
Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation

 ,
,  ,
, 
Abstract
:1. Introduction
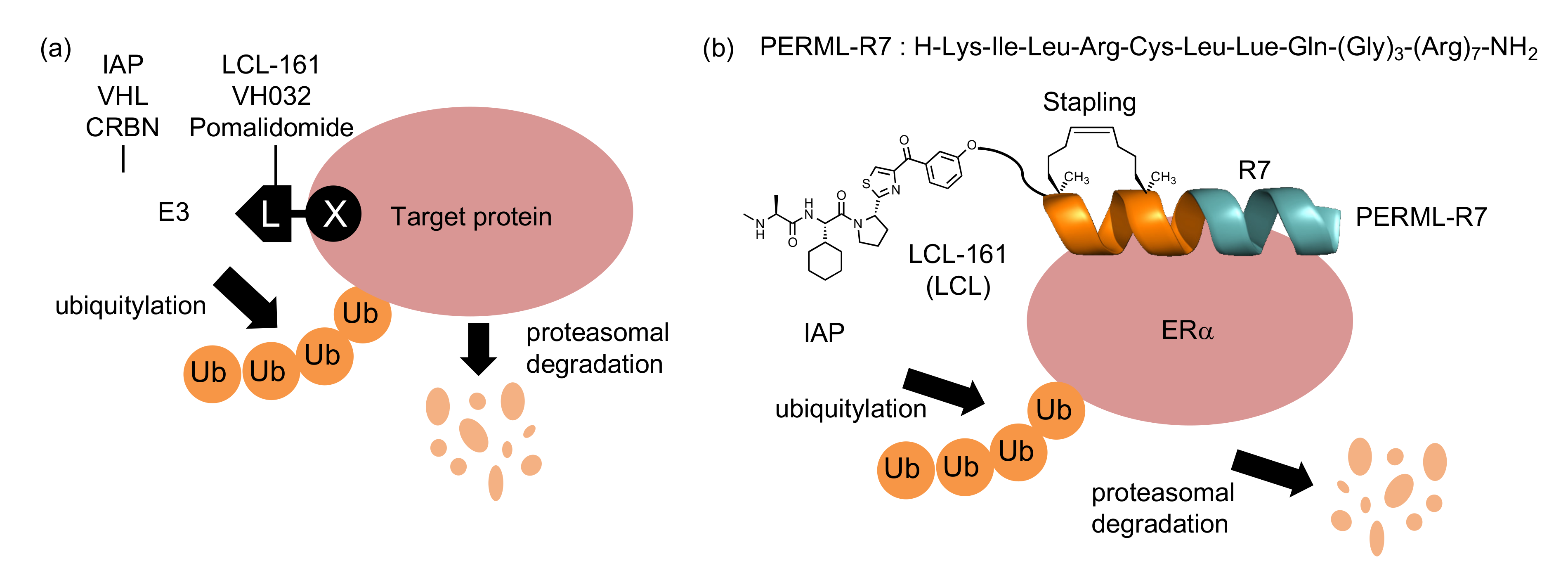
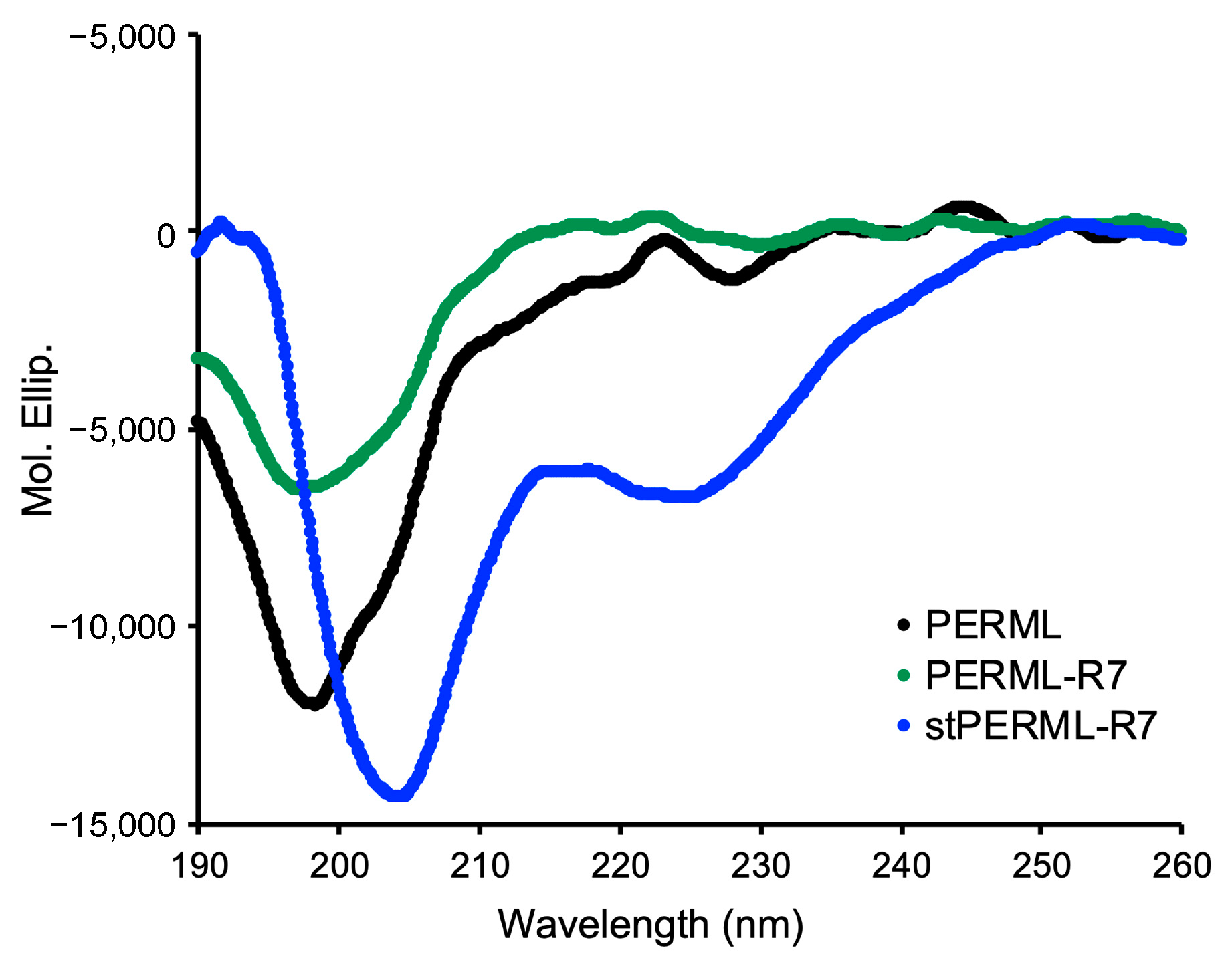
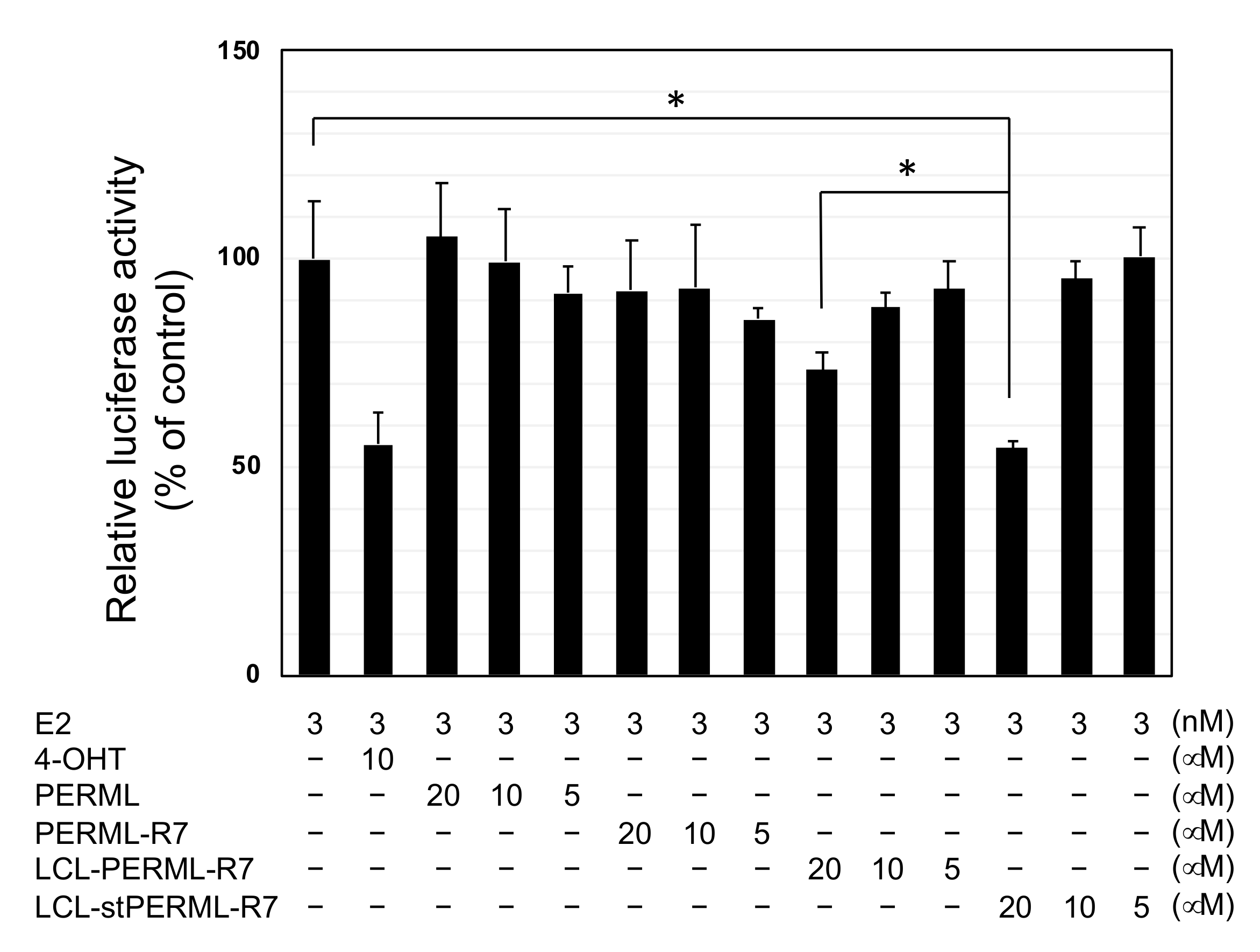
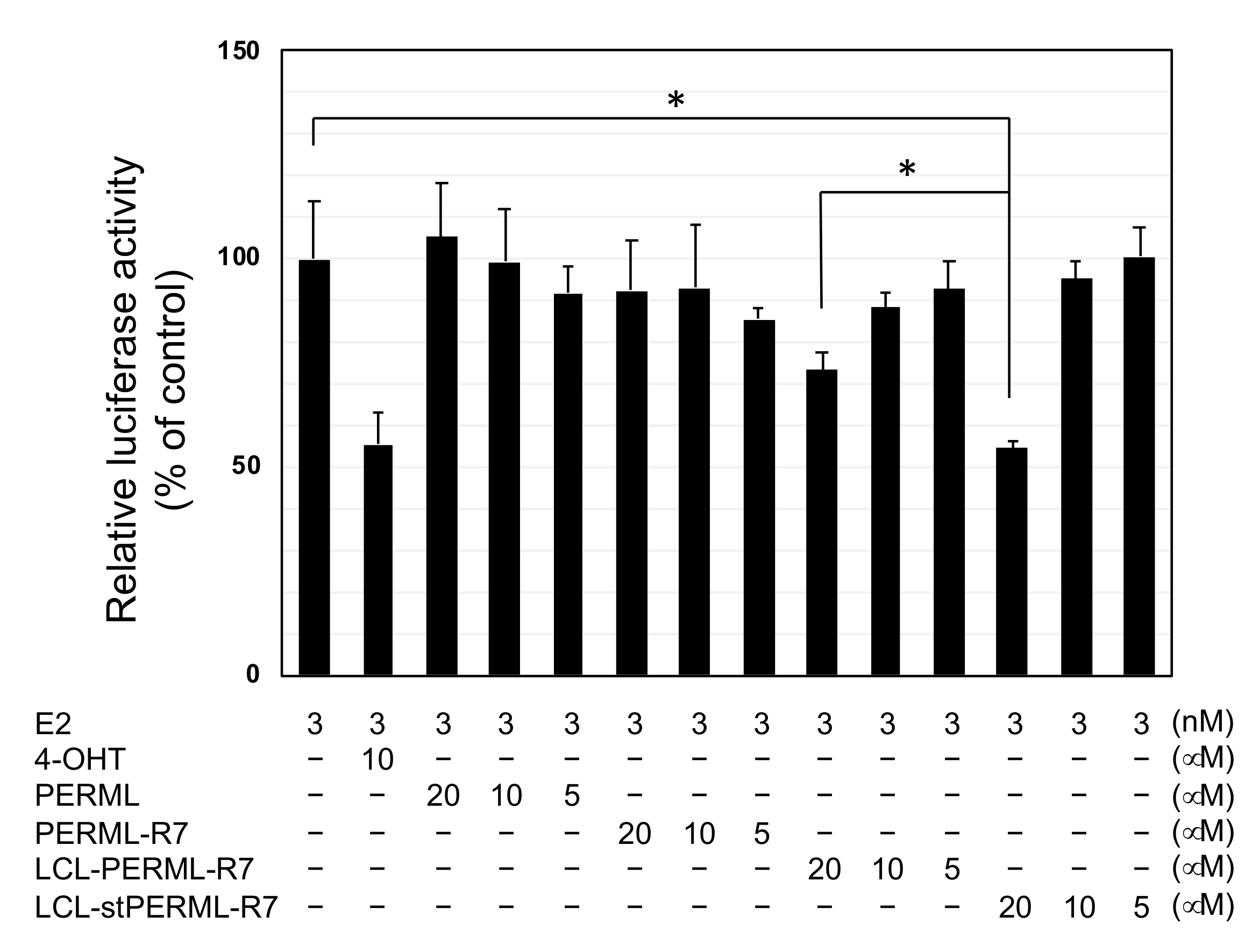
2. Results
3. Discussion
4. Materials and Methods
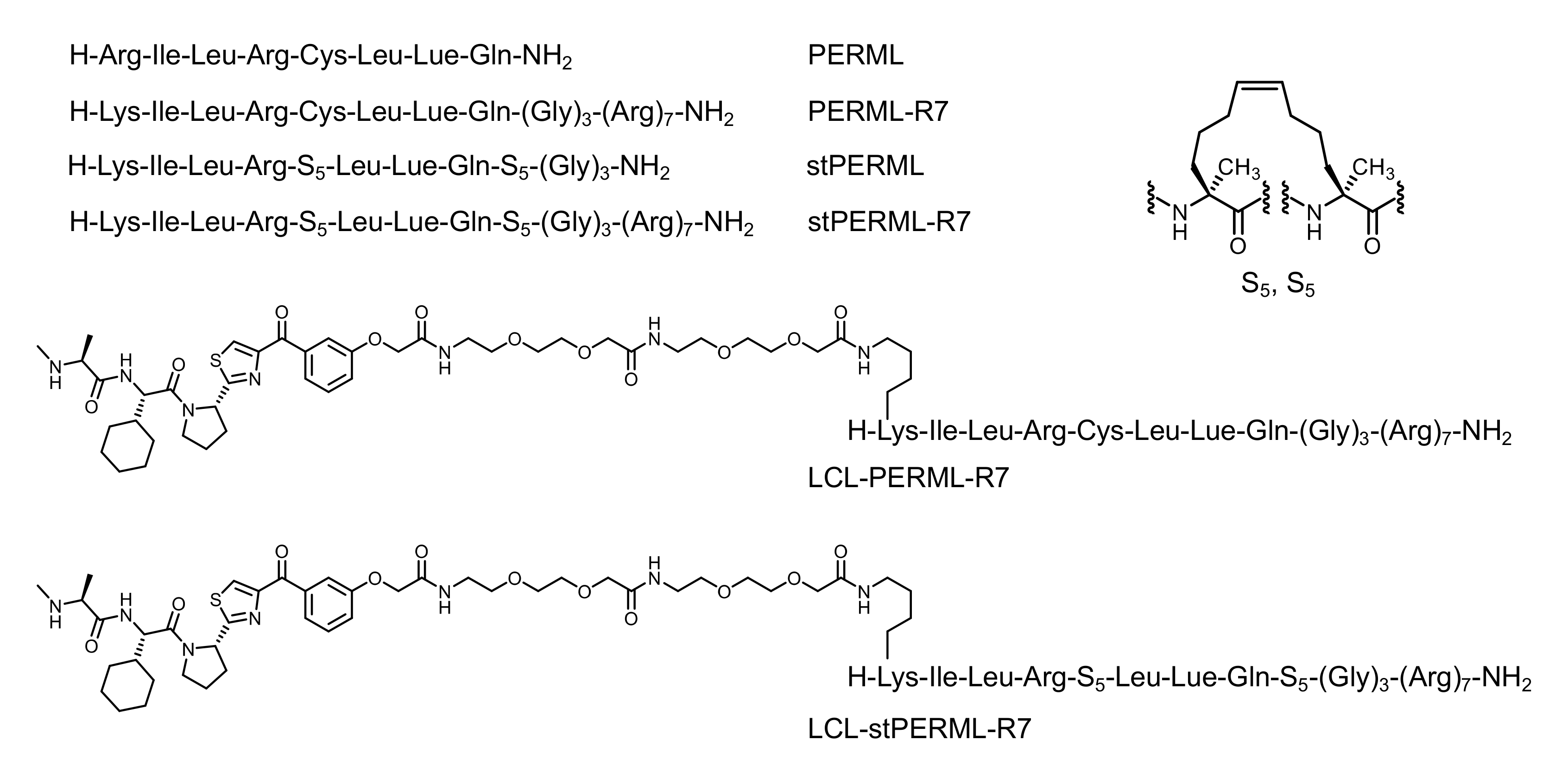
4.1. Synthesis and Characterization of the Peptides
4.1.1. Synthesis of PERML
4.1.2. Synthesis of PERML-R7
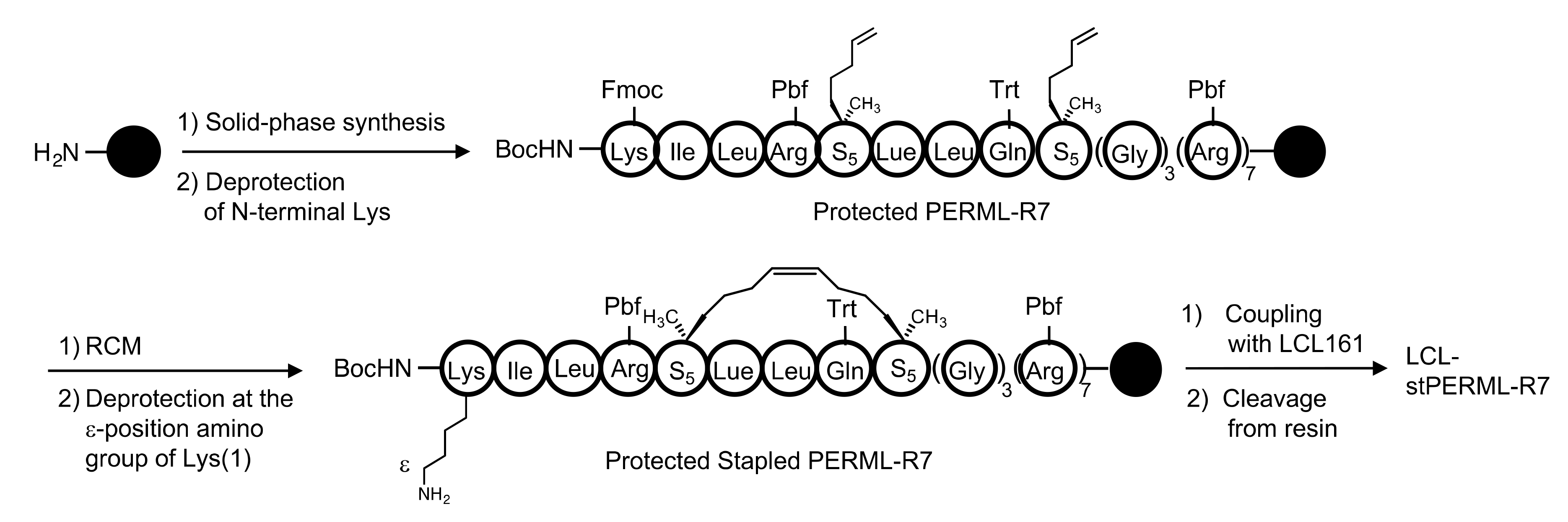
4.1.3. Synthesis of stPERML
4.1.4. Synthesis of stPERML-R7
4.1.5. Synthesis of LCL-PERML-R7
4.1.6. Synthesis of LCL-stPERML-R7
4.1.7. Synthesis of FAM-PERML
4.2. Binding Affinity to ERα
4.3. Cell Culture
4.4. Western Blot Analysis
