
Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
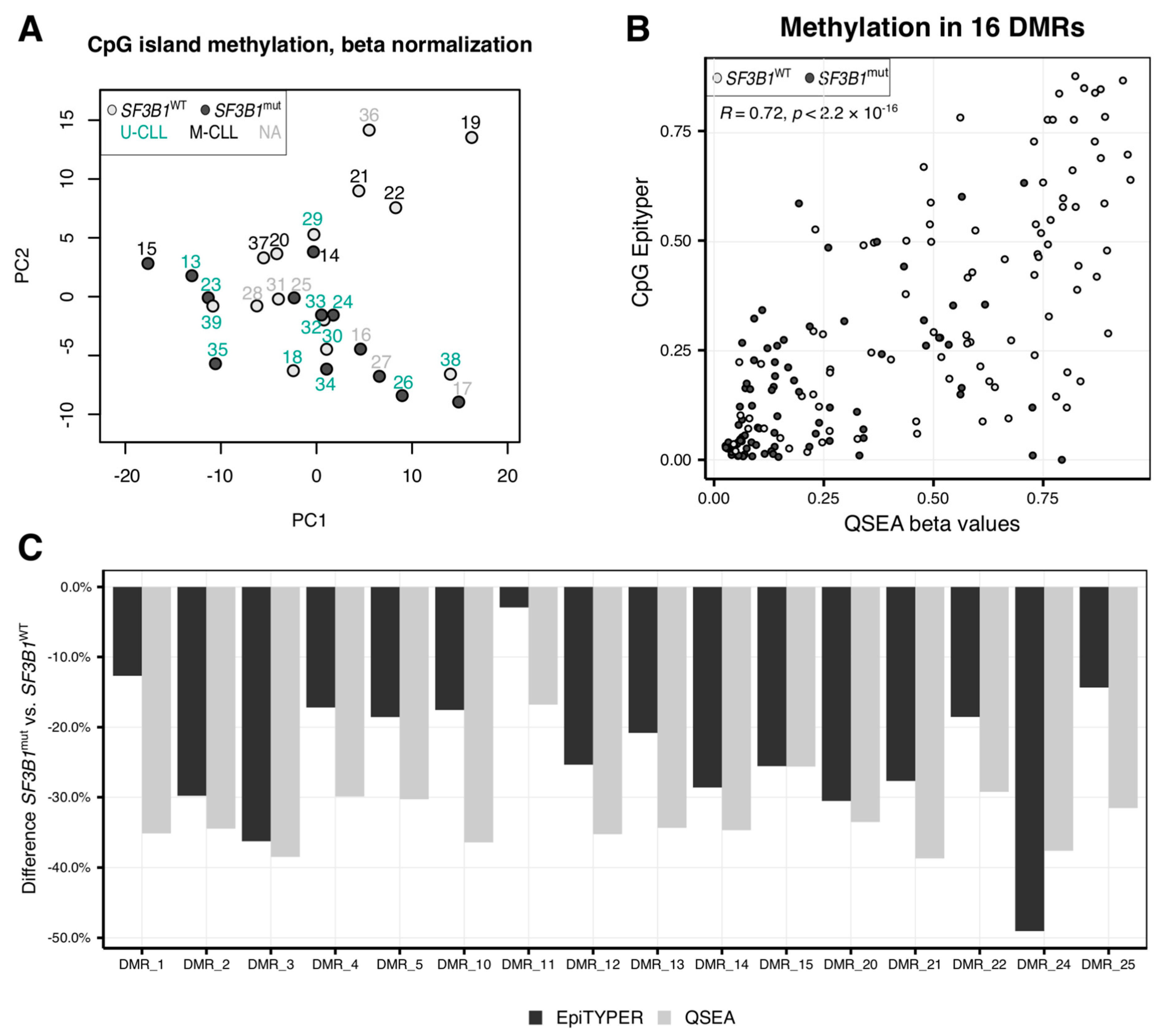
2.1. CLL Patients with SF3B1 Mutations Feature Local Hypomethylations
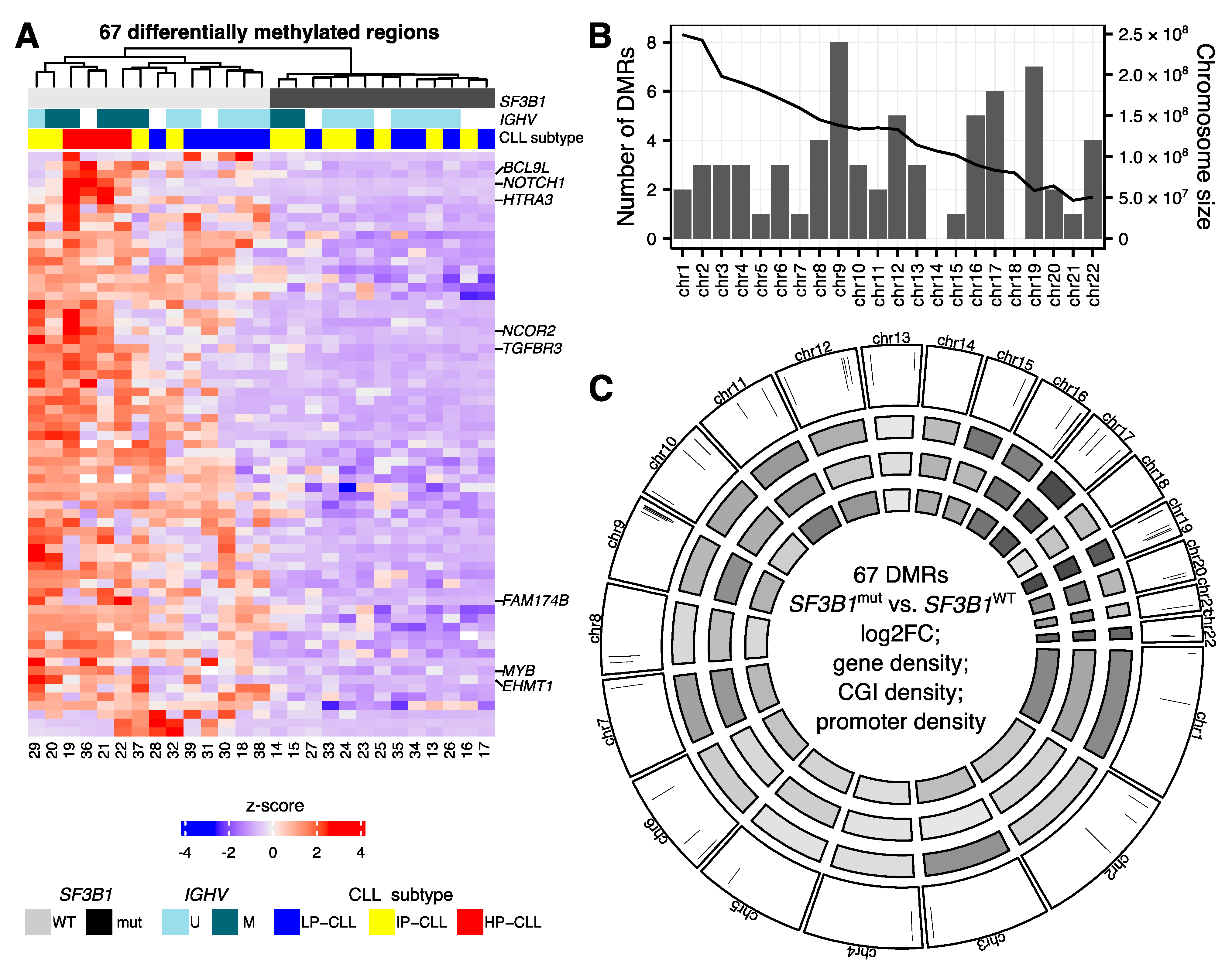
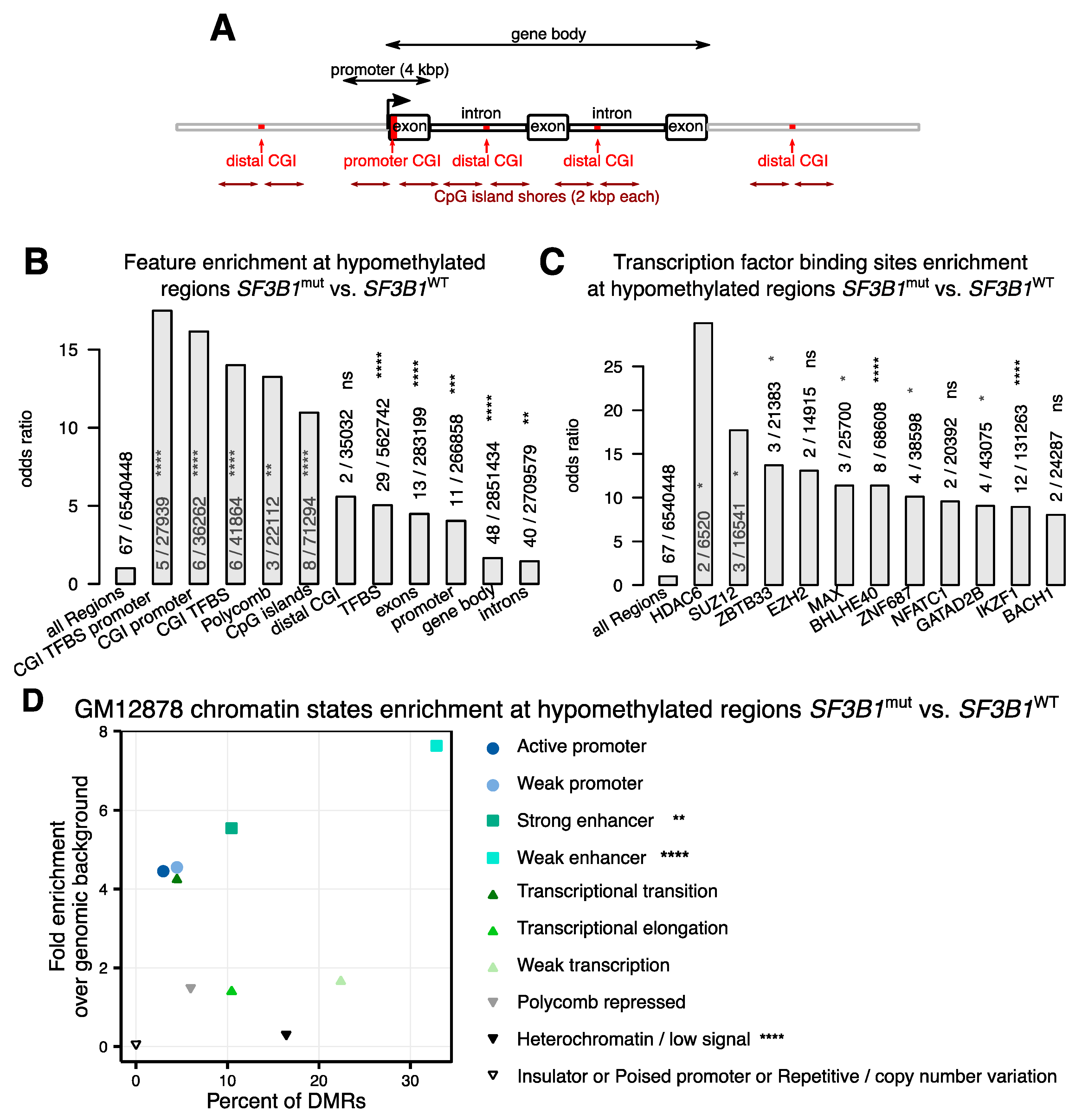
2.2. Hypomethylations in CLL Patients with SF3B1 Mutation Are Enriched in Gene Bodies and Subtelomeric Regions
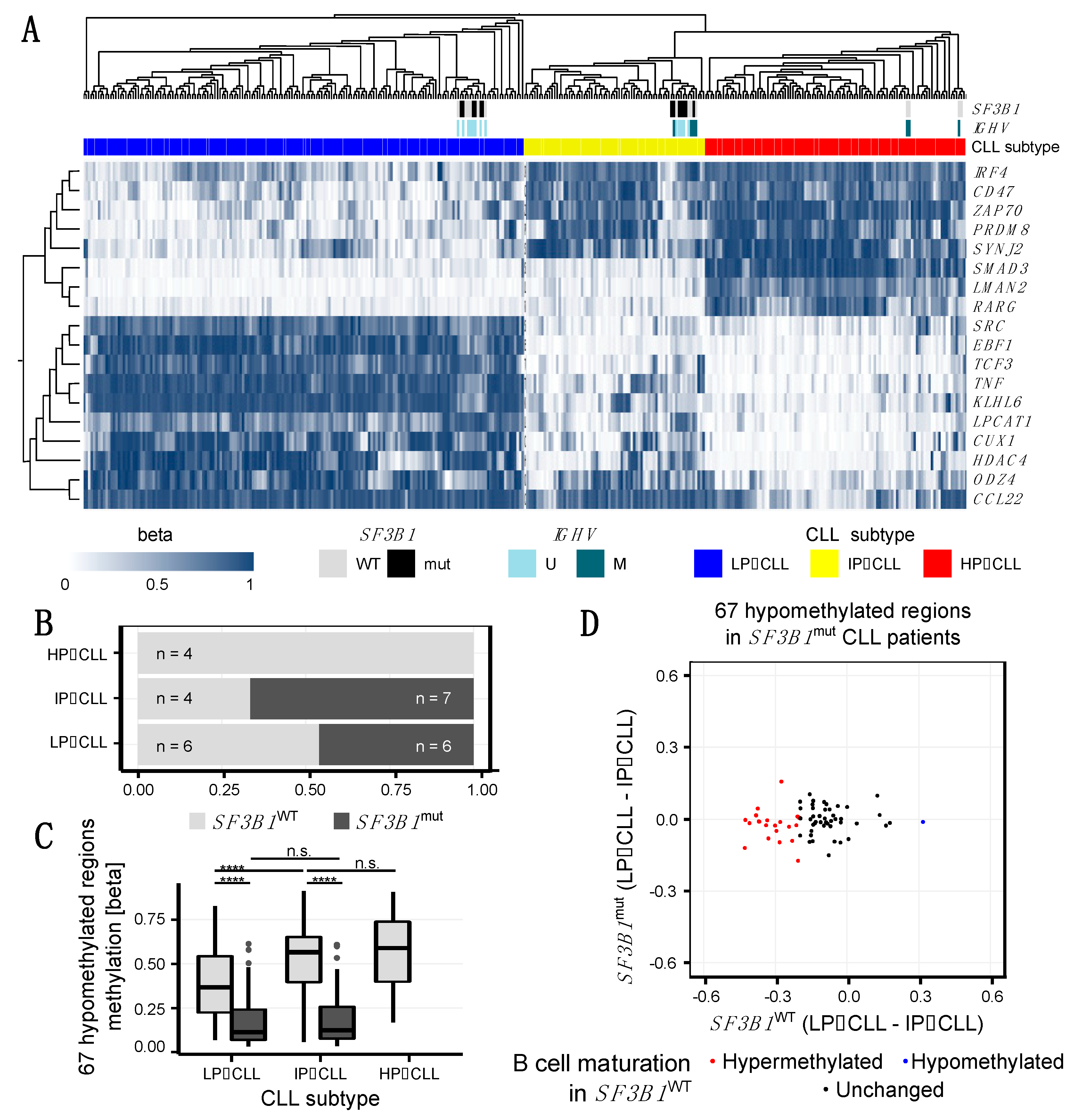
2.3. SF3B1mut Is Associated with Aberrant Methylation and Is Partially Related to the Developmental B Cell Epigenetic State
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Methylated DNA Immunoprecipitation Sequencing (MeDIP-Seq)
4.3. Sequencing Reads Processing
4.4. Differential Methylation Analysis
4.5. RNA-Seq and Differential Expression Analysis
4.6. Bisulfite Mass Spectrometry (BS-MS) with Agena Bioscience EpiTYPER-Assay
4.7. Gene Set Enrichment Analysis
4.8. Motif Enrichment Analysis
4.9. Assessing the CLL Subtype
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Guièze, R.; Robbe, P.; Clifford, R.; de Guibert, S.; Pereira, B.; Timbs, A.; Dilhuydy, M.S.; Cabes, M.; Ysebaert, L.; Burns, A.; et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood 2015, 126, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef]
- Ljungström, V.; Cortese, D.; Young, E.; Pandzic, T.; Mansouri, L.; Plevova, K.; Ntoufa, S.; Baliakas, P.; Clifford, R.; Sutton, L.A.; et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: Clinical impact of recurrent RPS15 mutations. Blood 2016, 127, 1007–1016. