Wakabayashi, G.; Maesawa, C. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol. Rep. 2012, 28, 867–873. [Google Scholar] [CrossRef]
- Wang, Y.; Toh, H.C.; Chow, P.; Chung, A.Y.; Meyers, D.J.; Cole, P.A.; Ooi, L.L.; Lee, C.G. MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms. FASEB J. 2012, 26, 3032–3041. [Google Scholar] [CrossRef]
- Zhu, Y.; Ming, Q. Expression and prognostic roles of PABPC1 in hepatocellular carcinoma. Int. J. Surg. 2020, 84, 3–12. [Google Scholar]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Zhang, T.; Delestienne, N.; Huez, G.; Kruys, V.; Gueydan, C. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J. Cell Sci. 2005, 118 Pt 23, 5453–5463. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, R.; Yang, F.; Cheng, R.; Chen, X.; Cui, S.; Gu, Y.; Sun, W.; You, C.; Liu, Z.; et al. miR-19a promotes colorectal cancer proliferation and migration by targeting TIA1. Mol. Cancer 2017, 16, 53. [Google Scholar] [CrossRef]
- Sanchez-Jimenez, C.; Ludena, M.D.; Izquierdo, J.M. T-cell intracellular antigens function as tumor suppressor genes. Cell Death Dis. 2015, 6, e1669. [Google Scholar] [CrossRef]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef]
- Frankel, J.; Turf, R.M.; Nichols, D. Complications of calcaneal osteotomies. Clin. Podiatr. Med. Surg. 1991, 8, 409–423. [Google Scholar]
- Subramaniam, K.; Ooi, L.L.; Hui, K.M. Transcriptional down-regulation of IGFBP-3 in human hepatocellular carcinoma cells is mediated by the binding of TIA-1 to its AT-rich element in the 3′-untranslated region. Cancer Lett. 2010, 297, 259–268. [Google Scholar] [CrossRef]
- Yu, A.T.; Berasain, C.; Bhatia, S.; Rivera, K.; Liu, B.; Rigo, F.; Pappin, D.J.; Spector, D.L. PHAROH lncRNA regulates Myc translation in hepatocellular carcinoma via sequestering TIAR. Elife 2021, 10, e68263. [Google Scholar] [CrossRef]
- Shih, J.W.; Wang, W.T.; Tsai, T.Y.; Kuo, C.Y.; Li, H.K.; Wu Lee, Y.H. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 2012, 441, 119–129. [Google Scholar] [CrossRef]
- Mo, J.; Liang, H.; Su, C.; Li, P.; Chen, J.; Zhang, B. DDX3X: Structure, physiologic functions and cancer. Mol. Cancer 2021, 20, 38. [Google Scholar] [CrossRef]
- Chao, C.H.; Chen, C.M.; Cheng, P.L.; Shih, J.W.; Tsou, A.P.; Lee, Y.H. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 2006, 66, 6579–6588. [Google Scholar] [CrossRef]
- Li, H.K.; Mai, R.T.; Huang, H.D.; Chou, C.H.; Chang, Y.A.; Chang, Y.W.; You, L.R.; Chen, C.M.; Lee, Y.H. DDX3 Represses Stemness by Epigenetically Modulating Tumor-suppressive miRNAs in Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 28637. [Google Scholar] [CrossRef]
- Wang, W.T.; Tsai, T.Y.; Chao, C.H.; Lai, B.Y.; Wu Lee, Y.H. Y-Box Binding Protein 1 Stabilizes Hepatitis C Virus NS5A via Phosphorylation-Mediated Interaction with NS5A To Regulate Viral Propagation. J. Virol. 2015, 89, 11584–11602. [Google Scholar] [CrossRef]
- Chang, P.C.; Chi, C.W.; Chau, G.Y.; Li, F.Y.; Tsai, Y.H.; Wu, J.C.; Wu Lee, Y.H. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene 2006, 25, 1991–2003. [Google Scholar] [CrossRef]
- Byrd, A.K.; Zybailov, B.L.; Maddukuri, L.; Gao, J.; Marecki, J.C.; Jaiswal, M.; Bell, M.R.; Griffin, W.C.; Reed, M.R.; Chib, S.; et al. Evidence That G-quadruplex DNA Accumulates in the Cytoplasm and Participates in Stress Granule Assembly in Response to Oxidative Stress. J. Biol. Chem. 2016, 291, 18041–18057. [Google Scholar] [CrossRef]
