
Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Nuclear Receptors
2. Peroxisome Proliferator-Activated Receptors
3. Pleiotropic Effect of PPARs
4. Working Hypothesis of the NR Ligand Superfamily
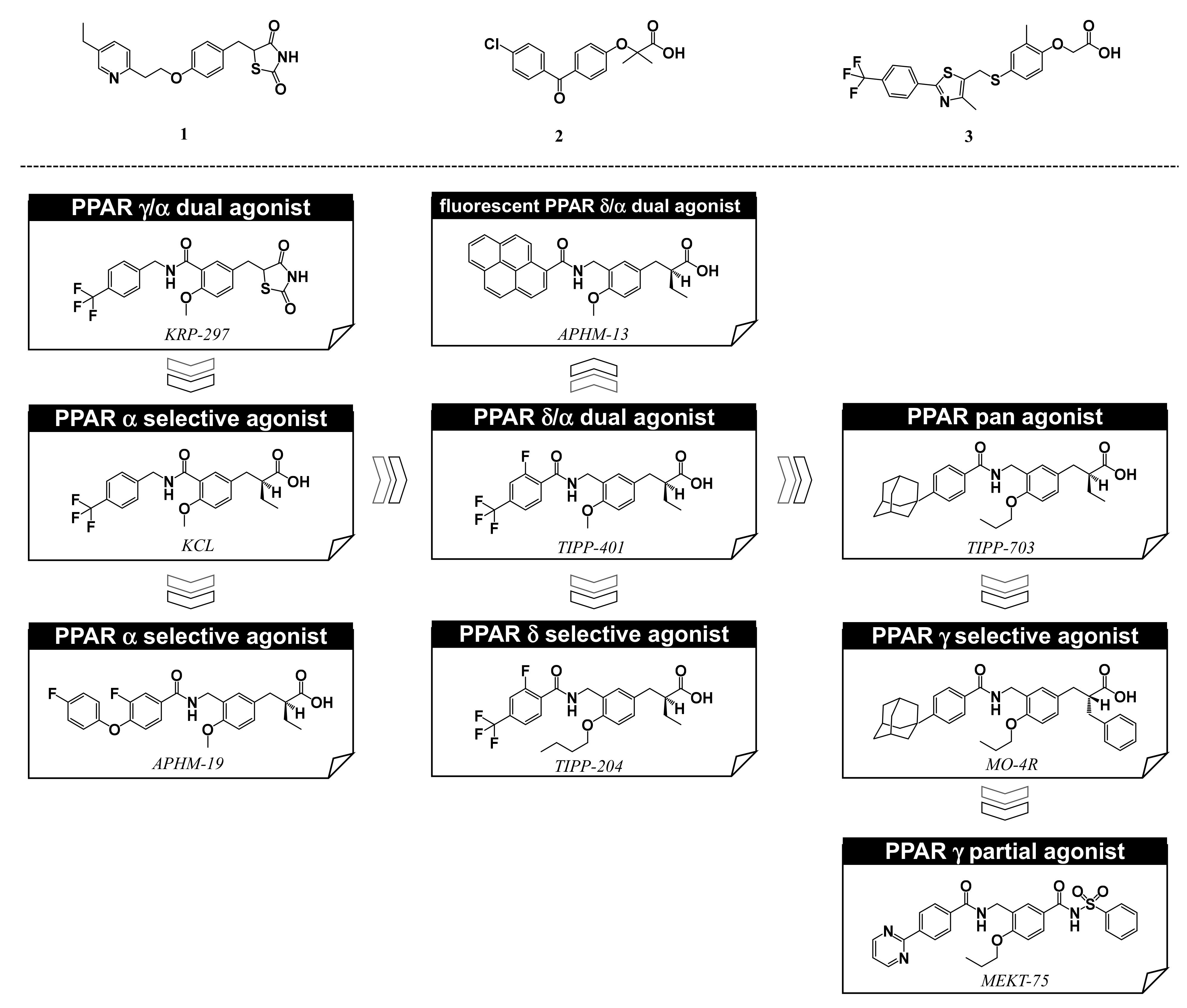
5. Synthesis of Our Ligands
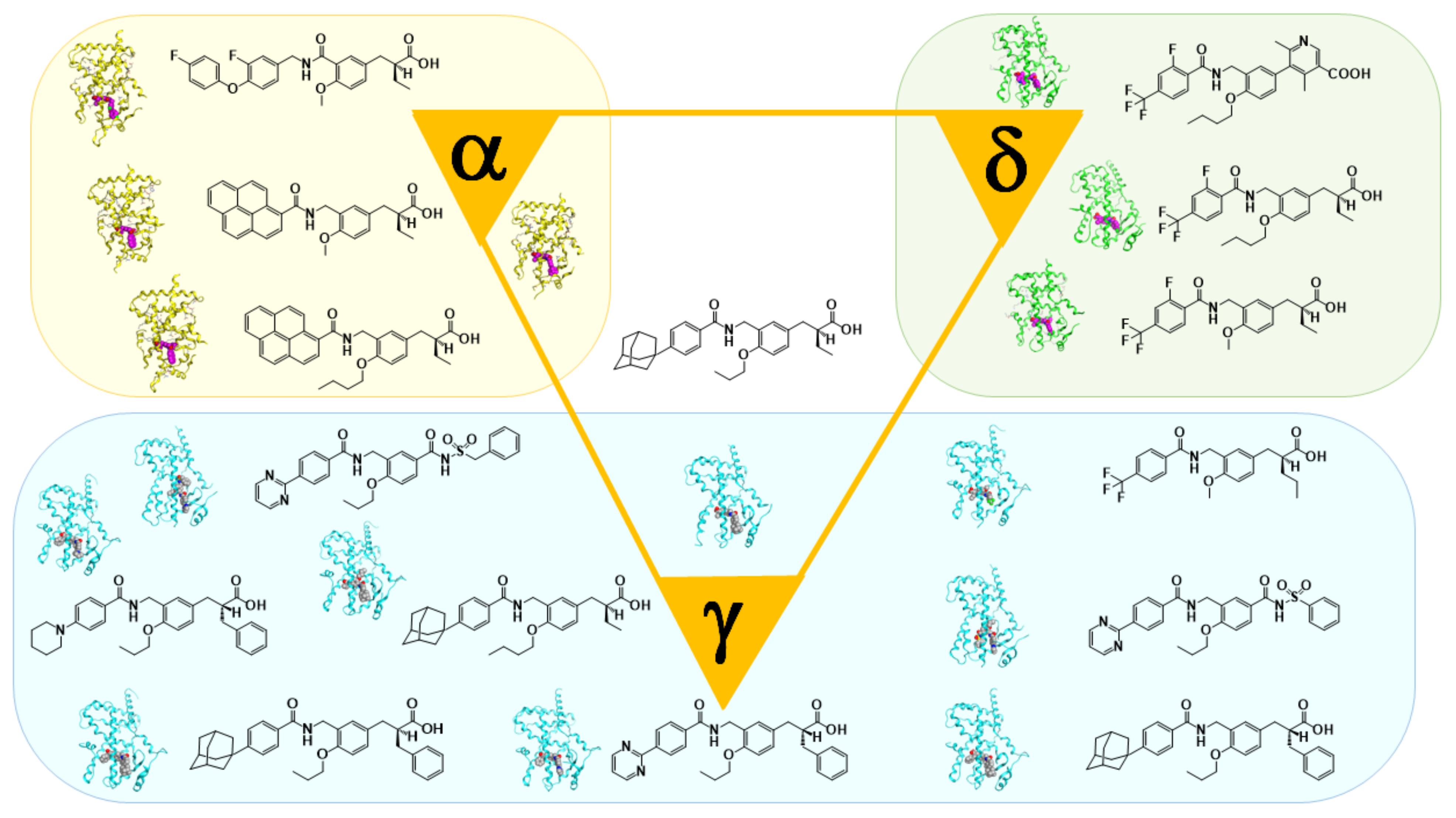
6. PPARα-Selective Agonist: From KCL to APHM-19

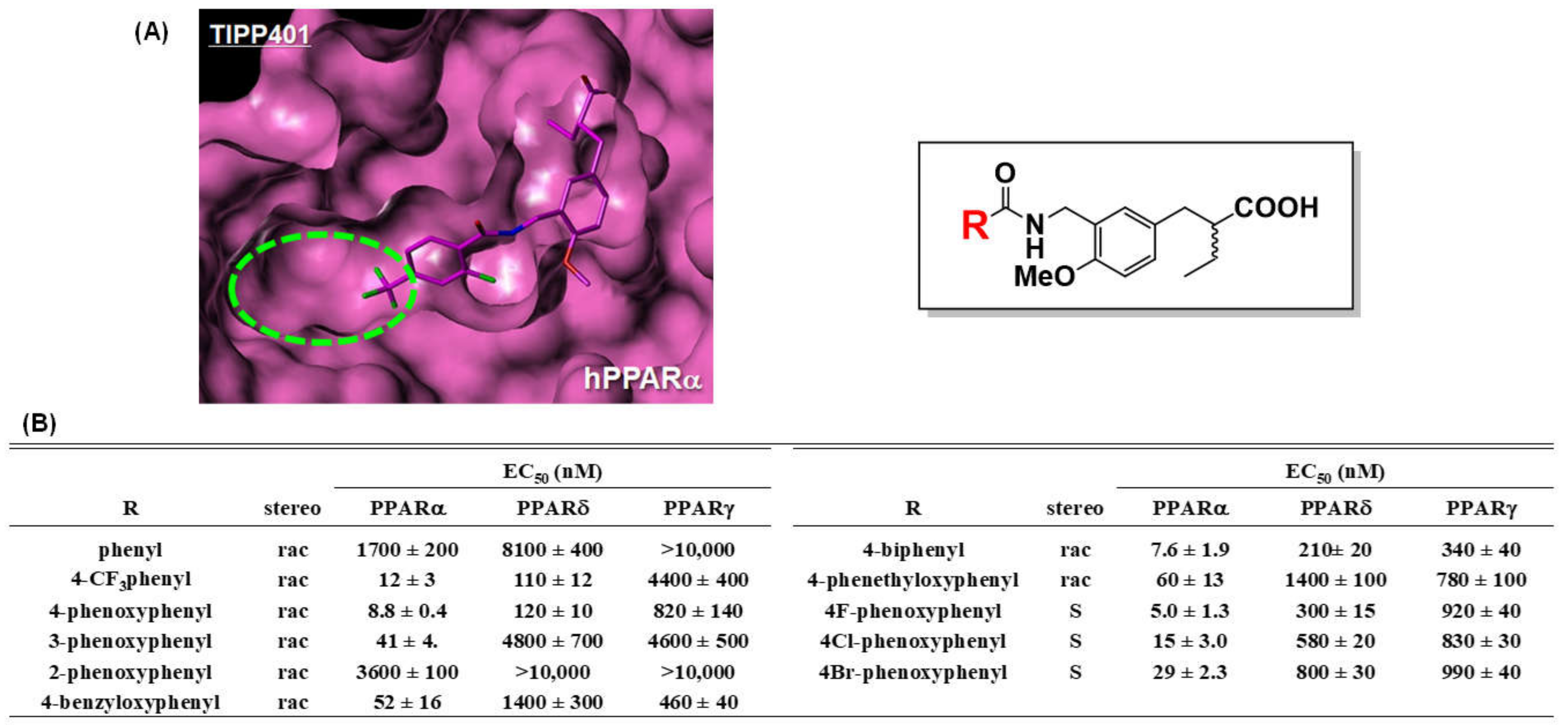
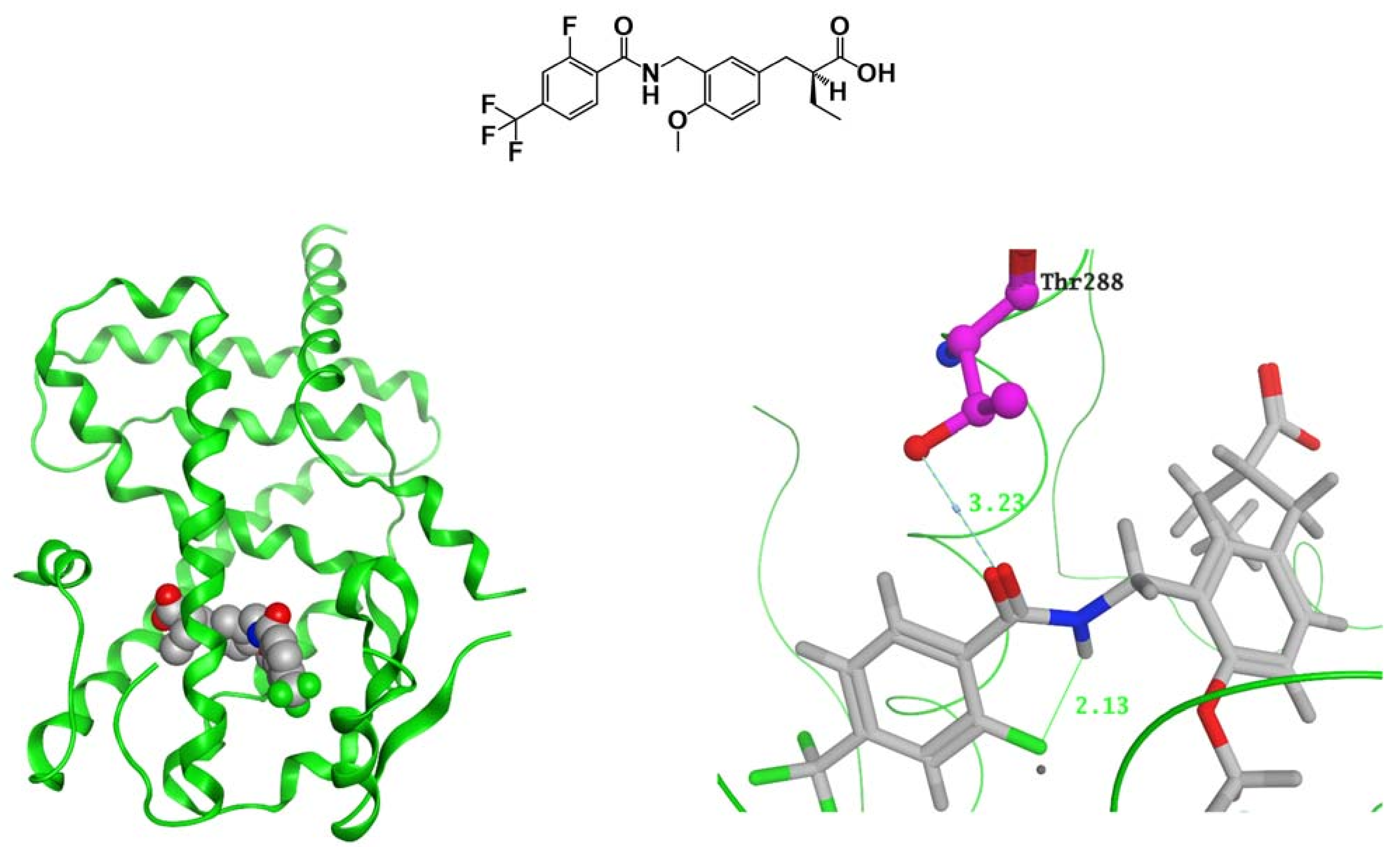
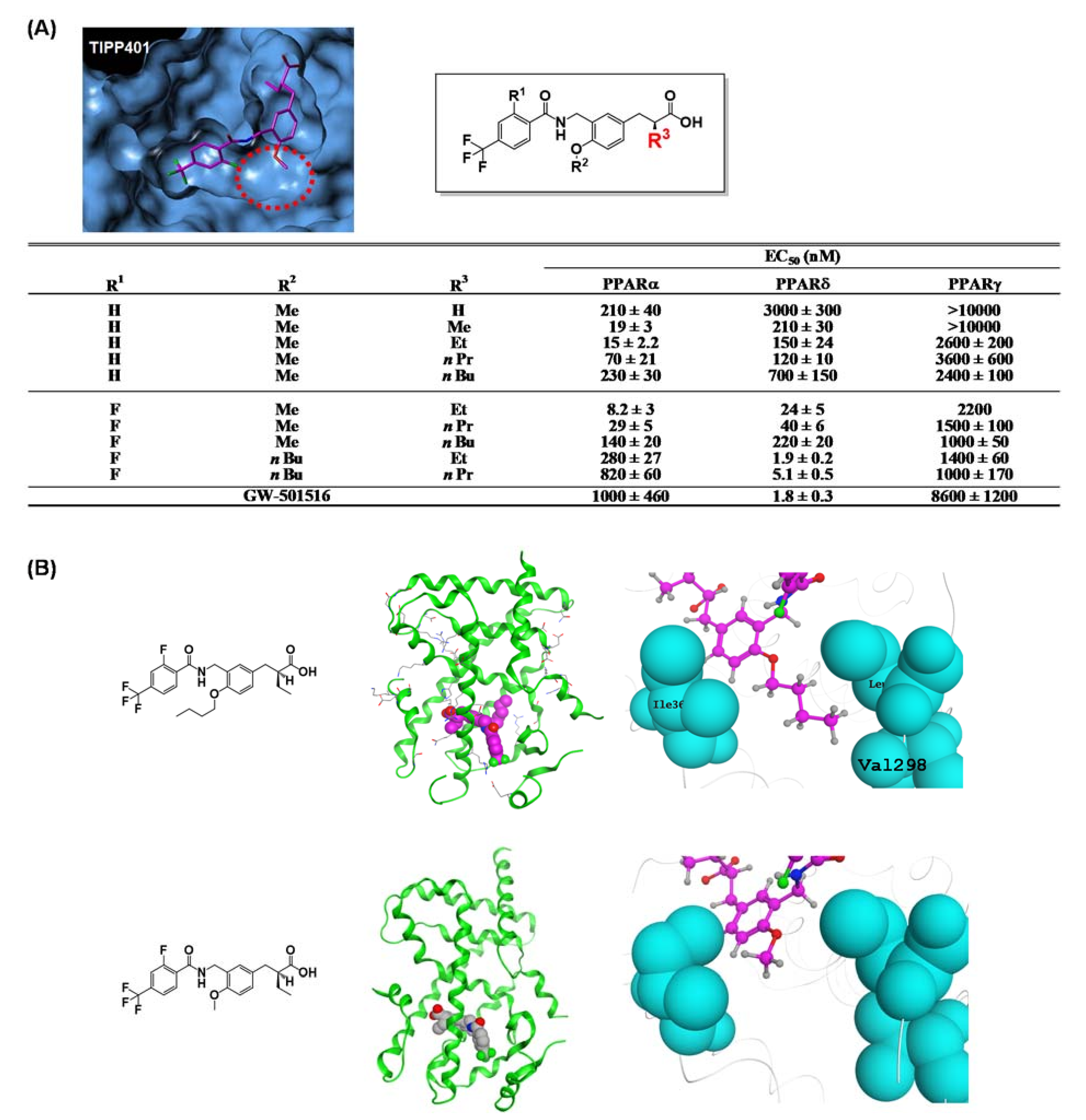
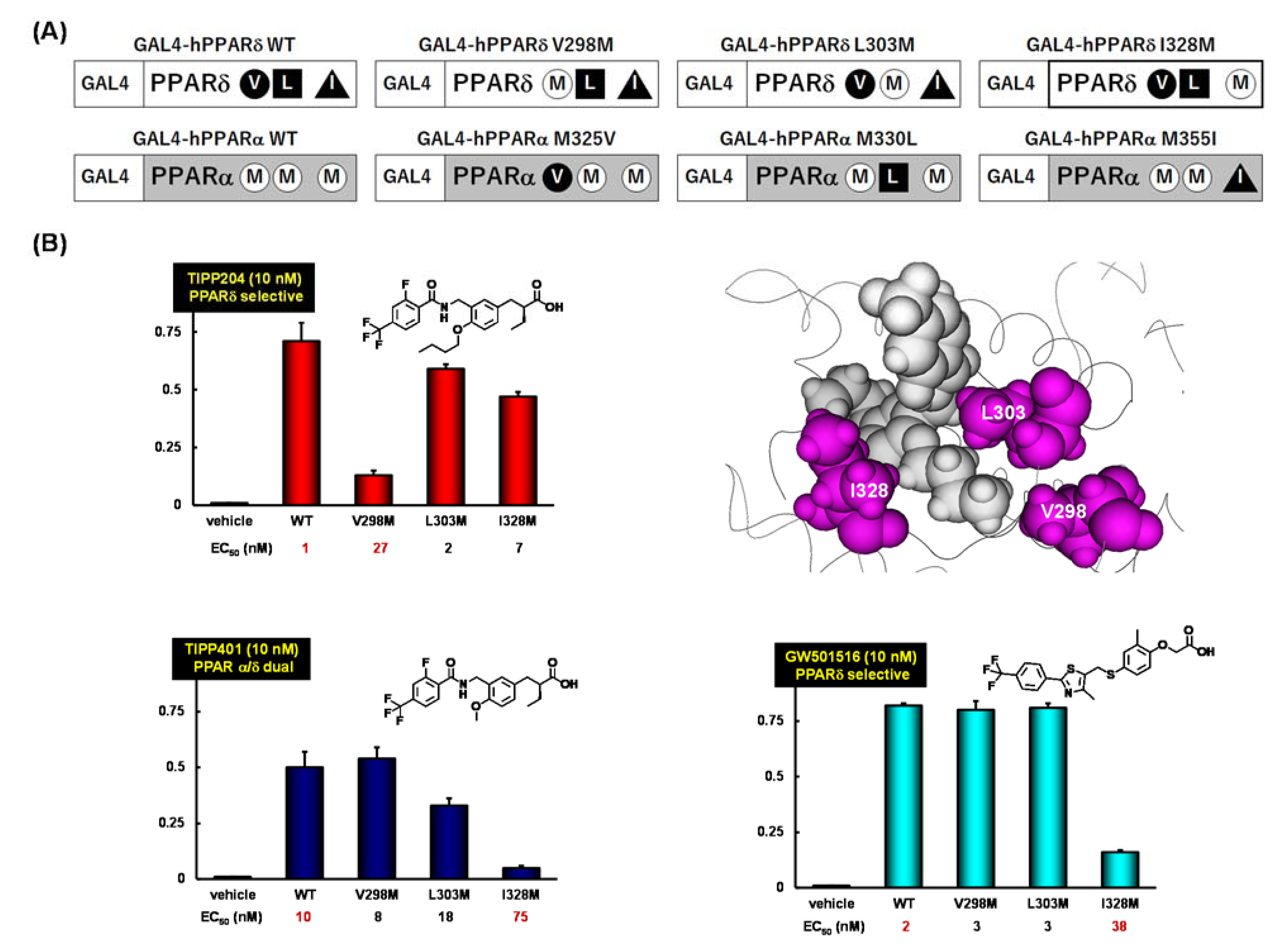
7. PARα/δ-Dual Agonist: TIPP-401

8. PPARδ-Selective Agonist: TIPP-204

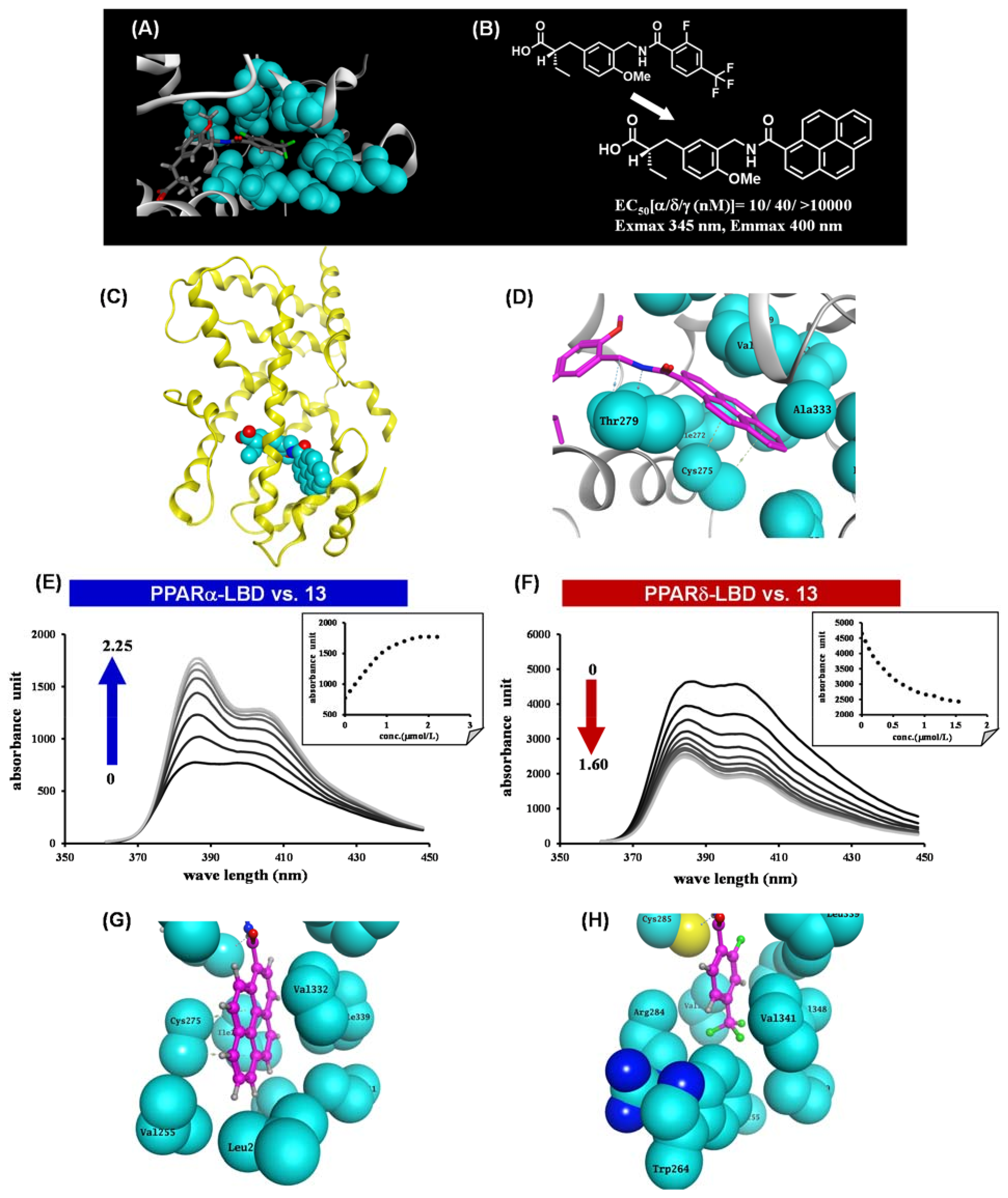
9. Fluorescent PPARα/δ Dual Agonist: APHM-13

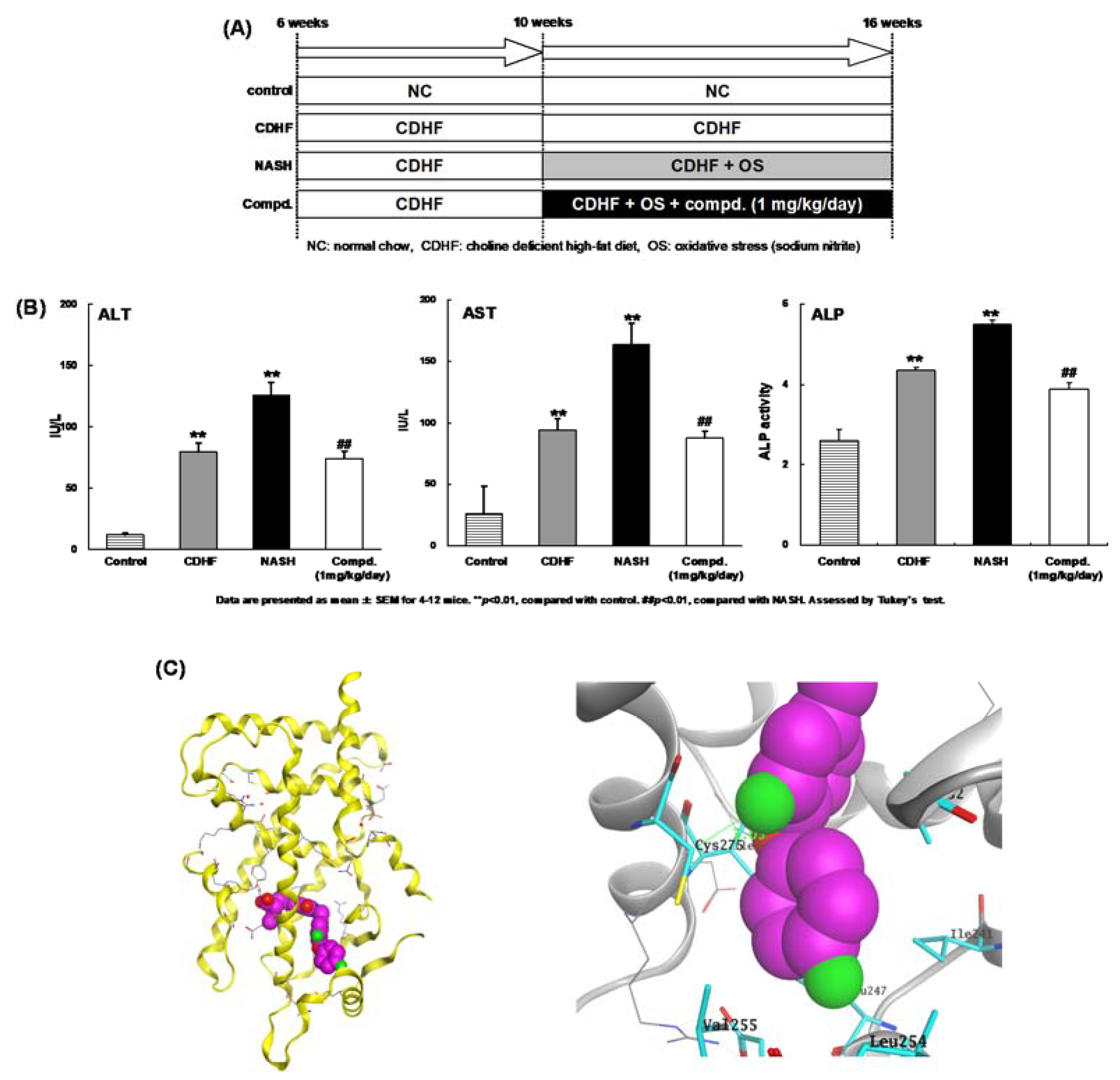
10. PPAR Pan Agonist: TIPP-703

11. PPARγ-Selective Agonist: MO-4R

12. PPARγ-Partial Agonist: MEKT-21

13. Structural Basis of Full and Partial PPARγ Agonists
14. Concluding Remarks and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADRP | adipocyte differentiation-related protein |
| AF-1 | transactivation function-1 |
| AF-2 | transcriptional activation function-2 |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| ANGPTL | angiopoietin-like protein |
| AST | aspartate aminotransferase |
| CPT1A | carnitine palmitoyltransferase 1A |
| DR1 | hexameric AGGTCA recognition motif, separated by one nucleotide |
| EPA | eicosapentaenoic acid |
| G1 | gap 1 |
| GAL4 | galactosidase 4 |
| H12 | helix 12 |
| HDL | high-density lipoprotein |
| HMGCS2 | HMG-CoA synthase 2 |
| LBD | ligand binding domain |
| NASH | nonalcoholic steato-hepatitis |
| NCoA | nuclear receptor corepressor |
| NCoR | nuclear receptor corepressor |
| NRs | nuclear receptors |
| PDB | protein data bank |
| PPARs | peroxisome proliferator-activated receptors |
| PPRE | peroxisome proliferator responsive element |
| RARα | retinoic acid receptor alpha |
| RXR | retinoid X receptor |
| RMSD | root mean square deviation |
| SMRT | silencing mediator for retinoid or thyroid-hormone receptor |
| SRC-1 | steroid Receptor Co-Activator-1 |
| THF | tetrahydrofurane |
| TNF-α | tumor necrosis factor-alpha |
| TZD | thiazolidinediones |
| ADRP | adipocyte differentiation-related protein |
| AF-1 | transactivation function-1 |
| AF-2 | transcriptional activation function-2 |
| ALP | alkaline phosphatase |
| ALT | alanine aminotransferase |
| ANGPTL | angiopoietin-like protein |
| AST | aspartate aminotransferase |
| CPT1A | carnitine palmitoyltransferase 1A |
| DR1 | hexameric AGGTCA recognition motif, separated by one nucleotide |
| EPA | eicosapentaenoic acid |
| G1 | gap 1 |
| GAL4 | galactosidase 4 |
| H12 | helix 12 |
| HDL | high-density lipoprotein |
| HMGCS2 | HMG-CoA synthase 2 |
| LBD | ligand binding domain |
| NASH | nonalcoholic steato-hepatitis |
| NCoA | nuclear receptor corepressor |
| NCoR | nuclear receptor corepressor |
| NRs | nuclear receptors |
| PDB | protein data bank |
| PPARs | peroxisome proliferator-activated receptors |
| PPRE | peroxisome proliferator responsive element |
References