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Schnaiter, A.; Paschka, P.; Zenz, T.; Rossi, M.; Döhner, K.; Bühler, A.; Böttcher, S.; Ritgen, M.; Kneba, M.; et al. Gene Mutations and Treatment Outcome in Chronic Lymphocytic Leukemia. Blood 2014, 123, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Kfir, N.; Lev-Maor, G.; Glaich, O.; Alajem, A.; Datta, A.; Sze, S.K.; Meshorer, E.; Ast, G. SF3B1 Association with Chromatin Determines Splicing Outcomes. Cell Rep. 2015, 11, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef]
- Ntziachristos, P.; Abdel-Wahab, O.; Aifantis, I. Emerging concepts of epigenetic dysregulation in hematological malignancies. Nat. Immunol. 2016, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Cavellán, E.; Asp, P.; Percipalle, P.; Farrants, A.K.Ö. The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J. Biol. Chem. 2006, 281, 16264–16271. [Google Scholar] [CrossRef]
- Isono, K.; Mizutani-Koseki, Y.; Komori, T.; Schmidt-Zachmann, M.S.; Koseki, H. Mammalian Polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 2005, 19, 536–541. [Google Scholar] [CrossRef]
- Bartholdy, B.A.; Wang, X.; Yan, X.J.; Pascual, M.; Fan, M.; Barrientos, J.; Allen, S.L.; Martinez-Climent, J.A.; Rai, K.R.; Chiorazzi, N.; et al. CLL intraclonal fractions exhibit established and recently acquired patterns of DNA methylation. Blood Adv. 2020, 4, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Cahill, N.; Rosenquist, R. Uncovering the DNA methylome in chronic lymphocytic leukemia. Epigenetics 2013, 8, 138–148. [Google Scholar] [CrossRef]