- Tao, E.W.; Cheng, W.Y.; Li, W.L.; Yu, J.; Gao, Q.Y. tiRNAs: A novel class of small noncoding RNAs that helps cells respond to stressors and plays roles in cancer progression. J. Cell Physiol. 2020, 235, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.M.; Achorn, C.; Kedersha, N.L.; Anderson, P.J.; Ivanov, P. YB-1 regulates tiRNA-induced Stress Granule formation but not translational repression. Nucleic Acids Res. 2016, 44, 6949–6960. [Google Scholar] [CrossRef]
- Hisai, H.; Kato, J.; Kobune, M.; Murakami, T.; Miyanishi, K.; Takahashi, M.; Yoshizaki, N.; Takimoto, R.; Terui, T.; Niitsu, Y. Increased expression of angiogenin in hepatocellular carcinoma in correlation with tumor vascularity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 4852–4859. [Google Scholar]
- Hengjuan, L.V.; Liu, G.; Kun, L.I.; Mingqiu, L.I.; Zhang, D. Angiogenin regulates epithelial-mesenchymal transition of hepatocellular carcinoma through upregulation of HMGA2. Pharmazie 2019, 74, 301–304. [Google Scholar]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Anders, M.; Chelysheva, I.; Goebel, I.; Trenkner, T.; Zhou, J.; Mao, Y.; Verzini, S.; Qian, S.B.; Ignatova, Z. Dynamic m(6)A methylation facilitates mRNA triaging to stress granules. Life Sci. Alliance 2018, 1, e201800113. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef]
- Fu, Y.; Zhuang, X. m(6)A-binding YTHDF proteins promote stress granule formation. Nat. Chem. Biol. 2020, 16, 955–963. [Google Scholar] [CrossRef]
- Wang, P.; Wang, X.; Zheng, L.; Zhuang, C. Gene Signatures and Prognostic Values of m6A Regulators in Hepatocellular Carcinoma. Front. Genet. 2020, 11, 540186. [Google Scholar] [CrossRef]
- Huang, H.; Bai, Y.; Lu, X.; Xu, Y.; Zhao, H.; Sang, X. N6-methyladenosine associated prognostic model in hepatocellular carcinoma. Ann. Transl. Med. 2020, 8, 633. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Chen, J.; Chen, D.; Yang, B.; He, B.; Hu, W.; Zhang, Y.; Liu, H.; Dai, L.; et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer 2019, 18, 127. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Shi, Y.; Liu, J.; Lin, L.; Chen, X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am. J. Transl. Res. 2019, 11, 6084–6092. [Google Scholar] [PubMed]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef]
- Shi, Y.; Zhuang, Y.; Zhang, J.; Chen, M.; Wu, S. METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner. Cancer Manag. Res. 2020, 12, 13173–13184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, Y.; Chen, J.; Peng, C.; Zhang, Y.; Tong, R.; Cheng, Q.; Yang, B.; Feng, X.; Lu, Y.; et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m(6)A-guided epigenetic inhibition of LYPD1. Mol. Cancer 2020, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qin, J.; Gao, T.; Li, C.; He, B.; Pan, B.; Xu, X.; Chen, X.; Zeng, K.; Xu, M.; et al. YTHDF1 Facilitates the Progression of Hepatocellular Carcinoma by Promoting FZD5 mRNA Translation in an m6A-Dependent Manner. Mol. Ther. Nucleic Acids 2020, 22, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cao, M.; Gao, F.; He, X. YTHDF1 promotes hepatocellular carcinoma progression via activating PI3K/AKT/mTOR signaling pathway and inducing epithelial-mesenchymal transition. Exp. Hematol. Oncol. 2021, 10, 35. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, Y.; Mao, Q.; Jiang, X.; Jiang, W.; Chen, J.; Xu, W.; Zhong, L.; Sun, X. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018, 21, 859–868. [Google Scholar] [CrossRef]
