
R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde



Abstract
1. Introduction
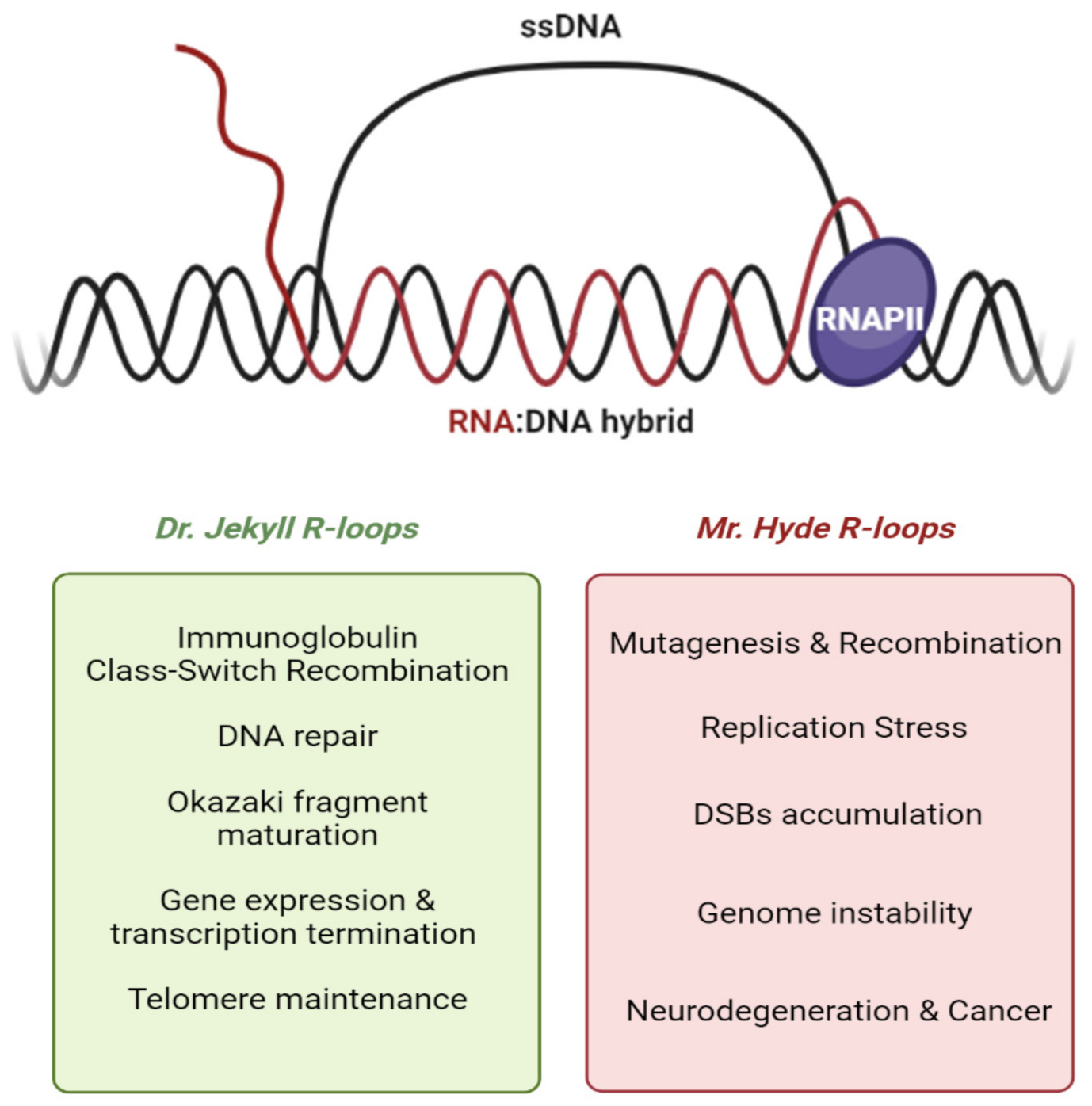
2. Dual Nature of R-Loops
3. R-Loops Accumulation: Why and Where
4. Prevention Mechanisms to Avoid R-Loop Accumulation
5. Resolving Mechanisms to Remove R-Loops
6. The Enigmatic Role of RNA:DNA Hybrids in DNA Repair
7. Role of Epigenetic Marks in R-Loop Homeostasis
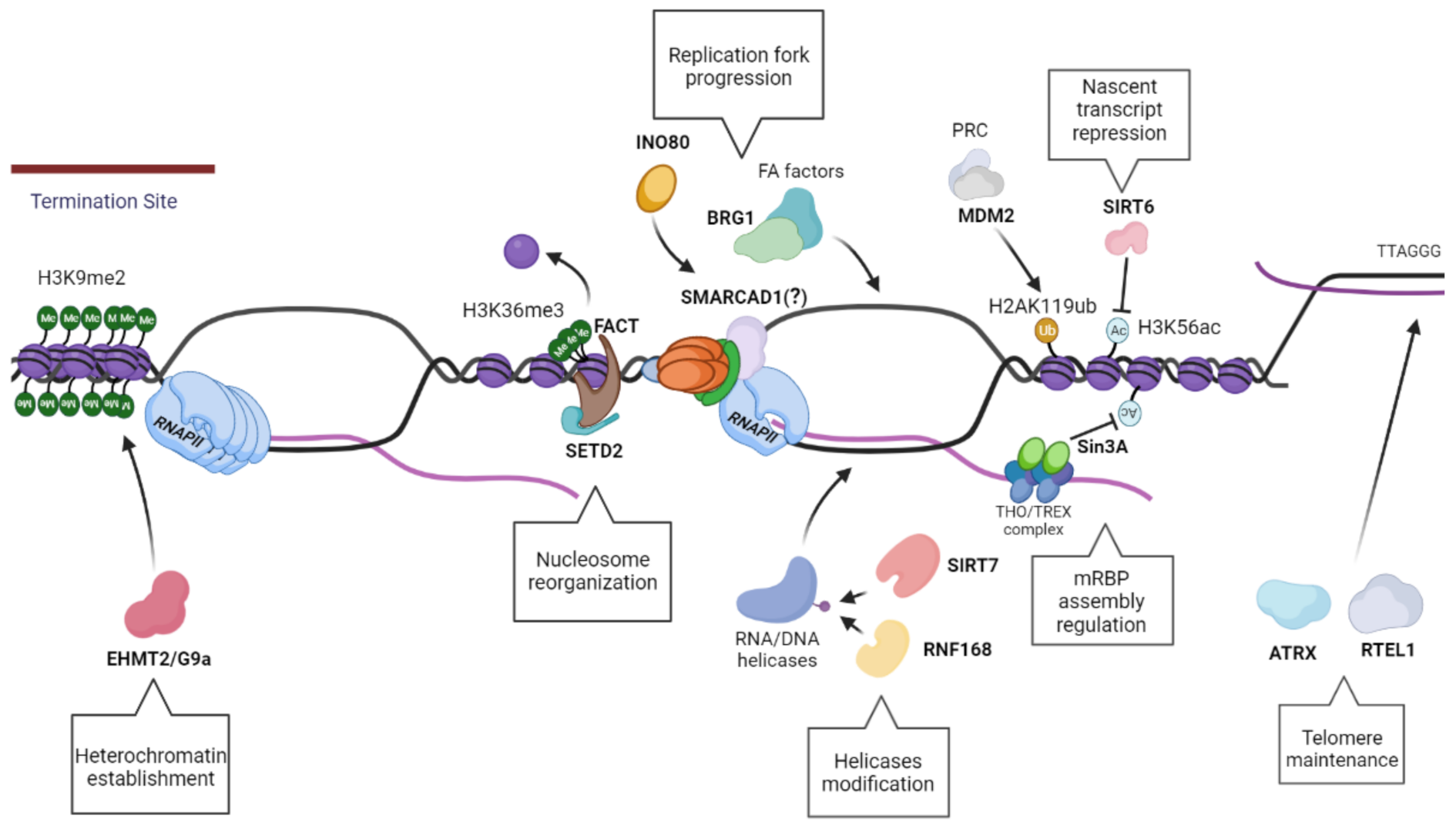
8. The Role of the Chro-Mates Part I: Chromatin Modifiers
8.1. EHMT2/G9a
8.2. SIRT6
8.3. SIRT7
8.4. Sin3A
8.5. RNF168
9. The Role of the Chro-Mates Part II: Chromatin Remodelers
9.1. MDM2
9.2. ATRX
9.3. FACT/SETD2
9.4. INO80
9.5. BRG1
9.6. Fft3/SMARCAD1
10. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 53BP1 | p53 Binding Protein 1 |
| AGS | Aicardi-Goutières Syndrome |
| AID | Activation-Induced Deaminase |
| ALS4 | Amyotrophic Lateral Sclerosis type 4 |
| ALT | Alternative Lengthening of Telomeres |
| AOA2 | Ataxia with Oculomotor Apraxia type 2 |
| ASF1 | Anti-silencing function 1 protein |
| ATM | Ataxia Telangectasia Mutated |
| ATR | Ataxia Telangectasia and Rad3-related protein |
| BAF | BRG1-associated factor |
| BAMBI | BMP and Activin Membrane-Bound Inhibitor |
| BAP1 | BRCA1-associated protein 1 |
| BER | Base Excision Repair |
| CAF-1 | Chromatin assembly factor-1 |
| cBAF | Canonical BAF |
| CBP | cAMP-response element binding protein |
| CD | Co-directional |
| CFSs | Common Fragile Sites |
| CPEO | Chronic Progressive External Ophthalmoplegia |
| CSB | Cockayne syndrome group B |
| CSR | Class-Switch Recombination |
| DIP-seq | DNA immunoprecipitation sequencing |
| DRIP-Seq | DNA–RNA Immuno-Precipitation sequencing |
| DRIPc-Seq | DNA-RNA Immuno-Precipitation followed by cDNA conversion coupled to high-throughput sequencing |
| DSB | Double-strand break |
| ERFSs | Early Replicating Fragile Sites |
| ESCs | Mouse embryonic stem cells |
| FA | Fanconi Anemia |
| FACT | Facilitates chromatin transcription |
| G4 | G-quadruplex |
| GLP | G9a-like protein |
| HAT | Histone acetyltransferase |
| HBD | Hybrid binding domain |
| HO | Head-on |
| HP1 | Heterochromatin protein 1 |
| HR | Homologous Recombination |
| ICLs | Inter-strand crosslinks |
| Ig | Immunoglobulin |
| iPSCs | induced pluripotent stem cells |
| m6A | N6-methyladenosine |
| MEFs | Mouse Embryonic Fibroblasts |
| METTL3 | Methyltransferase-like protein 3 |
| miRNA | microRNA |
| MMEJ | Micro-homology-Mediated End Joining |
| MMR | Mismatch Repair |
| MRN | Mre11-Rad50-Nbs1 |
| mRNP | messenger ribonucleoprotein |
| ncBAF | Non-canonical BAF |
| NHEJ | Non-Homologous End-Joining |
| PAF-1 | RNAPII-Associated Factor 1 |
| PCNA | Proliferating cell nuclear antigen |
| PRC | Polycomb Repressor Complex |
| PTMs | Post-translational modifications |
| RBPs | RNA-Binding Proteins |
| rDNA | ribosomal DNA |
| REZ | R-loop Elongation Zones |
| RFSs | Rare Fragile Sites |
| RIZ | R-loop Initiation Zones |
| RLFS | R-loop Forming Sequence |
| RNAPII | RNA polymerase II |
| RNF168 | Ring Finger protein 168 |
| rNMPs | ribonucleotide monophosphates |
| R-T | Replication-Transcription |
| SETX | Senataxin |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| TA-HRR | Transcription-Associated Homologous Recombination Repair |
| TAM | Transcription-associated mutagenesis |
| TAR | Transcription-associated recombination |
| TC-NER | Transcription-Coupled Nucleotide Excision Repair |
| TDRD3 | Tudor domain-containing protein 3 complex |
| TERRA | Telomeric-Repeat-containing RNA |
| TGFβ | Transforming growth factor beta |
| TonEBP | Tonicity-responsive enhancer binding protein |
| TOP1 | DNA topoisomerase 1 |
| TOP2A | DNA topoisomerase 2-alpha |
| TOP2B | DNA topoisomerase 2-beta |
| TOP3B | DNA topoisomerase 3 Beta |
| UFBs | Ultra-fine anaphase bridges |
| XPG | Xeroderma Pigmentosum group G |
| Xrn2 | 5′-3′ exoribonuclease 2 |
References
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double stranded DNA: Formation of R loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef]
- Robberson, D.L.; Kasamatsu, H.; Vinograd, J. Replication of mitochondrial DNA. Circular replicative intermediates in mouse L cells. Proc. Natl. Acad. Sci. USA 1972, 69, 737–741. [Google Scholar] [CrossRef]
- Gnatt, A.L.; Cramer, P.; Fu, J.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: An RNA polymerase II elongation complex at 3.3 A resolution. Science 2001, 292, 1876–1882. [Google Scholar] [CrossRef]
- Bushnell, D.A.; Westover, K.D.; Davis, R.E.; Kornberg, R.D. Structural Basis of Transcription: An RNA Polymerase II-TFIIB Cocrystal at 4.5 Angstroms. Science 2004, 303, 983–988. [Google Scholar] [CrossRef]
- Roy, D.; Lieber, M.R. G Clustering Is Important for the Initiation of Transcription-Induced R-Loops In Vitro, whereas High G Density without Clustering Is Sufficient Thereafter. Mol. Cell Biol. 2009, 29, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.F.; Wang, G.G. R-loops: Formation, function, and relevance to cell stress. Cell Stress 2019, 3, 38–46. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-Loop Formation Is a Distinctive Characteristic of Unmethylated Human CpG Island Promoters. Mol. Cell 2012, 30, 814–825. [Google Scholar] [CrossRef]