- Oakes, C.C.; Claus, R.; Gu, L.; Assenov, Y.; Hüllein, J.; Zucknick, M.; Bieg, M.; Brocks, D.; Bogatyrova, O.; Schmidt, C.R.; et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014, 4, 348–361. [Google Scholar] [CrossRef]
- Pastore, A.; Gaiti, F.; Lu, S.X.; Brand, R.M.; Kulm, S.; Chaligne, R.; Gu, H.; Huang, K.Y.; Stamenova, E.K.; Béguelin, W.; et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun. 2019, 10, 1874. [Google Scholar] [CrossRef]
- Wernig-Zorc, S.; Yadav, M.P.; Kopparapu, P.K.; Bemark, M.; Kristjansdottir, H.L.; Andersson, P.-O.; Kanduri, C.; Kanduri, M. Global distribution of DNA hydroxymethylation and DNA methylation in chronic lymphocytic leukemia. Epigenetics Chromatin 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Subhash, S.; Andersson, P.-O.; Kosalai, S.T.; Kanduri, C.; Kanduri, M. Global DNA methylation profiling reveals new insights into epigenetically deregulated protein coding and long noncoding RNAs in CLL. Clin. Epigenetics 2016, 8, 106. [Google Scholar] [CrossRef]
- Cahill, N.; Bergh, A.C.; Kanduri, M.; Göransson-Kultima, H.; Mansouri, L.; Isaksson, A.; Ryan, F.; Smedby, K.E.; Juliusson, G.; Sundström, C.; et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia 2013, 27, 150–158. [Google Scholar] [CrossRef]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef]
- Fabris, S.; Bollati, V.; Agnelli, L.; Morabito, F.; Motta, V.; Cutrona, G.; Matis, S.; Recchia, A.G.; Gigliotti, V.; Gentile, M.; et al. Biological and clinical relevance of quantitative global methylation of repetitive DNA sequences in chronic lymphocytic leukemia. Epigenetics 2011, 6, 188–194. [Google Scholar] [CrossRef][Green Version]