- Chawta, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the x-files. Science 2001, 294, 1866–1870. [Google Scholar]
- Banner, C.D.; Gottlicher, M.; Widmark, E.; Sjovall, J.; Rafter, J.J.; Gustafsson, J.A. A systematic analytical chemistry/cell assay approach to isolate activators of orphan nuclear receptors from biological extracts: Characterization of peroxisome proliferator-activated receptor activators in plasma. J. Lipid Res. 1993, 34, 1583–1591. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Dreyer, C.; Medin, J.; Mahfoudi, A.; Ozato, K.; Wahli, W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Auwerx, J. Role of PPAR in the pharmacological regulation of lipoprotein metabolism by fibrates and thiazolidinediones. Curr. Pharm. Des. 1997, 3, 1–14. [Google Scholar]
- Lim, H.; Gupta, R.A.; Ma, W.G.; Paria, B.C.; Moller, D.E.; Morrow, J.D.; DuBois, R.N.; Trzaskos, J.M.; Dey, S.K. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARδ. Genes Dev. 1999, 13, 1561–1574. [Google Scholar] [CrossRef]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ) synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ionka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Investig. 1998, 101, 1354–1361. [Google Scholar] [CrossRef]
- Huang, J.-W.; Shiau, C.-W.; Yang, J.; Wang, D.-S.; Chiu, H.-C.; Chen, C.-S.; Chen, A.C.-Y. Development of small-molecule cyclin D1-ablative agents. J. Med. Chem. 2006, 49, 4684–4689. [Google Scholar] [CrossRef]
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Miyachi, H. Nuclear receptor antagonists designed based on the helix-folding inhibition hypothesis. Bioorg. Med. Chem. 2005, 13, 5080–5093. [Google Scholar] [CrossRef]
- Nomura, M.; Tanase, T.; Ide, T.; Tsunoda, M.; Suzuki, M.; Uchiki, H.; Murakami, K.; Miyachi, H. Design, Synthesis and Evaluation of Substituted Phenylpropanoic Acid Derivatives as Human Peroxisome Proliferator-Activated Receptor Activators; Discovery of Potent and Human PPARα Subtype-Selective Activators. J. Med. Chem. 2003, 46, 3581–3599. [Google Scholar] [CrossRef]
- Oyama, T.; Kamata, S.; Ishii, I.; Miyachi, H. Crystal structures of the human peroxisome proliferator-activated receptor (PPAR)a ligand-binding domain in complexes with a series of phenylpropanoic acid derivatives generated by a ligand-exchange soaking method. Bio. Pharm. Bull. 2021. accepted for publication. [Google Scholar]
- Kasuga, J.; Yamasaki, D.; Araya, Y.; Nakagawa, A.; Makishima, M.; Doi, T.; Hashimoto, Y.; Miyachi, H. Design, synthesis and evaluation of a novel series of α-substituted phenylpropanoic acid derivatives as human peroxisome proliferator-activated receptor (PPAR) α/δ dual agonists for the treatment of metabolic syndrome. Bioorg. Med. Chem. 2006, 14, 8405–8414. [Google Scholar] [CrossRef] [PubMed]