- Bian, S.; Ni, W.; Zhu, M.; Song, Q.; Zhang, J.; Ni, R.; Zheng, W. Identification and Validation of the N6-Methyladenosine RNA Methylation Regulator YTHDF1 as a Novel Prognostic Marker and Potential Target for Hepatocellular Carcinoma. Front. Mol. Biosci. 2020, 7, 604766. [Google Scholar] [CrossRef]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019, 442, 252–261. [Google Scholar] [CrossRef]
- Shao, X.Y.; Dong, J.; Zhang, H.; Wu, Y.S.; Zheng, L. Systematic Analyses of the Role of the Reader Protein of N (6)-Methyladenosine RNA Methylation, YTH Domain Family 2, in Liver Hepatocellular Carcinoma. Front. Mol. Biosci. 2020, 7, 577460. [Google Scholar] [CrossRef]
- Wang, M.; Wei, K.; Qian, B.; Feiler, S.; Lemekhova, A.; Buchler, M.W.; Hoffmann, K. HSP70-eIF4G Interaction Promotes Protein Synthesis and Cell Proliferation in Hepatocellular Carcinoma. Cancers 2020, 12, 2262. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127 Pt 18, 3877–3883. [Google Scholar] [CrossRef]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef]
- Liu, Y.; Hei, Y.; Shu, Q.; Dong, J.; Gao, Y.; Fu, H.; Zheng, X.; Yang, G. VCP/p97, down-regulated by microRNA-129-5p, could regulate the progression of hepatocellular carcinoma. PLoS ONE 2012, 7, e35800. [Google Scholar] [CrossRef]
- Wang, B.; Maxwell, B.A.; Joo, J.H.; Gwon, Y.; Messing, J.; Mishra, A.; Shaw, T.I.; Ward, A.L.; Quan, H.; Sakurada, S.M.; et al. ULK1 and ULK2 Regulate Stress Granule Disassembly Through Phosphorylation and Activation of VCP/p97. Mol. Cell 2019, 74, 742–757.e8. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yu, H.; Zhang, X.; Shen, X.; Zhang, K.; Sheng, H.; Dai, S.; Gao, H. UNC51-like kinase 1 as a potential prognostic biomarker for hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2013, 6, 711–717. [Google Scholar]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Bakheet, T.; Frevel, M.; Williams, B.R.; Greer, W.; Khabar, K.S. ARED: Human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001, 29, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.; Williams, B.R.; Khabar, K.S. ARED 2.0: An update of AU-rich element mRNA database. Nucleic Acids Res. 2003, 31, 421–423. [Google Scholar] [CrossRef]
- Legrand, N.; Dixon, D.A.; Sobolewski, C. AU-rich element-binding proteins in colorectal cancer. World J. Gastrointest. Oncol. 2019, 11, 71–90. [Google Scholar] [CrossRef] [PubMed]
- DuBois, R.N.; McLane, M.W.; Ryder, K.; Lau, L.F.; Nathans, D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 1990, 265, 19185–19191. [Google Scholar] [CrossRef]
- Fu, R.; Olsen, M.T.; Webb, K.; Bennett, E.J.; Lykke-Andersen, J. Recruitment of the 4EHP-GYF2 cap-binding complex to tetraproline motifs of tristetraprolin promotes repression and degradation of mRNAs with AU-rich elements. RNA 2016, 22, 373–382. [Google Scholar] [CrossRef]