- Oakes, C.C.; Seifert, M.; Assenov, Y.; Gu, L.; Przekopowitz, M.; Ruppert, A.S.; Wang, Q.; Imbusch, C.D.; Serva, A.; Koser, S.D.; et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet. 2016, 48, 253–264. [Google Scholar] [CrossRef]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef]
- Gaiti, F.; Chaligne, R.; Gu, H.; Brand, R.M.; Kothen-Hill, S.; Schulman, R.C.; Grigorev, K.; Risso, D.; Kim, K.T.; Pastore, A.; et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 2019, 569, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.W.; Lin, M.J.; Lee, T.L.; Wen, S.C.; Chen, X.; Shen, C.K. Two major forms of DNA (cytosine-5) methyltransferase in human somatic tissues. Proc. Natl. Acad. Sci. USA 1999, 96, 9751–9756. [Google Scholar] [CrossRef] [PubMed]
- Franchina, M.; Hooper, J.; Kay, P.H. Five novel alternatively spliced transcripts of DNA (cytosine-5) methyltransferase 2 in human peripheral blood leukocytes. Int. J. Biochem. Cell Biol. 2001, 33, 1104–1115. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Velicescu, M.; Preciado-Lopez, M.A.; Gonzales, F.A.; Tsai, Y.C.; Liang, G.; Jones, P.A. Identification and characterization of alternatively spliced variants of DNA methyltransferase 3a in mammalian cells. Gene 2002, 298, 91–99. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.-Z.; Jiang, J.; Duan, C.-G. The Crosstalk Between Epigenetic Mechanisms and Alternative RNA Processing Regulation. Front. Genet. 2020, 11, 998. [Google Scholar] [CrossRef]