- Araya, Y.; Kasuga, J.; Toyota, K.; Hirakawa, Y.; Oyama, T.; Makishima, M.; Morikawa, K.; Hashimoto, Y.; Miyachi, H. Structure-Based Design and Synthesis of Fluorescent PPARα/δ Co-agonist and Its Application as a Probe for Fluorescent Polarization Assay of PPARδ Ligands. Chem. Pharm. Bull. 2008, 56, 1357–1359. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, J.; Nakagome, I.; Aoyama, A.; Sako, K.; Ishizawa, M.; Ogura, M.; Makishima, M.; Hirono, S.; Hashimoto, Y.; Miyachi, H. Design, synthesis, and evaluation of potent, structurally novel peroxisome proliferator-activated receptor (PPAR) δ-selective agonists. Bioorg. Med. Chem. 2007, 15, 5177–5190. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, J.; Yamasaki, D.; Ogura, K.; Shimizu, M.; Sato, M.; Makishima, M.; Doi, T.; Hashimoto, Y.; Miyachi, H. SAR-oriented discovery of peroxisome proliferator-activated receptor pan agonist with a 4-adamantylphenyl group as a hydrophobic tail. Bioorg. Med. Chem. Lett. 2008, 18, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Oyama, T.; Nakagome, I.; Satoh, M.; Nishio, Y.; Nobusada, H.; Hirono, S.; Morikawa, K.; Hashimoto, Y.; Miyachi, H. Design, Synthesis, and Structural Analysis of Phenylpropanoic Acid-Type PPARγ-Selective Agonists: Discovery of Reversed Stereochemistry-Activity Relationship. J. Med. Chem. 2011, 54, 331–341. [Google Scholar] [CrossRef]
- Ohashi, M.; Oyama, T.; Putranto, E.W.; Waku, T.; Nobusada, H.; Kataoka, K.; Matsuno, K.; Yashiro, M.; Morikawa, K.; Huh, N.H.; et al. Design and synthesis of a series of α-benzyl phenylpropanoic acid-type peroxisome proliferator-activated receptor (PPAR) γ partial agonists with improved aqueous solubility. Bioorg. Med. Chem. 2013, 21, 2319–2332. [Google Scholar] [CrossRef]
- Ohashi, M.; Oyama, T.; Miyachi, H. Different structures of the two peroxisome proliferator-activated receptor γ (PPARγ) ligand-binding domains in homodimeric complex with partial agonist, but not full agonist. Bioorg. Med. Chem. Lett. 2015, 25, 2639–2644. [Google Scholar] [CrossRef]
- Oyama, T.; Toyota, K.; Waku, T.; Hirakawa, Y.; Nagasawa, N.; Kasuga, J.; Hashimoto, Y.; Miyachi, H.; Morikawa, K. Adaptability and selectivity of human peroxisome proliferator-activated receptor (PPAR) pan agonists revealed from crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 786–795. [Google Scholar] [CrossRef]
- Kuwabara, N.; Oyama, T.; Tomioka, D.; Ohashi, M.; Yanagisawa, J.; Shimizu, T.; Miyachi, H. Peroxisome proliferator-activated receptors (PPARs) have multiple binding points that accommodate ligands in various conformations: Phenylpropanoic acid-type PPAR ligands bind to PPAR in different conformations, depending on the subtype. J. Med. Chem. 2012, 55, 893–902. [Google Scholar] [CrossRef]
- Ohashi, M.; Gamo, K.; Oyama, T.; Miyachi, H. Peroxisome proliferator-activated receptor γ (PPARγ) has multiple binding points that accommodate ligands in various conformations: Structurally similar PPARγ partial agonists bind to PPARγ LBD in different conformations. Bioorg. Med. Chem. Lett. 2015, 25, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric alkylation reaction of chiral imide enolates. A practical approach to the enantioselective synthesis of R-substituted carboxylic acid derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739. [Google Scholar] [CrossRef]