- Chien, Y.H.; Davidson, N. Rna-DNA Hybrids Are More Stable Than DNA-DNA Duplexes in Concentrated Perchlorate and Trichloroacetate Solutions. Nucleic Acids Res. 1978, 5, 1627–1637. [Google Scholar] [CrossRef]
- Sugimoto, N.; Nakano, S.; Katoh, M.; Matsumura, A.; Nakamuta, H.; Ohmichi, T.; Yoneyama, M.; Sasaki, M. Thermodynamic Parameters to Predict Stability of Rna/DNA Hybrid Duplexes. Biochemistry 1995, 34, 11211–11216. [Google Scholar] [CrossRef]
- Roy, D.; Zhang, Z.; Lu, Z.; Hsieh, C.-L.; Lieber, M.R. Competition between the RNA Transcript and the Nontemplate DNAStrand during R-Loop Formation In Vitro: A Nick Can Serve as a Strong R-Loop Initiation Site. Mol. Cell Biol. 2010, 20, 1998. [Google Scholar] [CrossRef]
- Wongsurawat, T.; Jenjaroenpun, P.; Kwoh, C.K.; Kuznetsov, V. Quantitative model of R-loop forming structures reveals a novel level of RNA-DNA interactome complexity. Nucleic Acids Res. 2012, 40, e16. [Google Scholar] [CrossRef]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell 2017, 170, 787–799.e18. [Google Scholar] [CrossRef]
- Leela, J.K.; Syeda, A.H.; Anupama, K.; Gowrishankar, J. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally Formed DNA: RNA Hybrids Mediate Transcription Elongation Impairment and Transcription-Associated Recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Xu, H.; Li, K.; Fan, Y.X.; Liu, Y.; Yang, X.R.; Sun, Q.W. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat. Plants 2017, 3, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, K.; Li, S.; Hou, Q.C.; Zhang, Y.S.; Liu, K.P.; Sun, Q.W. The R-Loop Atlas of Arabidopsis Development and Responses to Environmental Stimuli. Plant Cell 2020, 32, 888–903. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC skew at the 59 and 39 ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.Q.; Chedin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef]
- Chedin, F.; Benham, C.J.; Musier-Forsyth, K. Emerging roles for R-loop structures in the management of topological stress. J. Biol. Chem. 2020, 295, 4684–4695. [Google Scholar] [CrossRef]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.L. Genome instabilities arising from ribonucleotides in DNA. DNA Repair 2017, 56, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Clausen, A.R.; Clark, A.B.; Kunkel, T.A. Ribonucleotides Are Signals for Mismatch Repair of Leading-Strand Replication Errors. Mol. Cell 2013, 50, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.M.; Conlin, M.P.; Carvajal-Garcia, J.; Luedeman, M.E.; Luthman, A.J.; Small, G.W.; Ramsden, D.A. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 2018, 361, 1126–1129. [Google Scholar] [CrossRef]
- Westover, K.D.; Bushnell, D.A.; Kornberg, R.D. Structural basis of transcription: Nucleotide selection by rotation in the RNA polymerase II active center. Cell 2004, 119, 481–489. [Google Scholar] [CrossRef]
- Xin, L.; David, A.B.; Roger, D.K. RNA Polymerase II Transcription: Structure and Mechanism. Biochim. Biophys. Acta 2013, 1829, 2–8. [Google Scholar] [CrossRef]
- Yu, K.; Chedin, F.; Hsieh, C.L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef]
- Yu, K.; Lieber, M.R. Current insights into the mechanism of mammalian immunoglobulin class switch recombination. Crit. Rev. Biochem. Mol. 2019, 54, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Schrader, C.E. IgH Chain Class Switch Recombination: Mechanism and Regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef]
- Rawal, C.C.; Zardoni, L.; Di Terlizzi, M.; Galati, E.; Brambati, A.; Lazzaro, F.; Liberi, G.; Pellicioli, A. Senataxin Ortholog Sen1 Limits DNA:RNA Hybrid Accumulation at DNA Double-Strand Breaks to Control End Resection and Repair Fidelity. Cell Rep. 2020, 31, 107603. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Gromak, N. Out of Balance: R-loops in Human Disease. PLoS Genet. 2014, 10, 4630. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 24, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, Y.A.; Fernando, C.M.; Tran, E.J. The balancing act of R-loop biology: The good, the bad, and the ugly. J. Biol. Chem. 2020, 295, 905–913. [Google Scholar] [CrossRef]
- Chedin, F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA: DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef]
- Duquette, M.L.; Handa, P.; Vincent, J.A.; Taylor, A.F.; Maizels, N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004, 18, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Maffia, A.; Ranise, C.; Sabbioneda, S. From R-loops to G-quadruplexes: Emerging new threats for the replication fork. Int. J. Mol. Sci. 2020, 21, 1506. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guedin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [PubMed]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex-R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef]