- Lienhard, M.; Grasse, S.; Rolff, J.; Frese, S.; Schirmer, U.; Becker, M.; Börno, S.; Timmermann, B.; Chavez, L.; Sültmann, H.; et al. QSEA-modelling of genome-wide DNA methylation from sequencing enrichment experiments. Nucleic Acids Res. 2017, 45, e44. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, S.; Oleś, M.; Lu, J.; Sellner, L.; Anders, S.; Velten, B.; Wu, B.; Hüllein, J.; da Silva Liberio, M.; Walther, T.; et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Investig. 2018, 128, 427–445. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wierzbinska, J.A.; Toth, R.; Ishaque, N.; Rippe, K.; Mallm, J.P.; Klett, L.C.; Mertens, D.; Zenz, T.; Hielscher, T.; Seifert, M.; et al. Methylome-based cell-of-origin modeling (Methyl-COOM) identifies aberrant expression of immune regulatory molecules in CLL. Genome Med. 2020, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Grimwood, J.; Gordon, L.A.; Olsen, A.; Terry, A.; Schmutz, J.; Lamerdin, J.; Hellsten, U.; Goodstein, D.; Couronne, O.; Tran-Gyamil, M.; et al. The DNA sequence and biology of human chromosome 19. Nature 2004, 428, 529–535. [Google Scholar] [CrossRef]
- Humphray, S.J.; Oliver, K.; Hunt, A.R.; Plumb, R.W.; Loveland, J.E.; Howe, K.L.; Andrews, T.D.; Searle, S.; Hunt, S.E.; Scott, C.E.; et al. DNA sequence and analysis of human chromosome 9. Nature 2004, 429, 369–374. [Google Scholar] [CrossRef]
- ENCODE Uniform TFBS Track. Available online: http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeAwgTfbsUniform/ (accessed on 9 December 2020).