- Daniel, D.; Andrew, A.S. Reductive N-alkylation of amides, carbamates and ureas. Tetrahedron Lett. 1999, 40, 2295–2298. [Google Scholar]
- Murakami, K.; Tobe, K.; Ide, T.; Mochizuki, T.; Ohashi, M.; Akanuma, Y.; Yazaki, Y.; Kadowaki, T. A novel insulin sensitizer acts as a coligand for peroxisome proliferator activated receptor-α (PPAR-α) and PPAR-γ: Effect of PPAR-α activation on abnormal lipid metabolism in liver of Zucker fatty rats. Diabetes 1998, 47, 1841–1847. [Google Scholar] [CrossRef]
- Nomura, M.; Kinoshita, S.; Satoh, H.; Maeda, T.; Murakami, K.; Tsunoda, M.; Miyachi, H.; Awano, K. (3-substituted benzyl)thiazolidine-2, 4-diones as structurally new antihyperglycemic agents. Bioorg. Med. Chem. Lett. 1999, 9, 533–538. [Google Scholar] [CrossRef]
- Yajima, K.; Hirose, H.; Fujita, H.; Seto, Y.; Fujita, H.; Ukeda, K.; Miyashita, K.; Kawai, T.; Yamamoto, Y.; Ogawa, T.; et al. Combination therapy with PPARγ and PPARα agonists increases glucose-stimulated insulin secretion in db/db mice. Am. J. Physiol. 2003, 284, E966–E971. [Google Scholar]
- Nomura, M.; Tanase, T.; Miyachi, H. Efficient asymmetric synthesis of (S)-2-ethylphenylpropanoic acid derivative, a selective agonist for human peroxisome proliferator-activated receptor α. Bioorg. Med. Chem. Lett. 2002, 12, 2101–2104. [Google Scholar] [CrossRef]
- Miyachi, H.; Nomura, M.; Tanase, M.; Suzuki, M.; Murakami, K.; Awano, K. Enantio-dependent binding and transactivation of optically active phenylpropanoic acid derivatives at human peroxisome proliferator-activated receptor α. Bioorg. Med. Chem. Lett. 2002, 12, 333–335. [Google Scholar] [CrossRef]
- Miyachi, H.; Nomura, M.; Tanase, T.; Takahashi, Y.; Ide, T.; Tsunoda, M.; Murakami, K.; Awano, K. Design, synthesis and evaluation of substituted phenylpropanoic acid derivatives as peroxisome proliferator-activated receptor (PPAR) activators: Novel human PPARα-selective activators. Bioorg. Med. Chem. Lett. 2002, 12, 77–80. [Google Scholar] [CrossRef]
- Miyachi, H.; Uchiki, H. Analysis of the critical structural determinant(s) of species-selective peroxisome proliferator-activated receptor α (PPARα)-activation by phenylpropanoic acid-type PPARα agonists. Bioorg. Med. Chem. Lett. 2003, 13, 3145–3149. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Ishii, I. Preparation of co-crystals of human PPARα-LBD and ligand for high-resolution X-ray crystallography. STAR Protoc. 2021, 2, 100364. [Google Scholar] [CrossRef] [PubMed]
- Takayama, F.; Egashira, T.; Kawasaki, H.; Mankura, M.; Nakamoto, K.; Okada, S.; Mori, A. A Novel Animal Model of Nonalcoholic Steatohepatitis (NASH): Hypoxemia Enhances the Development of NASH. J. Clin. Biochem. Nutr. 2009, 45, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Takayama, F.; Mankura, M.; Hidaka, Y.; Egashira, T.; Ogino, T.; Kawasaki, H.; Mori, A. Beneficial Effects of Fermented Green Tea Extract in a Rat Model of Non-alcoholic Steatohepatitis. J. Clin. Biochem. Nutr. 2009, 44, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Day, C.D.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tokushige, K.; Takakura, M.; Tsuchiya-Matsushita, N.; Taniai, M.; Hashimoto, E.; Shiratori, K. Influence of TNF gene polymorphism in Japanese patients with NASH and simple steatosis. J. Hepatol. 2007, 46, 1104–1110. [Google Scholar] [CrossRef]