- Lim, G.; Hohng, S. Single-molecule fluorescence studies on cotranscriptional G-quadruplex formation coupled with R-loop formation. Nucleic Acids Res. 2020, 48, 9195–9203. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Nardini, E.; Pellicioli, A.; Liberi, G. The dark side of RNA:DNA hybrids. Mutation Research. Rev. Mutat. Res. 2020, 784, 108300. [Google Scholar] [CrossRef]
- Brambati, A.; Colosio, A.; Zardoni, L.; Galanti, L.; Liberi, G. Replication and transcription on a collision course: Eukaryotic regulation mechanisms and implications for DNA stability. Front. Genet. 2015, 6, 166. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Merrikh, H. The Clash of Macromolecular Titans: Replication-Transcription Conflicts in Bacteria. Annu. Rev. Microbiol. 2018, 72, 71–88. [Google Scholar] [CrossRef]
- Rondón, A.G.; Aguilera, A. What causes an RNA-DNA hybrid to compromise genome integrity? DNA Repair 2019, 81, 102660. [Google Scholar] [CrossRef]
- Lin, Y.L.; Pasero, P. Transcription-Replication Conflicts: Orientation Matters. Cell 2017, 170, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Achar, Y.J.; Piccini, D.; Galanti, L.; Colosio, A.; Foiani, M.; Liberi, G. Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res. 2018, 46, 1227–1239. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [PubMed]
- French, S. Consequences of replication fork movement through transcription units in vivo. Science 1992, 258, 1362–1365. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell Biol. 2005, 25, 888–895. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Replication Fork Stalling at Natural Impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 2021, 8, 618157. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome Fragile Sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef]
- Sutherland, G.R. Rare fragile sites. Cytogenet. Genome Res. 2003, 100, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Marquis Gacy, A.; Goellner, G.; Juranić, N.; Macura, S.; McMurray, C.T. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Courtot, L.; Hoffmann, J.S.; Bergoglio, V. The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef]
- Voutsinos, V.; Munk, S.H.N.; Oestergaard, V.H. Common chromosomal fragile sites—Conserved failure stories. Genes 2018, 9, 580. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Chan, K.L.; Hickson, I.D. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol. 2011, 22, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Aguilera, A. The connection between transcription and genomic instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Sánchez, M.S.; Barroso, S.; Gómez-González, B.; Luna, R.; Aguilera, A. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011, 7, e1002386. [Google Scholar] [CrossRef] [PubMed]
- González-Aguilera, C.; Tous, C.; Gómez-González, B.; Huertas, P.; Luna, R.; Aguilera, A. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol. Biol. Cell 2008, 19, 4310–4318. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. mRNA processing and genomic instability. Nat. Struct. Mol. Biol. 2005, 12, 737–738. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing Replication Stress to Maintain Genome Stability: Resolving Conflicts between Replication and Transcription. Mol. Cell 2012, 45, 710–718. [Google Scholar] [CrossRef]
- Keszthelyi, A.; Minchell, N.E.; Baxter, J. The Causes and Consequences of Topological Stress during DNA Replication. Genes 2016, 7, 134. [Google Scholar] [CrossRef]
- Doksani, Y.; Bermejo, R.; Fiorani, S.; Haber, J.E.; Foiani, M. Replicon Dynamics, Dormant Origin Firing, and Terminal Fork Integrity after Double-Strand Break Formation. Cell 2009, 137, 247–258. [Google Scholar] [CrossRef]
- Yang, Y.Z.; McBride, K.M.; Hensley, S.; Lu, Y.; Chedin, F.; Bedford, M.T. Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation. Mol. Cell 2014, 53, 484–497. [Google Scholar] [CrossRef]
- Bermejo, R.; Doksani, Y.; Capra, T.; Katou, Y.M.; Tanaka, H.; Shirahige, K.; Foiani, M. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007, 21, 1921–1936. [Google Scholar] [CrossRef]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication Termination at Eukaryotic Chromosomes Is Mediated by Top2 and Occurs at Genomic Loci Containing Pausing Elements. Mol. Cell 2010, 39, 595–605. [Google Scholar] [CrossRef]
- Drolet, M.; Phoenix, P.; Menzel, R.; Massé, E.; Liu, L.F.; Crouch, R.J. Overexpression of RNase H partially complements the growth defect of an Escherichia coli ΔtopA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. USA 1995, 92, 3526–3530. [Google Scholar] [CrossRef]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100. [Google Scholar] [CrossRef]