- Hsieh, H.H.; Chen, Y.A.; Chang, Y.J.; Wang, H.H.; Yu, Y.H.; Lin, S.W.; Huang, Y.J.; Lin, S.; Chang, C.J. The functional characterization of phosphorylation of tristetraprolin at C-terminal NOT1-binding domain. J. Inflamm. 2021, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.E.; Kuwano, Y.; Alkharouf, N.; Blackshear, P.J.; Gorospe, M.; Wilson, G.M. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009, 69, 5168–5176. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Sanduja, S.; Bemis-Standoli, K.; Pena, E.A.; Price, R.L.; Dixon, D.A. The mRNA binding proteins HuR and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis. Gastroenterology 2009, 136, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Dolicka, D.; Sobolewski, C.; Gjorgjieva, M.; Correia de Sousa, M.; Berthou, F.; De Vito, C.; Colin, D.J.; Bejuy, O.; Fournier, M.; Maeder, C.; et al. Tristetraprolin Promotes Hepatic Inflammation and Tumor Initiation but Restrains Cancer Progression to Malignancy. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 597–621. [Google Scholar] [CrossRef]
- Stoecklin, G.; Stubbs, T.; Kedersha, N.; Wax, S.; Rigby, W.F.; Blackwell, T.K.; Anderson, P. MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004, 23, 1313–1324. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef]
- Krohler, T.; Kessler, S.M.; Hosseini, K.; List, M.; Barghash, A.; Patial, S.; Laggai, S.; Gemperlein, K.; Haybaeck, J.; Muller, R.; et al. The mRNA-binding Protein TTP/ZFP36 in Hepatocarcinogenesis and Hepatocellular Carcinoma. Cancers 2019, 11, 1754. [Google Scholar] [CrossRef]
- Wu, J.C.; Luo, S.Z.; Liu, T.; Lu, L.G.; Xu, M.Y. linc-SCRG1 accelerates liver fibrosis by decreasing RNA-binding protein tristetraprolin. FASEB J. 2019, 33, 2105–2115. [Google Scholar] [CrossRef]
- Tran, D.D.H.; Koch, A.; Allister, A.; Saran, S.; Ewald, F.; Koch, M.; Nashan, B.; Tamura, T. Treatment with MAPKAP2 (MK2) inhibitor and DNA methylation inhibitor, 5-aza dC, synergistically triggers apoptosis in hepatocellular carcinoma (HCC) via tristetraprolin (TTP). Cell. Signal. 2016, 28, 1872–1880. [Google Scholar] [CrossRef]
- Sohn, B.H.; Park, I.Y.; Lee, J.J.; Yang, S.J.; Jang, Y.J.; Park, K.C.; Kim, D.J.; Lee, D.C.; Sohn, H.A.; Kim, T.W.; et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology 2010, 138, 1898–1908. [Google Scholar] [CrossRef]
- Sanduja, S.; Blanco, F.F.; Dixon, D.A. The roles of TTP and BRF proteins in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2011, 2, 42–57. [Google Scholar] [CrossRef]
- Blackshear, P.J. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 2002, 30 Pt 6, 945–952. [Google Scholar] [CrossRef]
- Suk, F.M.; Chang, C.C.; Lin, R.J.; Lin, S.Y.; Liu, S.C.; Jau, C.F.; Liang, Y.C. ZFP36L1 and ZFP36L2 inhibit cell proliferation in a cyclin D-dependent and p53-independent manner. Sci. Rep. 2018, 8, 2742. [Google Scholar] [CrossRef]
- Loh, X.Y.; Sun, Q.Y.; Ding, L.W.; Mayakonda, A.; Venkatachalam, N.; Yeo, M.S.; Silva, T.C.; Xiao, J.F.; Doan, N.B.; Said, J.W.; et al. RNA-Binding Protein ZFP36L1 Suppresses Hypoxia and Cell-Cycle Signaling. Cancer Res. 2020, 80, 219–233. [Google Scholar] [CrossRef]
- Zhong, S.; Machida, K.; Tsukamoto, H.; Johnson, D.L. Alcohol induces RNA polymerase III-dependent transcription through c-Jun by co-regulating TATA-binding protein (TBP) and Brf1 expression. J. Biol. Chem. 2011, 286, 2393–2401. [Google Scholar] [CrossRef]
- Tarling, E.J.; Clifford, B.L.; Cheng, J.; Morand, P.; Cheng, A.; Lester, E.; Sallam, T.; Turner, M.; de Aguiar Vallim, T.Q. RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J. Clin. Investig. 2017, 127, 3741–3754. [Google Scholar] [CrossRef]