- Burt, A.D.; Mutton, A.; Day, C.P. Diagnosis and interpretation of steatosis and steatohepatitis. Semin. Diagn. Pathol. 1998, 15, 246–258. [Google Scholar]
- Holoman, J.; Glasa, J.; Galbavy, S.; Danis, D.; Molnarova, A.; Kazar, J.; Bednarova, A.; Misianik, J. Serum markers of liver fibrogenesis, and liver histology findings in patients with chronic liver diseases. Bratisl. Lek. Listy. 2002, 103, 70–75. [Google Scholar] [PubMed]
- Masaro, C.; Acosta, E.; Ortiz, J.A.; Marrero, P.F.; Hegardt, F.G.; Haro, D. Control of Human Muscle-type Carnitine Palmitoyltransferase I Gene Transcription by Peroxisome Proliferator-activated Receptor. J. Biol. Chem. 1998, 273, 8560–8563. [Google Scholar] [CrossRef]
- Mandard, S.; Zandbergen, F.; Tan, N.S.; Escher, P.; Patsouris, D.; Koenig, W.; Kleemann, R.; Bakker, A.; Veenman, F.; Wahli, W.; et al. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 2004, 279, 34411–34420. [Google Scholar] [CrossRef]
- Bassene, C.E.; Suzenet, F.; Hennuyer, N.; Staels, B.; Caignard, D.H.; Dacquet, C.; Renard, P.; Guillaumet, G. Studies towards the conception of new selective PPARβ/δ ligands. Bioorg. Med. Chem. Lett. 2006, 16, 4528–4532. [Google Scholar] [CrossRef] [PubMed]
- Weigand, S.; Bischoff, H.; Dittrich-Wengenroth, E.; Heckroth, H.; Lang, D.; Vaupel, A.; Woltering, M. Minor structural modifications convert a selective PPARα agonist into a potent, highly selective PPARδ agonist. Bioorg. Med. Chem. Lett. 2005, 15, 4619–4623. [Google Scholar] [CrossRef]
- Epple, R.; Azimioara, M.; Russo, R.; Bursulaya, B.; Tian, S.S.; Gerken, A.; Iskandar, M. 1,3,5-trisubstituted aryls as highly selective PPARδ agonists. Bioorg. Med. Chem. Lett. 2006, 16, 2969–2973. [Google Scholar] [CrossRef]
- Epple, R.; Azimioara, M.; Russo, R.; Xie, Y.; Wang, X.; Cow, C.; Wityak, J.; Karanewsky, D.; Bursulaya, B.; Kreusch, A.; et al. 3,4,5-trisubstituted isoxazoles as novel PPARδ agonists: Part 2. Bioorg. Med. Chem. Lett. 2006, 16, 5488–5492. [Google Scholar] [CrossRef] [PubMed]
- Epple, R.; Russo, R.; Azimioara, M.; Cow, C.; Xie, Y.; Wang, X.; Wityak, J.; Karanewsky, D.; Gerken, A.; Iskandar, M.; et al. 3,4,5-trisubstituted isoxazoles as novel PPARδ agonists: Part 1. Bioorg. Med. Chem. Lett. 2006, 16, 4376–4380. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator- activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Smith, D.S.; Eremin, S.A. Fluorescence polarization immunoassays and related methods for simple, high-throughput screening of small molecules. Anal. Bioanal. Chem. 2008, 391, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A. Probing protein folding and conformational transitions with fluorescence. Chem. Rev. 2006, 106, 1769–1784. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Staels, B.; Duriez, P. The role of fibric acids in atherosclerosis. Curr. Atheroscler. Rep. 2001, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Lee, S.H.; Eling, T.E.; Baek, S.J. A novel peroxisome proliferator–activated receptor γ ligand, MCC-555, induces apoptosis via posttranscriptional regulation of NAG-1 in colorectal cancer cells. Mol. Cancer Ther. 2006, 5, 1352–1361. [Google Scholar] [CrossRef]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef] [PubMed]