- Suzuki, Y.; Holmes, J.B.; Cerritelli, S.M.; Sakhuja, K.; Minczuk, M.; Holt, I.J.; Crouch, R.J. An Upstream Open Reading Frame and the Context of the Two AUG Codons Affect the Abundance of Mitochondrial and Nuclear RNase H1. Mol. Cell Biol. 2010, 30, 5123–5134. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.G.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Reyes, A.; Melchionda, L.; Nasca, A.; Carrara, F.; Lamantea, E.; Zanolini, A.; Lamperti, C.; Fang, M.Y.; Zhang, J.G.; Ronchi, D.; et al. RNASEH1 Mutations Impair mtDNA Replication and Cause Adult-Onset Mitochondrial Encephalomyopathy. Am. J. Hum. Genet. 2015, 97, 186–193. [Google Scholar] [CrossRef]
- Sachdev, A.; Fratter, C.; McMullan, T.F.W. Novel mutation in the RNASEH1 gene in a chronic progressive external ophthalmoplegia patient. Can. J. Ophthalmol. 2018, 53, e203–e205. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.; Sparks, J.L.; Rychlik, M.; Nowotny, M.; Burgers, P.M.; Crouch, R.J.; Cerritelli, S.M. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 2013, 41, 3130–3143. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef]
- Lockhart, A.; Pires, V.B.; Bento, F.; Kellner, V.; Luke-Glaser, S.; Yakoub, G.; Ulrich, H.D.; Luke, B. RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cell Rep. 2019, 29, 2890–2900.e5. [Google Scholar] [CrossRef]
- Marnef, A.; Legube, G. m(6)A RNA modification as a new player in R-loop regulation. Nat Genet. 2020, 52, 27–28. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Fu, J.R.; Zhou, Y.F. A Review in Research Progress Concerning m6A Methylation and Immunoregulation. Front. Immunol. 2019, 10, 922. [Google Scholar] [CrossRef]
- Abakir, A.; Giles, T.C.; Cristini, A.; Foster, J.M.; Dai, N.; Starczak, M.; Rubio-Roland, A.; Li, M.M.; Eleftheriou, M.; Crutchley, J.; et al. N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat. Genet. 2020, 52, 48–55. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Q.L.; Xu, W.; Zhang, Y.C.; Yang, Y.; Ju, L.F.; Chen, J.; Chen, Y.S.; Li, K.; Ren, J.; et al. m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res. 2019, 29, 1035–1038.e5. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Cheon, N.Y.; Park, H.; Jeong, G.W.; Ye, B.J.; Yoo, E.J.; Lee, J.H.; Hur, J.H.; Lee, E.A.; Kim, H.K.; et al. TonEBP recognizes R-loops and initiates m6A RNA methylation for R-loop resolution. Nucleic Acids Res. 2021, 49, 269–284. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 2017, 67, 19–29.e3. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair. Cell 2018, 175, 558–570.e11. [Google Scholar] [CrossRef] [PubMed]
- Song, C.L.; Hotz-Wagenblatt, A.; Voit, R.; Grummt, I. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Gene Dev. 2017, 31, 1370–1381. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Chang, E.Y.C.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.Q.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop- associated genome instabil. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef]
- Okamoto, Y.; Hejna, J.; Takata, M. Regulation of R-loops and genome instability in Fanconi anemia. J. Biochem. 2019, 165, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Yuce, O.; West, S.C. Senataxin, Defective in the Neurodegenerative Disorder Ataxia with Oculomotor Apraxia 2, Lies at the Interface of Transcription and the DNA Damage Response. Mol. Cell Biol. 2013, 33, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.K.; Linder, P. DExD/H box RNA helicases: From generic motors to specific dissociation functions. Mol. Cell 2001, 8, 251–262. [Google Scholar] [CrossRef]
- Santoriello, C.; Sporrij, A.; Yang, S.; Flynn, R.A.; Henriques, T.; Dorjsuren, B.; Greig, E.C.; McCall, W.; Stanhope, M.E.; Fazio, M.; et al. RNA helicase DDX21 mediates nucleotide stress responses in neural crest and melanoma cells. Nat. Cell Biol. 2020, 22, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Uchida, C.; Anderson, S.F.; Lee, C.G.; Hurwitz, J.; Parvin, J.D.; Montminy, M. RNA helicase a mediates association of CBP with RNA polymerase II. Cell 1997, 90, 1107–1112. [Google Scholar] [CrossRef]
- Anderson, S.E.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Grosse, F. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 2011, 10, 654–665. [Google Scholar] [CrossRef]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during Telomere Maintenance in ALT Cells. PLoS ONE 2014, 9, e103819. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 2253. [Google Scholar] [CrossRef]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.C.; Stirling, P.C. Replication fork protection factors controlling R-loop bypass and suppression. Genes 2017, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nienninuszczy, J.; Shin-ya, K.; Niedzwiedz, W. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J. Cell Biol. 2013, 201, 33–48. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbson, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Gómez-González, B.; Grzechnik, P.; Rondón, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Appanah, R.; Lones, E.C.; Aiello, U.; Libri, D.; De Piccoli, G. Sen1 Is Recruited to Replication Forks via Ctf4 and Mrc1 and Promotes Genome Stability. Cell Rep. 2020, 30, 2094–2105.