- Benjamin, D.; Schmidlin, M.; Min, L.; Gross, B.; Moroni, C. BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites. Mol. Cell. Biol. 2006, 26, 9497–9507. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA stability. Cell. Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8, e1372. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Investig. 2001, 108, 1657–1665. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. Landmark Ed. 2012, 17, 189–205. [Google Scholar] [CrossRef]
- Von Roretz, C.; Di Marco, S.; Mazroui, R.; Gallouzi, I.E. Turnover of AU-rich-containing mRNAs during stress: A matter of survival. Wiley Interdiscip. Rev. RNA 2011, 2, 336–347. [Google Scholar] [CrossRef]
- Embade, N.; Fernandez-Ramos, D.; Varela-Rey, M.; Beraza, N.; Sini, M.; Gutierrez de Juan, V.; Woodhoo, A.; Martinez-Lopez, N.; Rodriguez-Iruretagoyena, B.; Bustamante, F.J.; et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 2012, 55, 1237–1248. [Google Scholar] [CrossRef]
- Zhu, H.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.H.; Ao, X.; Feng, L.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol. Cancer Res. 2015, 13, 809–818. [Google Scholar] [CrossRef]
- Vazquez-Chantada, M.; Fernandez-Ramos, D.; Embade, N.; Martinez-Lopez, N.; Varela-Rey, M.; Woodhoo, A.; Luka, Z.; Wagner, C.; Anglim, P.P.; Finnell, R.H.; et al. HuR/methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology 2010, 138, 1943–1953. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [PubMed]
- Moraes, K.C.; Monteiro, C.J.; Pacheco-Soares, C. A novel function for CUGBP2 in controlling the pro-inflammatory stimulus in H9c2 cells: Subcellular trafficking of messenger molecules. Cell Biol. Int. 2013, 37, 1129–1138. [Google Scholar] [CrossRef]
- Anant, S.; Henderson, J.O.; Mukhopadhyay, D.; Navaratnam, N.; Kennedy, S.; Min, J.; Davidson, N.O. Novel role for RNA-binding protein CUGBP2 in mammalian RNA editing. CUGBP2 modulates C to U editing of apolipoprotein B mRNA by interacting with apobec-1 and ACF, the apobec-1 complementation factor. J. Biol. Chem. 2001, 276, 47338–47351. [Google Scholar] [CrossRef]
- Morales, A.; Mari, M.; Garcia-Ruiz, C.; Colell, A.; Fernandez-Checa, J.C. Hepatocarcinogenesis and ceramide/cholesterol metabolism. Anticancer Agents Med. Chem. 2012, 12, 364–375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, G.; Jeong, Y.S.; Kim, D.W.; Kwak, M.J.; Koh, J.; Joo, E.W.; Lee, J.S.; Kah, S.; Sim, Y.E.; Yim, S.Y. Clinical significance of APOB inactivation in hepatocellular carcinoma. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Suzuki, H.; Takeuchi, M.; Sugiyama, A.; Alam, A.K.; Vu, L.T.; Sekiyama, Y.; Dam, H.C.; Ohki, S.Y.; Tsukahara, T. Alternative splicing produces structural and functional changes in CUGBP2. BMC Biochem. 2012, 13, 6. [Google Scholar] [CrossRef]
- Ohyama, T.; Nagata, T.; Tsuda, K.; Kobayashi, N.; Imai, T.; Okano, H.; Yamazaki, T.; Katahira, M. Structure of Musashi1 in a complex with target RNA: The role of aromatic stacking interactions. Nucleic Acids Res. 2012, 40, 3218–3231. [Google Scholar] [CrossRef]
- Nahas, G.R.; Murthy, R.G.; Patel, S.A.; Ganta, T.; Greco, S.J.; Rameshwar, P. The RNA-binding protein Musashi 1 stabilizes the oncotachykinin 1 mRNA in breast cancer cells to promote cell growth. FASEB J. 2016, 30, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, K.; Yang, Y.; Li, H.; Wang, Z.; Xu, X. Musashi-1 positively regulates growth and proliferation of hepatoma cells in vitro. Nan Fang Yi Ke Da Xue Xue Bao 2019, 39, 1436–1442. [Google Scholar] [PubMed]