4.5. Reporter Gene Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crews, C.M. Targeting the Undruggable Proteome: The Small Molecules of My Dreams. Chem. Biol. 2010, 17, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.J.; Ciulli, A. Molecular recognition of ternary complexes: A new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017, 61, 505–516. [Google Scholar]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Ohoka, N.; Shibata, N. SNIPERs—Hijacking IAP activity to induce protein degradation. Drug Discov. Today Technol. 2019, 31, 35–42. [Google Scholar] [CrossRef]
- Okuhira, K.; Demizu, Y.; Hattori, T.; Ohoka, N.; Shibata, N.; Nishimaki-Mogami, T.; Okuda, H.; Kurihara, M.; Naito, M. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013, 104, 1492–1498. [Google Scholar] [CrossRef]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Bulatov, E.; Ciulli, A. Targeting Cullin-RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. [Google Scholar] [CrossRef] [Green Version]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 24, 248. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 2020, 27, 66–73. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.W.; Duckett, C.S. Reawakening the cellular death program in neoplasia through the therapeutic blockade of IAP function. J. Clin. Invest. 2005, 115, 2673–2678. [Google Scholar] [CrossRef] [Green Version]
- Silke, J.; Meier, P. Inhibitor of Apoptosis (IAP) Proteins–Modulators of Cell Death and Inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef]
- Jin, J.; Wu, Y.; Chen, J.; Shen, Y.; Zhang, L.; Zhang, H.; Chen, L.; Yuan, H.; Chen, H.; Zhang, W.; et al. The peptide PROTAC modality: A novel strategy for targeted protein ubiquitination. Theranostics 2020, 10, 10141–10153. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Kutchukian, P.S.; Yang, J.S.; Verdine, G.L.; Shakhnvich, E.I. All-Atom Model for Stabilization of α-Helical Structure in Peptides by Hydrocarbon Staples. J. Am. Chem. Soc. 2009, 131, 4622–4627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Deng, Q.; Zhao, H.; Xie, M.; Chen, L.; Yin, F.; Qin, X.; Zheng, W.; Zhao, Y.; Li, Z. Development of Stabilized Peptide-Based PROTACs against Estrogen Receptor α. ACS Chem. Biol. 2018, 13, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Yue, N.; Gong, J.; Liu, C.; Li, Q.; Zhou, J.; Huang, W.; Qian, H. Development of cell-permeable peptide-based PROTACs targeting estrogen receptor α. Eur. J. Med. Chem. 2020, 187, 111967. [Google Scholar] [CrossRef] [PubMed]
- Demizu, Y.; Ohoka, N.; Nagakubo, T.; Yamashita, H.; Misawa, T.; Okuhira, K.; Naito, M.; Kurihara, M. Development of a peptide-based inducer of nuclear receptors degradation. Bioorg. Med. Chem. Lett. 2016, 26, 2655–2658. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, H.; Ohoka, N.; Naito, M.; Demizu, Y. Design and synthesis of peptide-based chimeric molecules to induce degradation of the estrogen and androgen receptors. Bioorg. Med. Chem. 2020, 28, 115595. [Google Scholar] [CrossRef]
- Galande, A.K.; Bramlett, K.S.; Trent, J.O.; Burris, T.P.; Wittliff, J.L.; Spatola, A.F. Potent Inhibitors of LXXLL-Based Protein–Protein Interactions. ChemBioChem 2005, 6, 1991–1998. [Google Scholar] [CrossRef]