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kirkness, E.F.; Caballero, O.L.; Galante, P.A.; Parmigiani, R.B.; Edsall, L.; Kuan, S.; Ye, Z.; Levy, S.; Vasconcelos, A.T.R.; et al. Systematic detection of putative tumor suppressor genes through the combined use of exome and transcriptome sequencing. Genome Biol. 2010, 11, R114. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.C.; Klur, S.; Watanabe, M.; Németh, A.H.; Le Ber, I.; Moniz, J.C.; Tranchant, C.; Aubourg, P.; Tazim, M.; Schols, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Cho, E.; Wahnich, A.; Gao, F.; Becherel, O.J.; Wang, X.; Fike, F.; Chen, L.; Criscuolo, C.; De Michele, G.; et al. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum. Mol. Genet. 2014, 23, 4758–4769. [Google Scholar] [CrossRef]
- Suraweera, A.; Becherel, O.J.; Chen, P.; Rundle, N.; Woods, R.; Nakamura, J.; Gatei, M.; Criscuolo, C.; Filla, A.; Chessa, L.; et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007, 177, 969–979. [Google Scholar] [CrossRef]
- Yeo, A.J.; Becherel, O.J.; Luff, J.E.; Cullen, J.K.; Wongsurawat, T.; Jenjaroenpun, P.; Kuznetsov, V.A.; McKinnon, P.J.; Lavin, M.F. R-loops in proliferating cells but not in the brain: Implications for AOA2 and other autosomal recessive ataxias. PLoS ONE 2014, 9, e90219. [Google Scholar] [CrossRef]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol. Cell 2018, 69, 426–437.e7. [Google Scholar] [CrossRef]
- Mersaoui, S.Y.; Yu, Z.B.; Coulombe, Y.; Karam, M.; Busatto, F.F.; Masson, J.Y.; Richard, S. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019, 38, e100986. [Google Scholar] [CrossRef]
- Sessa, G.; Gomez-Gonzalez, B.; Silva, S.; Perez-Calero, C.; Beaurepere, R.; Barroso, S.; Martineau, S.; Martin, C.; Ehlén, Å.; Martínez, J.S.; et al. BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repairdouble dagger. EMBO J. 2021, 40, e106018. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Whelan, D.R.; Howard, S.M.; Vitelli, V.; Renaudin, X.; Adamowicz, M.; Iannelli, F.; Jones-Weinert, C.W.; Lee, M.; Matti, V.; et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 2018, 9, 5376. [Google Scholar] [CrossRef]
- Lu, W.T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA: RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 2016, 167, 1001–1013.e7. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Puget, N.; Lin, Y.-L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Marnef, A.; Legube, G. R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol. 2021, 23, 305–313. [Google Scholar] [CrossRef] [PubMed]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef]
- Barroso, S.; Herrera-Moyano, E.; Munoz, S.; Garcia-Rubio, M.; Gomez-Gonzalez, B.; Aguilera, A. The DNA damage response acts as a safeguard against harmful DNA-RNA hybrids of different origins. EMBO Rep. 2019, 20, e47250. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.P.; Wade-Martins, R.; Gromak, N. R-loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef]
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondó, A.G.; Barroso, S.; Andú, E.; Nica Pé Rez-Alegre, M.; García-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Komar, D.; Juszczynski, P. Rebelled epigenome: Histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin. Epigenetics 2020, 12, 147. [Google Scholar] [CrossRef]
- Chen, C.C.L.; Goyal, P.; Karimi, M.M.; Abildgaard, M.H.; Kimura, H.; Lorincz, M.C. H3S10ph broadly marks early-replicating domains in interphase ESCs and shows reciprocal antagonism with H3K9me2. Genome Res. 2018, 28, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Tachibana, M. H3K9 methyltransferase G9a and the related molecule GLP. Gene Dev. 2011, 25, 781–788. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, Y.; Walker, D.L.; Dong, H.; Conley, C.; Johansen, J.; Johansen, K.M. JIL-1: A novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol. Cell 1999, 4, 129–135. [Google Scholar] [CrossRef]
- Zhang, W.G.; Deng, H.; Bao, X.; Lerach, S.; Girton, J.; Johansen, J.; Johansen, K.M. The JIL-1 histone H3S10 kinase regulates dimethyl H3K9 modifications and heterochromatic spreading in Drosophila. Development 2006, 133, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, L.; Wang, Q.; Hu, Y.; Zhao, W.; Gautam, M.; Li, L. H3K9 Demethylation-Induced R-Loop Accumulation Is Linked to Disorganized Nucleoli. Front Genet. 2020, 11, 43. [Google Scholar] [CrossRef]
- Feldman, J.L.; Peterson, C.L. Yeast Sirtuin Family Members Maintain Transcription Homeostasis to Ensure Genome Stability. Cell Rep. 2019, 27, 2978–2989.e5. [Google Scholar] [CrossRef]
- Che, J.; Smith, S.; Kim, Y.J.; Shim, E.Y.; Myung, K.; Lee, S.E. Hyper-Acetylation of Histone H3K56 Limits Break-Induced Replication by Inhibiting Extensive Repair Synthesis. PLoS Genet. 2015, 11, e1004990. [Google Scholar] [CrossRef]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef] [PubMed]