- Zandbergen, F.; Mandard, S.; Escher, P.; Tan, N.S.; Patsouris, D.; Jatkoe, T.; Rojas-Caro, S.; Madore, S.; Wahli, W.; Tafuri, S.; et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem. J. 2005, 392, 313–324. [Google Scholar] [CrossRef]
- Tachibana, K.; Kobayashi, Y.; Tanaka, T.; Tagami, M.; Sugiyama, A.; Katayama, T.; Ueda, C.; Yamasaki, D.; Ishimoto, K.; Sumitomo, M.; et al. Gene expression profiling of potential peroxisome proliferator-activated receptor (PPAR) target genes in human hepatoblastoma cell lines inducibly expressing different PPAR isoforms. Nucl. Recept. 2005, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Samudio, I.; Liu, S.; Abdelrahim, M.; Safe, S. Peroxisome Proliferator-Activated Receptor-Dependent Activation of p21 in Panc-28 Pancreatic Cancer Cells Involves Sp1 and Sp4 Proteins. Endocrinology 2004, 145, 5774–5785. [Google Scholar] [CrossRef]
- Ming, M.; Yu, J.P.; Meng, X.Z.; Zhou, Y.H.; Yu, H.G.; Luo, H.S. Effect of ligand troglitazone on peroxisome proliferator-activated receptor γ expression and cellular growth in human colon cancer cells. World J. Gastroenterol. 2006, 7, 7263–7270. [Google Scholar] [CrossRef]
- Kasuga, J.; Ishikawa, M.; Yonehara, M.; Makishima, M.; Hashimoto, Y.; Miyachi, H. Improvement of water-solubility of biarylcarboxylic acid peroxisome proliferator-activated receptor (PPAR) δ-selective partial agonists by disruption of molecular planarity/symmetry. Bioorg. Med. Chem. 2010, 18, 7164–7173. [Google Scholar] [CrossRef]
- Yashiro, M.; Chung, Y.S.; Nishimura, S.; Inoue, T.; Sowa, M. Peritoneal metastatic models for human scirrhous gastric carcinoma in nude mice. Clin. Exp. Metastasis 1996, 14, 43–54. [Google Scholar] [CrossRef]
- Takahashi, N.; Okumura, T.; Motomura, W.; Fujimoto, Y.; Kawabata, I.; Kohgo, Y. Activation of PPARγ inhibits cell growth and induces apoptosis in human gastric cancer cells. FEBS Lett. 1999, 455, 135–139. [Google Scholar] [CrossRef]
- Cheon, C.W.; Kim, D.H.; Cho, Y.H.; Kim, J.H. Effects of ciglitazone and troglitazone on the proliferation of human stomach cancer cells. World J. Gastroenterol. 2009, 15, 310–320. [Google Scholar] [CrossRef]
- Leo, C.; Yang, X.; Liu, J.; Li, H.; Chen, J.D. Role of Retinoid Receptor Coactivator Pockets in Cofactor Recruitment and Transcriptional Regulation. J. Biol. Chem. 2001, 276, 23127–23134. [Google Scholar] [CrossRef] [PubMed]
- Benko, S.; Love, J.D.; Beládi, M.; Schwabe, J.W.; Nagy, L. Molecular Determinants of the Balance between Co-repressor and Co-activator Recruitment to the Retinoic Acid Receptor. J. Biol. Chem. 2003, 278, 43797–43806. [Google Scholar] [CrossRef]