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R.; et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.Y.; Whitfield, T.W.; Greven, M.C.; Pierce, B.G.; Dong, X.; Kundaje, A.; Cheng, Y.; et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012, 22, 1798–1812. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, J.; Iyer, S.; Lin, X.Y.; Greven, M.C.; Kim, B.H.; Moore, J.; Pierce, B.G.; Dong, X.; Virgil, D.; et al. Factorbook.org: A Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013, 41, D171–D176. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Nichogiannopoulou, A.; Wu, L.; Sun, L.; Sharpe, A.H.; Bigby, M.; Georgopoulos, K. Selective defects in the development of the fetal and adult lymphoid system in mice with an Ikaros null mutation. Immunity 1996, 5, 537–549. [Google Scholar] [CrossRef]
- Cook, M.E.; Jarjour, N.N.; Lin, C.-C.; Edelson, B.T. Transcription Factor Bhlhe40 in Immunity and Autoimmunity. Trends Immunol. 2020, 41, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Wu, H.; Feinberg, A.P. A species-generalized probabilistic model-based definition of CpG islands. Mamm. Genome 2009, 20, 674–680. [Google Scholar] [CrossRef]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef]
- Ernst, J.; Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010, 28, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.D.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011, 473, 43–49. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Amarasinghe, H.E.; Kadalayil, L.; Beattie, A.; Forster, J.; Blakemore, S.J.; Parker, H.; Bryant, D.; Larrayoz, M.; Clifford, R.; et al. Clinical significance of DNA methylation in chronic lymphocytic leukemia patients: Results from 3 UK clinical trials. Blood Adv. 2019, 3, 2474–2481. [Google Scholar] [CrossRef]
- Bhoi, S.; Ljungström, V.; Baliakas, P.; Mattsson, M.; Smedby, K.E.; Juliusson, G.; Rosenquist, R.; Mansouri, L. Prognostic impact of epigenetic classification in chronic lymphocytic leukemia: The case of subset #2. Epigenetics 2016, 11, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Queirós, A.C.; Villamor, N.; Clot, G.; Martinez-Trillos, A.; Kulis, M.; Navarro, A.; Penas, E.M.M.; Jayne, S.; Majid, A.; Richter, J.; et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2015, 29, 598–605. [Google Scholar] [CrossRef]
- Shayevitch, R.; Askayo, D.; Keydar, I.; Ast, G. The importance of DNA methylation of exons on alternative splicing. RNA 2018, 24, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tian, Y.; Wang, J.; Sun, Z.; Zhu, Y. Genome-wide analysis reveals the association between alternative splicing and DNA methylation across human solid tumors. BMC Med. Genomics 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef]
- Yoshimi, A.; Lin, K.-T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.-W.; Micol, J.-B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef]
- Ferry, L.; Fournier, A.; Tsusaka, T.; Adelmant, G.; Shimazu, T.; Matano, S.; Kirsh, O.; Amouroux, R.; Dohmae, N.; Suzuki, T.; et al. Methylation of DNA Ligase 1 by G9a/GLP Recruits UHRF1 to Replicating DNA and Regulates DNA Methylation. Mol. Cell 2017, 67, 550–565.e5. [Google Scholar] [CrossRef]
- Goodman, S.J.; Cytrynbaum, C.; Chung, B.H.-Y.; Chater-Diehl, E.; Aziz, C.; Turinsky, A.L.; Kellam, B.; Keller, M.; Ko, J.M.; Caluseriu, O.; et al. EHMT1 pathogenic variants and 9q34.3 microdeletions share altered DNA methylation patterns in patients with Kleefstra syndrome. J. Transl. Genet. Genomics 2020, 4, 144–158. [Google Scholar] [CrossRef]
- Mansouri, L.; Grabowski, P.; Degerman, S.; Svenson, U.; Gunnarsson, R.; Cahill, N.; Smedby, K.E.; Geisler, C.; Juliusson, G.; Roos, G.; et al. Short telomere length is associated with NOTCH1/SF3B1/TP53 aberrations and poor outcome in newly diagnosed chronic lymphocytic leukemia patients. Am. J. Hematol. 2013, 88, 647–651. [Google Scholar] [CrossRef]
- Jebaraj, B.M.C.; Stilgenbauer, S. Telomere Dysfunction in Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 10, 612665. [Google Scholar] [CrossRef]