- Salas-Armenteros, I.; Pérez-Calero, C.; Bayona-Feliu, A.; Tumini, E.; Luna, R.; Aguilera, A. Human THO–Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 2017, 36, 3532–3547. [Google Scholar] [CrossRef]
- Patel, P.S.; Abraham, K.J.; Guturi, K.K.N.; Halaby, M.J.; Khan, Z.; Palomero, L.; Ho, B.; Duan, S.; St-Germain, J.; Algouneh, A.; et al. RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J. Clin. Investig. 2021, 131, e140105. [Google Scholar] [CrossRef]
- Klusmann, I.; Wohlberedt, K.; Magerhans, A.; Teloni, F.; Korbel, J.O.; Altmeyer, M.; Dobbelstein, M. Chromatin modifiers Mdm2 and RNF2 prevent RNA: DNA hybrids that impair DNA replication. Proc. Natl. Acad. Sci. USA 2018, 115, E11311–E113E20. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.Y.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86–101.e16. [Google Scholar] [CrossRef]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human FACT chromatinreorganizing complexes solve R-loop-mediated transcription-replication conflicts. Gene Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef]
- Carvalho, S.; Raposo, A.C.; Martins, F.B.; Grosso, A.R.; Sridhara, S.C.; Rino, J.; Carmo-Fonseca, M.; de Almeida, S.F. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013, 41, 2881–2893. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gerhold, C.B.; Tosi, A.; Hustedt, N.; Seeber, A.; Sack, R.; Herzog, F.; Pasero, P.; Shimada, K.; Hopfner, K.P.; et al. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Gene Dev. 2016, 30, 337–354. [Google Scholar] [CrossRef]
- Prendergast, L.; McClurg, U.L.; Hristova, R.; Berlinguer-Palmini, R.; Greener, S.; Veitch, K.; Hernandez, I.; Pasero, P.; Rico, D.; Higgins, J.M.G.; et al. Resolution of R-loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat. Commun. 2020, 11, 4534. [Google Scholar] [CrossRef]
- Bayona-Feliu, A.; Barroso, S.; Munoz, S.; Aguilera, A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription-replication conflicts. Nat. Genet. 2021, 53, 1050–1063. [Google Scholar] [CrossRef]
- Neves-Costa, A.; Will, W.R.; Vetter, A.T.; Miller, J.R.; Varga-Weisz, P. The SNF2-Family Member Fun30 Promotes Gene Silencing in Heterochromatic Loci. PLoS ONE 2009, 4, e8111. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, S.P.; Barki, L.; Neves-Costa, A.; Santos, F.; Dean, W.; Hawkes, N.; Choudhary, P.; Will, W.R.; Webster, J.; Oxley, D.; et al. Maintenance of Silent Chromatin through Replication Requires SWI/SNF-like Chromatin Remodeler SMARCAD1. Mol. Cell 2011, 42, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.; Zofall, M.; Balachandran, V.; Thillainadesan, G.; Sugiyama, T.; Wheeler, D.; Zhou, M.; Grewal, S.I. SNF2 Family Protein Fft3 Suppresses Nucleosome Turnover to Promote Epigenetic Inheritance and Proper Replication. Mol. Cell 2017, 66, 50–62.e6. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.S.Y.; van Toorn, M.; Gaggioli, V.; Paes Dias, M.; Zhu, Y.; Manolika, E.M.; Zhao, W.; van der Does, M.; Mukherjee, C.; Gonçalves, J.G.; et al. SMARCAD1-mediated active replication fork stability maintains genome integrity. Sci. Adv. 2021, 7, eabe7804. [Google Scholar] [CrossRef]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Powell, W.T.; Coulson, R.L.; Gonzales, M.L.; Crary, F.K.; Wong, S.S.; Adams, S.; Ach, R.A.; Tsang, P.; Yamada, N.A.; Yasui, D.H.; et al. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc. Natl. Acad. Sci. USA 2013, 110, 13938–13943. [Google Scholar] [CrossRef]
- Nakama, M.; Kawakami, K.; Kajitani, T.; Urano, T.; Murakami, Y. DNA-RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 2012, 17, 218–233. [Google Scholar] [CrossRef]
- Azmi, I.F.; Watanabe, S.; Maloney, M.F.; Kang, S.; Belsky, J.A.; MacAlpine, D.M.; Peterson, C.L.; Bell, S.P. Nucleosomes influence multiple steps during replication initiation. eLife 2017, 6, e22512. [Google Scholar] [CrossRef] [PubMed]
- MacAlpine, D.M.; Almouzni, G. Chromatin and DNA replication. Csh. Perspect. Biol. 2013, 5, a010207. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef]
- Michishita, E.; McCord, R.A.; Boxer, L.D.; Barber, M.F.; Hong, T.; Gozani, O.; Chua, K.F. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009, 8, 2664–2666. [Google Scholar] [CrossRef]
- Sebastián, C.; Zwaans, B.M.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Celic, I.; Masumoto, H.; Griffith, W.P.; Meluh, P.; Cotter, R.J.; Boeke, J.D.; Verreault, A. The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone H3 lysine 56 deacetylation. Curr. Biol. 2006, 16, 1280–1289. [Google Scholar] [CrossRef]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Greco, T.M.; Boonmee, A.; Miteva, Y.; Cristea, I.M. Functional Proteomics Establishes the Interaction of SIRT7 with Chromatin Remodeling Complexes and Expands Its Role in Regulation of RNA Polymerase I Transcription. Mol. Cell Proteom. 2012, 11, 60–76. [Google Scholar] [CrossRef]
- Sträßer, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondón, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and Multiple RNA Biogenesis Factors Cooperate to Prevent RNA:DNA Hybrids from Generating Genome Instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef]