- Yamagishi, K.; Yamamoto, K.; Mochizuki, Y.; Nakano, T.; Yamada, S.; Tokiwa, H. Flexible ligand recognition of peroxisome proliferator-activated receptor-γ (PPARγ). Bioorg. Med. Chem. Lett. 2010, 20, 3344–3347. [Google Scholar] [CrossRef]
- Pochetti, G.; Godio, C.; Mitro, N.; Caruso, D.; Galmozzi, A.; Scurati, S.; Loiodis, F.; Fratiolla, G.; Tortollera, P.; Lagfezza, A.; et al. Insights into the mechanism of partial agonism: Crystal structures of the peroxisome proliferator-activated receptor γ ligand-binding domain in the complex with two enantiomeric ligands. J. Biol. Chem. 2007, 282, 17314–17324. [Google Scholar] [CrossRef] [PubMed]
- Carbonara, G.; Di Giovanni, C.; Fracchiolla, G.; Laghezza, A.; Lavecchia, A.; Loiodice, F.; Montanari, R.; Novellino, E.; Parente, M.; Piemontese, L.; et al. Synthesis, biological evaluation and molecular investigation of fluorinated peroxisome proliferator-activated receptors α/γ dual agonists. Bioorg. Med. Chem. 2012, 20, 2141–2151. [Google Scholar]
- Blanchard, S.G.; Cobb, J.E.; Collins, J.L.; Cooper, J.P.; Goreham, D.M.; Holmes, C.P.; Hull-Ryde, E.A.; Kliewer, S.A.; Lehmann, J.M.; Lenhard, J.M.; et al. Peroxisome proliferator-activated receptor γ ligand inhibits adipocyte differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 6102–6106. [Google Scholar]
- Chao, Y.S.; Chen, C.T.; Chen, H.Y.; Chen, X.; Goparaju, C.M.; Hsieh, H.P.; Hsu, J.T.; Huang, C.F.; Lee, H.J.; Liao, C.C.; et al. Structure-Based Drug Design of a Novel Family of PPARγ Partial Agonists: Virtual Screening, X-ray Crystallography, and in Vitro/in Vivo Biological Activities. J. Med. Chem. 2006, 49, 2703–2712. [Google Scholar]
- Abdalla, D.S.; Amato, A.A.; Ayers, S.D.; Baxter, J.D.; Brennan, R.G.; Carvalho, B.M.; Figueira, A.C.; Galdino, S.L.; Lima, M.C.; Lin, J.Z.; et al. GQ-16, a novel peroxisome proliferator-activated receptor (PPAR γ) ligand, promotes insulin sensitization without weight gain. J. Biol. Chem. 2012, 287, 28169–28179. [Google Scholar]
- Kawai, M.; Rosen, C.J. PPARγ: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629–636. [Google Scholar] [CrossRef]
- Taygerly, J.P.; McGee, L.R.; Rubenstein, S.M.; Houze, J.B.; Cushing, T.D.; Li, Y.; Motani, A.; Chen, J.L.; Frankmoelle, W.; Ye, G.; et al. Discovery of INT131: A selective PPARγ modulator that enhances insulin sensitivity. Bioorg. Med. Chem 2013, 21, 979–992. [Google Scholar] [CrossRef]
- Lu, W.; Lau, F.; Liu, K.; Wood, H.B.; Zhou, G.; Chen, Y.; Li, Y.; Akiyama, T.E.; Castriota, G.; Einstein, M.; et al. Benzimidazolones: A New Class of Selective Peroxisome Proliferator-Activated Receptor γ (PPARγ) Modulators. J. Med. Chem. 2011, 54, 8541–8554. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARg Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, S.; Tanatani, A.; Wakabayashi, K.; Makishima, M.; Imai, K.; Miyachi, H.; Nagasawa, K.; Hashimoto, Y. Ligands with a 3,3-diphenylpentane skeleton for nuclear vitamin D and androgen receptors: Dual activities and metabolic activation. Bioorg. Med. Chem. 2006, 14, 5489–5502. [Google Scholar] [CrossRef]