- Chiou, G.Y.; Yang, T.W.; Huang, C.C.; Tang, C.Y.; Yen, J.Y.; Tsai, M.C.; Chen, H.Y.; Fadhilah, N.; Lin, C.C.; Jong, Y.J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Wang, H.; Ning, Y.; Zheng, H.; Liu, S.; Yang, Y.; Zhou, M.; Fan, S. Understanding the roles of stress granule during chemotherapy for patients with malignant tumors. Am. J. Cancer Res. 2020, 10, 2226–2241. [Google Scholar]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Xu, Y.; Pang, L.; Wang, H.; Xu, C.; Shah, H.; Guo, P.; Shu, D.; Qian, S.Y. Specific delivery of delta-5-desaturase siRNA via RNA nanoparticles supplemented with dihomo-gamma-linolenic acid for colon cancer suppression. Redox Biol. 2019, 21, 101085. [Google Scholar] [CrossRef]
- Dai, H.; Li, M.; Yang, W.; Sun, X.; Wang, P.; Wang, X.; Su, J.; Wang, X.; Hu, X.; Zhao, M. Resveratrol inhibits the malignant progression of hepatocellular carcinoma via MARCH1-induced regulation of PTEN/AKT signaling. Aging 2020, 12, 11717–11731. [Google Scholar] [CrossRef]
- Tang, Y.; Cao, J.; Cai, Z.; An, H.; Li, Y.; Peng, Y.; Chen, N.; Luo, A.; Tao, H.; Li, K. Epigallocatechin gallate induces chemopreventive effects on rats with diethylnitrosamineinduced liver cancer via inhibition of cell division cycle 25A. Mol. Med. Rep. 2020, 22, 3873–3885. [Google Scholar]
- Shim, J.H.; Su, Z.Y.; Chae, J.I.; Kim, D.J.; Zhu, F.; Ma, W.Y.; Bode, A.M.; Yang, C.S.; Dong, Z. Epigallocatechin gallate suppresses lung cancer cell growth through Ras-GTPase-activating protein SH3 domain-binding protein 1. Cancer Prev. Res. 2010, 3, 670–679. [Google Scholar] [CrossRef]
- Oi, N.; Yuan, J.; Malakhova, M.; Luo, K.; Li, Y.; Ryu, J.; Zhang, L.; Bode, A.M.; Xu, Z.; Li, Y.; et al. Resveratrol induces apoptosis by directly targeting Ras-GTPase-activating protein SH3 domain-binding protein 1. Oncogene 2015, 34, 2660–2671. [Google Scholar] [CrossRef]
- Naito, Y.; Ushiroda, C.; Mizushima, K.; Inoue, R.; Yasukawa, Z.; Abe, A.; Takagi, T. Epigallocatechin-3-gallate (EGCG) attenuates non-alcoholic fatty liver disease via modulating the interaction between gut microbiota and bile acids. J. Clin. Biochem. Nutr. 2020, 67, 2–9. [Google Scholar] [CrossRef]
- Sojoodi, M.; Wei, L.; Erstad, D.J.; Yamada, S.; Fujii, T.; Hirschfield, H.; Kim, R.S.; Lauwers, G.Y.; Lanuti, M.; Hoshida, Y.; et al. Epigallocatechin Gallate Induces Hepatic Stellate Cell Senescence and Attenuates Development of Hepatocellular Carcinoma. Cancer Prev. Res. 2020, 13, 497–508. [Google Scholar] [CrossRef]
- Gho, Y.S.; Yoon, W.H.; Chae, C.B. Antiplasmin activity of a peptide that binds to the receptor-binding site of angiogenin. J. Biol. Chem. 2002, 277, 9690–9694. [Google Scholar] [CrossRef]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, G.H.; Xie, J.M.; Li, B.; Gao, Q.G. Rottlerin upregulates DDX3 expression in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 495, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; Niu, C.; Cheng, Y. Diosgenin increased DDX3 expression in hepatocellular carcinoma. Am. J. Transl. Res. 2018, 10, 3590–3599. [Google Scholar] [PubMed]
- Zhang, B.; Yin, X.; Sui, S. Resveratrol inhibited the progression of human hepatocellular carcinoma by inducing autophagy via regulating p53 and the phosphoinositide 3kinase/protein kinase B pathway. Oncol. Rep. 2018, 40, 2758–2765. [Google Scholar] [PubMed]