- Tardivon, D.; Antoszewski, M.; Zangger, N.; Nkosi, M.; Sordet-Dessimoz, J.; Hendriks, R.W.; Koch, U.; Radtke, F. Notch Signaling Promotes Disease Initiation and Progression in Murine Chronic Lymphocytic Leukemia. Blood 2020, 137, 3079–3092. [Google Scholar] [CrossRef]
- Fabbri, G.; Holmes, A.B.; Viganotti, M.; Scuoppo, C.; Belver, L.; Herranz, D.; Yan, X.-J.; Kieso, Y.; Rossi, D.; Gaidano, G.; et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2017, 114, E2911–E2919. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, B.; Baldoni, S.; Dorillo, E.; de Falco, F.; Rompietti, C.; Cecchini, D.; Cantelmi, M.G.; Sorcini, D.; Nogarotto, M.; Adamo, F.M.; et al. Decreased NOTCH1 Activation Correlates with Response to Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 7540–7553. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, F.; Bittolo, T.; Tissino, E.; Vit, F.; Vendramini, E.; Laurenti, L.; D’Arena, G.; Olivieri, J.; Pozzato, G.; Zaja, F.; et al. SF3B1-mutated chronic lymphocytic leukemia shows evidence of NOTCH1 pathway activation including CD20 downregulation. Haematologica 2020. Online ahead of print. [Google Scholar] [CrossRef]
- De Oliveira, V.C.; de Lacerda, M.P.; Moraes, B.B.M.; Gomes, C.P.; Maricato, J.T.; Souza, O.F.; Schenkman, S.; Pesquero, J.B.; Moretti, N.S.; Rodrigues, C.A.; et al. Deregulation of Ikaros expression in B-1 cells: New insights in the malignant transformation to chronic lymphocytic leukemia. J. Leukoc. Biol. 2019, 106, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Ferreirós-Vidal, I.; Carroll, T.; Taylor, B.; Terry, A.; Liang, Z.; Bruno, L.; Dharmalingam, G.; Khadayate, S.; Cobb, B.S.; Smale, S.T.; et al. Genome-wide identification of Ikaros targets elucidates its contribution to mouse B-cell lineage specification and pre-B-cell differentiation. Blood 2013, 121, 1769–1782. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G.J. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Fecteau, J.-F.; Corral, L.G.; Ghia, E.M.; Gaidarova, S.; Futalan, D.; Bharati, I.S.; Cathers, B.; Schwaederlé, M.; Cui, B.; Lopez-Girona, A.; et al. Lenalidomide inhibits the proliferation of CLL cells via a cereblon/p21(WAF1/Cip1)-dependent mechanism independent of functional p53. Blood 2014, 124, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; Fink, A.M.; Schilhabel, A.; Weiss, J.; Robrecht, S.; Eckert, R.; de la Serna, J.; Crespo, M.; Coscia, M.; Vitale, C.; et al. B-cell acute lymphoblastic leukemia in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 2021, 137, 2267–2271. [Google Scholar] [CrossRef]
- Yoshida, T.; Ng, S.Y.-M.; Zuniga-Pflucker, J.C.; Georgopoulos, K. Early hematopoietic lineage restrictions directed by Ikaros. Nat. Immunol. 2006, 7, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Kirstetter, P.; Thomas, M.; Dierich, A.; Kastner, P.; Chan, S. Ikaros is critical for B cell differentiation and function. Eur. J. Immunol. 2002, 32, 720–730. [Google Scholar] [CrossRef]
- Schwickert, T.A.; Tagoh, H.; Gültekin, S.; Dakic, A.; Axelsson, E.; Minnich, M.; Ebert, A.; Werner, B.; Roth, M.; Cimmino, L.; et al. Stage-specific control of early B cell development by the transcription factor Ikaros. Nat. Immunol. 2014, 15, 283–293. [Google Scholar] [CrossRef]
- Fiorcari, S.; Benatti, S.; Zucchetto, A.; Zucchini, P.; Gattei, V.; Luppi, M.; Marasca, R.; Maffei, R. Overexpression of CD49d in trisomy 12 chronic lymphocytic leukemia patients is mediated by IRF4 through induction of IKAROS. Leukemia 2019, 33, 1278–1302. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Fiorcari, S.; Benatti, S.; Atene, C.G.; Martinelli, S.; Zucchini, P.; Potenza, L.; Luppi, M.; Marasca, R. IRF4 modulates the response to BCR activation in chronic lymphocytic leukemia regulating IKAROS and SYK. Leukemia 2021, 35, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, B.; Payne, J.L.; Song, C.; Ge, Z.; Gowda, C.; Iyer, S.; Dhanyamraju, P.K.; Dorsam, G.; Reeves, M.E.; et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 2019, 33, 2720–2731. [Google Scholar] [CrossRef]
- Heizmann, B.; Kastner, P.; Chan, S. The Ikaros family in lymphocyte development. Curr. Opin. Immunol. 2018, 51, 14–23. [Google Scholar] [CrossRef]