- Asano, L.; Ito, I.; Kuwabara, N.; Waku, T.; Yanagisawa, J.; Miyachi, H.; Shimizu, T. Structural basis for vitamin D receptor agonism by novel non-secosteroidal ligands. FEBS Lett. 2013, 587, 957–963. [Google Scholar] [CrossRef]
- Kainuma, M.; Makishima, M.; Hashimoto, Y.; Miyachi, H. Design, synthesis, and evaluation of non-steroidal farnesoid X receptor (FXR) antagonist. Bioorg. Med. Chem. 2007, 15, 2587–2600. [Google Scholar] [CrossRef] [PubMed]
- Kainuma, M.; Kasuga, J.; Hosoda, S.; Wakabayashi, K.; Tanatani, A.; Nagasawa, K.; Miyachi, H.; Makishima, M.; Hashimoto, Y. Diphenylmethane skeleton as a multi-template for nuclear receptor ligands: Preparation of FXR and PPAR ligands. Bioorg. Med. Chem. Lett. 2006, 16, 3213–3218. [Google Scholar] [CrossRef]
- Kasuga, J.; Ishida, S.; Yamasaki, D.; Makishima, M.; Doi, T.; Hashimoto, Y.; Miyachi, H. Novel biphenylcarboxylic acid peroxisome proliferator-activated receptor (PPAR) α selective antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6595–6599. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Gamo, K.; Tanaka, Y.; Waki, M.; Beniyama, Y.; Matsuno, K.; Wada, J.; Tenta, M.; Eguchi, J.; Makishima, M.; et al. Structural design and synthesis of arylalkynyl amide-type peroxisome proliferator-activated receptor γ (PPARγ)-selective antagonists based on the helix12-folding inhibition hypothesis. Eur. J. Med. Chem. 2015, 27, 53–67. [Google Scholar] [CrossRef]
- Aoyama, A.; Aoyama, H.; Dodo, K.; Makishima, M.; Hashimoto, Y.; Miyachi, H. LXR Antagonists with a 5-Substituted Phenanthridin-6-one Skeleton: Synthesis and LXR Transrepression Activities of Conformationally Restricted Carba-T0901317 Analogs. Heterocycles 2008, 76, 137–142. [Google Scholar] [CrossRef]
- Kishida, K.; Aoyama, A.; Hashimoto, Y.; Miyachi, H. Design and synthesis of phthalimide-based fluorescent LXR antagonists. Chem. Pharm. Bull. 2010, 58, 1525–1528. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakagawa, A.; Uno, S.; Makishima, M.; Miyachi, H.; Hashimoto, Y. Progesterone receptor antagonists with a 3-phenylquinazoline-2,4-dione/2-phenylisoquinoline-1,3-dione skeleton. Bioorg. Med. Chem. 2008, 16, 7046–7054. [Google Scholar] [CrossRef] [PubMed]




























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyachi, H. Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2021, 22, 9223. https://doi.org/10.3390/ijms22179223
Miyachi H. Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors. International Journal of Molecular Sciences. 2021; 22(17):9223. https://doi.org/10.3390/ijms22179223
Chicago/Turabian StyleMiyachi, Hiroyuki. 2021. "Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors" International Journal of Molecular Sciences 22, no. 17: 9223. https://doi.org/10.3390/ijms22179223
APA StyleMiyachi, H. (2021). Structural Biology-Based Exploration of Subtype-Selective Agonists for Peroxisome Proliferator-Activated Receptors. International Journal of Molecular Sciences, 22(17), 9223. https://doi.org/10.3390/ijms22179223