- Won, H.R.; Lee, D.H.; Yeon, S.K.; Ryu, H.W.; Kim, G.W.; Kwon, S.H. HDAC6selective inhibitor synergistically enhances the anticancer activity of immunomodulatory drugs in multiple myeloma. Int. J. Oncol. 2019, 55, 499–512. [Google Scholar]
- Kaliszczak, M.; van Hechanova, E.; Li, Y.; Alsadah, H.; Parzych, K.; Auner, H.W.; Aboagye, E.O. The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 2018, 119, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhong, B.W.; Zhao, Z.R. ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma. J. Biol. Regul. Homeost. Agents 2018, 32, 851–858. [Google Scholar]
- Chen, M.C.; Lin, Y.C.; Liao, Y.H.; Liou, J.P.; Chen, C.H. MPT0G612, a Novel HDAC6 Inhibitor, Induces Apoptosis and Suppresses IFN-gamma-Induced Programmed Death-Ligand 1 in Human Colorectal Carcinoma Cells. Cancers 2019, 11, 1617. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Zhang, Y.; Fang, X.; Chen, N.; Zhou, X.; Wang, X. Sirt6 promotes tumorigenesis and drug resistance of diffuse large B-cell lymphoma by mediating PI3K/Akt signaling. J. Exp. Clin. Cancer Res. 2020, 39, 142. [Google Scholar] [CrossRef]
- Zhou, Q.; Lui, V.W.; Lau, C.P.; Cheng, S.H.; Ng, M.H.; Cai, Y.; Chan, S.L.; Yeo, W. Sustained antitumor activity by co-targeting mTOR and the microtubule with temsirolimus/vinblastine combination in hepatocellular carcinoma. Biochem. Pharm. 2012, 83, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Nagahara, T.; Matsumoto, K.; Murawaki, Y. The growth inhibition of liver cancer cells by paclitaxel and the involvement of extracellular signal-regulated kinase and apoptosis. Oncol. Rep. 2007, 17, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.; Cerofolini, L.; D’Agostino, V.G.; Zucal, C.; Fuccio, C.; Bonomo, I.; Dassi, E.; Giuntini, S.; Di Maio, D.; Vishwakarma, V.; et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. 2017, 45, 9514–9527. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Jiao, F.; Zhang, L.; Jiang, Y. Dihydrotanshinone Inhibits Hepatocellular Carcinoma by Suppressing the JAK2/STAT3 Pathway. Front. Pharm. 2021, 12, 654986. [Google Scholar] [CrossRef]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef]
- Yang, W.L.; Perillo, W.; Liou, D.; Marambaud, P.; Wang, P. AMPK inhibitor compound C suppresses cell proliferation by induction of apoptosis and autophagy in human colorectal cancer cells. J. Surg. Oncol. 2012, 106, 680–688. [Google Scholar] [CrossRef]
- Mahboubi, H.; Koromilas, A.E.; Stochaj, U. AMP Kinase Activation Alters Oxidant-Induced Stress Granule Assembly by Modulating Cell Signaling and Microtubule Organization. Mol. Pharm. 2016, 90, 460–468. [Google Scholar] [CrossRef]
- Ferrin, G.; Guerrero, M.; Amado, V.; Rodriguez-Peralvarez, M.; De la Mata, M. Activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef]
- Berzigotti, A.; Saran, U.; Dufour, J.F. Physical activity and liver diseases. Hepatology 2016, 63, 1026–1040. [Google Scholar] [CrossRef]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An Old Drug with New Applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef]
- Kwon, S.; Zhang, Y.; Matthias, P. The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 2007, 21, 3381–3394. [Google Scholar] [CrossRef]