- Bohgaki, M.; Bohgaki, T.; El Ghamrasni, S.; Srikumar, T.; Maire, G.; Panier, S.; Fradet-Turcotte, A.; Stewart, G.S.; Raught, B.; Hakem, A.; et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2013, 110, 20982–20987. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 Ubiquitinates K13-15 on H2A/H2AX to Drive DNA Damage Signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Chen, L.H.; Li, Z.Y.; Zwolinska, A.K.; Smith, M.A.; Cross, B.; Koomen, J.; Yuan, Z.M.; Jenuwein, T.; Marine, J.C.; Wright, K.L.; et al. MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO J. 2010, 29, 2538–2552. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.; Lozano, G.; et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol. Cell 2016, 61, 68–83. [Google Scholar] [CrossRef]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef]
- Law, M.J.; Lower, K.M.; Voon, H.P.J.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X Syndrome Protein Targets Tandem Repeats and Influences Allele-Specific Expression in a Size-Dependent Manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Reddel, R.R. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett. 2003, 194, 155–162. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef]
- Graf, M.; Bonetti, D.; Lockhart, A.; Serhal, K.; Kellner, V.; Maicher, A.; Jolivet, P.; Teixeira, M.T.; Luke, B. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 2017, 170, 72–85.e14. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]
- Montero, J.J.; Lopez-Silanes, I.; Megias, D.; Fraga, M.F.; Castells-Garcia, A.; Blasco, M.A. TERRA recruitment of polycomb to telomeres is essential for histone trymethylation marks at telomeric heterochromatin. Nat. Commun. 2018, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
- Doksani, Y.; de Lange, T. The Role of Double-Strand Break Repair Pathways at Functional and Dysfunctional Telomeres. Csh. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.D.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Ghisays, F.; Garzia, A.; Wang, H.; Canasto-Chibuque, C.; Hohl, M.; Savage, S.A.; Tuschl, T.; Petrini, J.H.J. RTEL1 influences the abundance and localization of TERRA RNA. Nat. Commun. 2021, 12, 3016. [Google Scholar] [CrossRef]
- Wu, W.; Bhowmick, R.; Vogel, I.; Ozer, O.; Ghisays, F.; Thakur, R.S.; Sanchez de Leon, E.; Richter, P.H.; Ren, L.; Petrini, J.H.; et al. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020, 27, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Segura-Bayona, S.; Margalef, P.; Marzec, P.; Ruis, P.; Hewitt, G.; Bellelli, R.; Patel, H.; Goldstone, R.; Poetsch, A.R.; et al. RTEL1 Regulates G4/R-Loops to Avert Replication-Transcription Collisions. Cell Rep. 2020, 33, 108546. [Google Scholar] [CrossRef]
- Knezetic, J.A.; Luse, D.S. The Presence of Nucleosomes on a DNA-Template Prevents Initiation by Rna Polymerase-II in vitro. Cell 1986, 45, 95–104. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Orphanides, G.; Lagrange, T.; Reinberg, D. The general transcription factors of RNA polymerase II. Gene Dev. 1996, 10, 2657–2683. [Google Scholar] [CrossRef]
- McCullough, L.L.; Connell, Z.; Xin, H.; Studitsky, V.M.; Feofanov, A.V.; Valieva, M.E.; Formosa, T. Functional roles of the DNA-binding HMGB domain in the histone chaperone FACT in nucleosome reorganization. J. Biol. Chem. 2018, 293, 6121–6133. [Google Scholar] [CrossRef]
- Winkler, D.D.; Luger, K. The histone chaperone FACT: Structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem. 2011, 286, 18369–18374. [Google Scholar] [CrossRef]
- Formosa, T.; Eriksson, P.; Wittmeyer, J.; Ginn, J.; Yu, Y.; Stillman, D.J. Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. EMBO J. 2001, 20, 3506–3517. [Google Scholar] [CrossRef] [PubMed]
- VanDemark, A.P.; Blanksma, M.; Ferris, E.; Heroux, A.; Hill, C.P.; Formosa, T. The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol. Cell 2006, 22, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Wittmeyer, J.; Formosa, T. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell Biol. 1997, 17, 4178–4190. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, E.; Bortfeld, M.; White, S.A.; Pidoux, A.L.; Ekwall, K.; Allshire, R.C.; Ladurner, A.G. The chromatin-remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Curr. Biol. 2007, 17, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Gene Dev. 2010, 24, 2133–2145. [Google Scholar] [CrossRef]
- Fleming, A.B.; Kao, C.F.; Hillyer, C.; Pikaart, M.; Osley, M.A. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 2008, 31, 57–66. [Google Scholar] [CrossRef]
- Bao, Y.H.; Shen, X.T. INO80 subfamily of chromatin remodeling complexes. Mutat. Res. Fund. Mol. Mech. 2007, 618, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Lafon, A.; Taranum, S.; Pietrocola, F.; Dingli, F.; Loew, D.; Brahma, S.; Bartholomew, B. INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation. Mol. Cell 2015, 60, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. B 2017, 372, 290. [Google Scholar] [CrossRef] [PubMed]