- Oravecz, A.; Apostolov, A.; Polak, K.; Jost, B.; Le Gras, S.; Chan, S.; Kastner, P. Ikaros mediates gene silencing in T cells through Polycomb repressive complex 2. Nat. Commun. 2015, 6, 8823. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Kashiwagi, M.; Yoshida, T.; Joshi, I.; Jena, N.; Somasundaram, R.; Emmanuel, A.O.; Sigvardsson, M.; Fitamant, J.; et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev. 2016, 30, 1971–1990. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 Transcription Factor Mediates β-Catenin-Induced Axis Formation in Xenopus Embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/β-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef]
- Mani, M.; Carrasco, D.E.; Zhang, Y.; Takada, K.; Gatt, M.E.; Dutta-Simmons, J.; Ikeda, H.; Diaz-Griffero, F.; Pena-Cruz, V.; Bertagnolli, M.; et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. [Google Scholar] [CrossRef]
- Wang, X.; Feng, M.; Xiao, T.; Guo, B.; Liu, D.; Liu, C.; Pei, J.; Liu, Q.; Xiao, Y.; Rosin-Arbesfeld, R.; et al. BCL9/BCL9L promotes tumorigenicity through immune-dependent and independent mechanisms in triple negative breast cancer. Oncogene 2021, 40, 2982–2997. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Keshet, I.; Schlesinger, Y.; Farkash, S.; Rand, E.; Hecht, M.; Segal, E.; Pikarski, E.; Young, R.A.; Niveleau, A.; Cedar, H.; et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006, 38, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef]
- Vollbrecht, C.; Mairinger, F.D.; Koitzsch, U.; Peifer, M.; Koenig, K.; Heukamp, L.C.; Crispatzu, G.; Wilden, L.; Kreuzer, K.A.; Hallek, M.; et al. Comprehensive analysis of disease-related genes in chronic lymphocytic leukemia by multiplex PCR-based next generation sequencing. PLoS ONE 2015, 10, e0129544. [Google Scholar] [CrossRef]
- Rosenquist, R.; Ghia, P.; Hadzidimitriou, A.; Sutton, L.A.; Agathangelidis, A.; Baliakas, P.; Darzentas, N.; Giudicelli, V.; Lefranc, M.P.; Langerak, A.W.; et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: Updated ERIC recommendations. Leukemia 2017, 31, 1477–1481. [Google Scholar] [CrossRef]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schübeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Haghighi, M.M.; Fan, A.X.-C.; Torbati, P.M.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. High-throughput hacking of the methylation patterns in breast cancer by in vitro transcription and thymidine-specific cleavage mass array on MALDI-TOF silico-chip. Mol. Cancer Res. 2008, 6, 1702–1709. [Google Scholar] [CrossRef]
- Ehrich, M.; Nelson, M.R.; Stanssens, P.; Zabeau, M.; Liloglou, T.; Xinarianos, G.; Cantor, C.R.; Field, J.K.; van den Boom, D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc. Natl. Acad. Sci. USA 2005, 102, 15785–15790. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Peterson, H.; Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g: Profiler. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacholewska, A.; Grimm, C.; Herling, C.D.; Lienhard, M.; Königs, A.; Timmermann, B.; Altmüller, J.; Mücke, O.; Reinhardt, H.C.; Plass, C.; et al. Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients. Int. J. Mol. Sci. 2021, 22, 9337. https://doi.org/10.3390/ijms22179337
Pacholewska A, Grimm C, Herling CD, Lienhard M, Königs A, Timmermann B, Altmüller J, Mücke O, Reinhardt HC, Plass C, et al. Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients. International Journal of Molecular Sciences. 2021; 22(17):9337. https://doi.org/10.3390/ijms22179337
Chicago/Turabian StylePacholewska, Alicja, Christina Grimm, Carmen D. Herling, Matthias Lienhard, Anja Königs, Bernd Timmermann, Janine Altmüller, Oliver Mücke, Hans Christian Reinhardt, Christoph Plass, and et al. 2021. "Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients" International Journal of Molecular Sciences 22, no. 17: 9337. https://doi.org/10.3390/ijms22179337
APA StylePacholewska, A., Grimm, C., Herling, C. D., Lienhard, M., Königs, A., Timmermann, B., Altmüller, J., Mücke, O., Reinhardt, H. C., Plass, C., Herwig, R., Hallek, M., & Schweiger, M. R. (2021). Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients. International Journal of Molecular Sciences, 22(17), 9337. https://doi.org/10.3390/ijms22179337