- Kaliszczak, M.; Trousil, S.; Aberg, O.; Perumal, M.; Nguyen, Q.D.; Aboagye, E.O. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br. J. Cancer 2013, 108, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Damonte, P.; Sociali, G.; Parenti, M.D.; Soncini, D.; Bauer, I.; Boero, S.; Grozio, A.; Holtey, M.V.; Piacente, F.; Becherini, P.; et al. SIRT6 inhibitors with salicylate-like structure show immunosuppressive and chemosensitizing effects. Bioorgan. Med. Chem. 2017, 25, 5849–5858. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-Related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar]
- Huang, F.; Wang, B.R.; Wang, Y.G. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 4643–4651. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Cermak, V.; Dostal, V.; Jelinek, M.; Libusova, L.; Kovar, J.; Rosel, D.; Brabek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.W.; Preet, R.; Dhir, T.; Dixon, D.A.; Brody, J.R. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip. Rev. RNA 2020, 11, e1581. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Target | Cell Models | Tested in HCC Cells * |
|---|---|---|---|
| Resveratrol | G3BP1 [163] | SK-MEL-5 human melanoma, HCT116 human colorectal carcinoma | yes [170] |
| EGCG (epigallocatechin-gallate) | G3BP1 [162] | H1299 and CL13 lung cancer cells | yes [165] |
| GAP161 | G3BP1/2 [58] | HCT116 human colorectal carcinoma | no |
| EMICORON | G4DNA [167] | BJ EHLT immortalized human fibroblasts, A90-LUC colorectal murine cells | no |
| chANG | Angiogenin [166] | HT1080 (human fibrosarcoma), HM7 (human colorectal carcinoma), NIH/3T3 (Mouse fibroblast) | no |
| Rottlerin | DDX3 [168] | QGY7703, SMMC7721 liver cancer cells | yes [168] |
| Diosgenin | DDX3 [169] | HepG2, SMMC-7721 human liver cancer cells | yes [169] |
| Compound-C | AMPKα [21] | COS7 cells monkey kidney fibroblasts | no |
| A452 | HDAC6 [171] | Multiple myeloma cells: MM.1S, H929, BM-MSCs, PCS-500-012 cell lines | no |
| C1A | HDAC6 [172] | Panel of cancer cell lines (colon, breast, endometrial, epidermal, lung, myeloma, neuroblastoma, ovarian, and prostate cancer cells) | no |
| ACY-1215 | HDAC6 [173] | Lymphoma cells: OCI-LY10 | no |
| MPT0G612 | HDAC6 [174] | Colon cancer cells: HCT-116, HT-29 and DLD-1 | no |
| OSS_128167 | SIRT6 [175] | Large B-Cell Lymphoma: DLBCL cells | no |
| Vinblastine | Microtubules [30] | CV-1 green monkey kidney fibroblasts | yes [176] |
| Nocodazole | Microtubules [30] | CV-1 green monkey kidney fibroblasts | no |
| Paclitaxel | Microtubules [30] | CV-1 green monkey kidney fibroblasts | yes [177] |
| Temsirolimus | mTOR inhibitor [176] | Hep3B, HepG2, Huh7 | ys [176] |
| DHTS | HuR [178] | Colon cancer cells, HCT116 | yes [179] |
| MS-444 | HuR [180] | Colon cancer cells, HCT116 | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolicka, D.; Foti, M.; Sobolewski, C. The Emerging Role of Stress Granules in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 9428. https://doi.org/10.3390/ijms22179428
Dolicka D, Foti M, Sobolewski C. The Emerging Role of Stress Granules in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(17):9428. https://doi.org/10.3390/ijms22179428
Chicago/Turabian StyleDolicka, Dobrochna, Michelangelo Foti, and Cyril Sobolewski. 2021. "The Emerging Role of Stress Granules in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 17: 9428. https://doi.org/10.3390/ijms22179428
APA StyleDolicka, D., Foti, M., & Sobolewski, C. (2021). The Emerging Role of Stress Granules in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(17), 9428. https://doi.org/10.3390/ijms22179428