- Euskirchen, G.M.; Auerbach, R.K.; Davidov, E.; Gianoulis, T.A.; Zhong, G.N.; Rozowsky, J.; Bhardwaj, N.; Gerstein, M.B.; Snyder, M. Diverse Roles and Interactions of the SWI/SNF Chromatin Remodeling Complex Revealed Using Global Approaches. PLoS Genet. 2011, 7, e1002008. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Adkins, N.L.; Swygert, S.G.; Kaur, P.; Niu, H.Y.; Grigoryev, S.A.; Sung, P.; Wang, H.; Peterson, C.L. Nucleosome-like, Single-stranded DNA (ssDNA)-Histone Octamer Complexes and the Implication for DNA Double Strand Break Repair. J. Biol. Chem. 2017, 292, 5271–5281. [Google Scholar] [CrossRef]
- Awad, S.; Ryan, D.; Prochasson, P.; Owen-Hughes, T.; Hassan, A.H. The Snf2 Homolog Fun30 Acts as a Homodimeric ATP-dependent Chromatin-remodeling Enzyme. J. Biol. Chem. 2010, 285, 9477–9484. [Google Scholar] [CrossRef]
- Byeon, B.; Wang, W.; Barski, A.; Ranallo, R.T.; Bao, K.; Schones, D.E.; Zhao, K.; Wu, C.; Wu, W.H. The ATP-dependent Chromatin Remodeling Enzyme Fun30 Represses Transcription by Sliding Promoter-proximal Nucleosomes. J. Biol. Chem. 2013, 288, 23182–23193. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Pandita, R.K.; Hambarde, S.; Mattoo, A.R.; Charaka, V.; Ahmed, K.M.; Iyer, S.P.; Hunt, C.R.; Pandita, T.K. SMARCAD1 Phosphorylation and Ubiquitination Are Required for Resection during DNA Double-Strand Break Repair. IScience 2018, 2, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef]
- Bi, X. Functions of chromatin remodeling factors in heterochromatin formation and maintenance. Sci. China Life Sci. 2012, 55, 89–96. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jasencakova, Z.; Groth, A. Broken Silence Restored-Remodeling Primes for Deacetylation at Replication Forks. Mol. Cell 2011, 42, 267–269. [Google Scholar] [CrossRef]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Pogliano, C.B.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 2020, 77, 528–541.e8. [Google Scholar] [CrossRef]
- Her, J.; Ray, C.; Altshuler, J.; Zheng, H.Y.; Bunting, S.F. 53BP1 Mediates ATR-Chk1 Signaling and Protects Replication Forks under Conditions of Replication Stress. Mol. Cell Biol. 2018, 38, e00472-17. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; de Boer, H.R.; Demmers, J.; van Vugt, M.A.T.M.; Ray Chaudhuri, A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.F.; Chen, L.P.; Peng, D.; Jiang, A.; He, Y.R.; Zeng, Y.R.; Chen, X.; Zhou, H.X.; Luo, X.T.; Liu, H.Y.; et al. METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol. Cell 2020, 79, 425–447.e7. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chro-Mates | Homologues | Activity | Role in R-Loop Processing | Ref. |
| EHMT2/G9a | DmeI (Drosophila melanogaster) | Modifier | Methylation of H3K9me2 at heterochromatin regions | [150,153,158] |
| SIRT6 | Sirt6 (Mus musculus) Hst3/4 (Saccharomyces cerevisiae) | Modifier | Deacetylation of H3K56ac | [159,160] |
| SIRT7 | Sirt7 (Mus musculus) | Modifier | Deacetylation of DDX21 Histone desuccinylation | [110,161,162] |
| Sin3A | Sin3 (Saccharomyces cerevisiae) | Modifier | Histone deacetylation through interaction with THO complex | [163] |
| RNF168 | Rnf168 (Mus musculus) | Modifier | Ubiquitination of DHX9 | [164] |
| MDM2 | Mdm2 (Mus musculus) | Remodeler | Ubiquitination of H2AK119 and genome expression | [165] |
| ATRX | Atrx (Mus musculus) | Remodeler | Antagonization of TERRA RNA at telomeric R-loops | [166,167] |
| FACT/SETD2 | yFACT/Set2 (Saccharomyces cerevisiae) | Remodeler | Nucleosome reassembly after RNAPII passage | [168,169] |
| INO80 | Ino80 (Saccharomyces cerevisiae) | Remodeler | Chromatin relaxation | [170,171] |
| BRG1 | Swi (Saccharomyces cerevisiae) | Remodeler | Regulation of chromatin accessibility | [172] |
| Fft3/SMARCAD1 | Fun30 (Saccharomyces cerevisiae) Fft3 (Schizosaccharomyces pombe) | Remodeler | Nucleosome turn-over Active fork protection | [173,174,175,176] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uruci, S.; Lo, C.S.Y.; Wheeler, D.; Taneja, N. R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde. Int. J. Mol. Sci. 2021, 22, 8850. https://doi.org/10.3390/ijms22168850
Uruci S, Lo CSY, Wheeler D, Taneja N. R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde. International Journal of Molecular Sciences. 2021; 22(16):8850. https://doi.org/10.3390/ijms22168850
Chicago/Turabian StyleUruci, Sidrit, Calvin Shun Yu Lo, David Wheeler, and Nitika Taneja. 2021. "R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde" International Journal of Molecular Sciences 22, no. 16: 8850. https://doi.org/10.3390/ijms22168850
APA StyleUruci, S., Lo, C. S. Y., Wheeler, D., & Taneja, N. (2021). R-Loops and Its Chro-Mates: The Strange Case of Dr. Jekyll and Mr. Hyde. International Journal of Molecular Sciences, 22(16), 8850. https://doi.org/10.3390/ijms22168850