- Gao, W. Peptide Antagonist of the Androgen Receptor. Curr. Pharm. Des. 2010, 16, 1106–1113. [Google Scholar] [CrossRef]
- Leduc, A.M.; Trent, J.O.; Wittliff, J.L.; Bramlett, K.S.; Briggs, S.L.; Chirgadze, N.Y.; Wang, Y.; Burris, T.P.; Spatola, A.F. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor–coactivator interactions. Proc. Natl. Acad. Sci. USA 2003, 100, 11273–11278. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | PERML | stPERML |
|---|---|---|
| IC50 (μM) | 0.7 ± 0.7 | 1.1 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoo, H.; Ohoka, N.; Takyo, M.; Ito, T.; Tsuchiya, K.; Kurohara, T.; Fukuhara, K.; Inoue, T.; Naito, M.; Demizu, Y. Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation. Int. J. Mol. Sci. 2021, 22, 8772. https://doi.org/10.3390/ijms22168772
Yokoo H, Ohoka N, Takyo M, Ito T, Tsuchiya K, Kurohara T, Fukuhara K, Inoue T, Naito M, Demizu Y. Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation. International Journal of Molecular Sciences. 2021; 22(16):8772. https://doi.org/10.3390/ijms22168772
Chicago/Turabian StyleYokoo, Hidetomo, Nobumichi Ohoka, Mami Takyo, Takahito Ito, Keisuke Tsuchiya, Takashi Kurohara, Kiyoshi Fukuhara, Takao Inoue, Mikihiko Naito, and Yosuke Demizu. 2021. "Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation" International Journal of Molecular Sciences 22, no. 16: 8772. https://doi.org/10.3390/ijms22168772
APA StyleYokoo, H., Ohoka, N., Takyo, M., Ito, T., Tsuchiya, K., Kurohara, T., Fukuhara, K., Inoue, T., Naito, M., & Demizu, Y. (2021). Peptide Stapling Improves the Sustainability of a Peptide-Based Chimeric Molecule That Induces Targeted Protein Degradation. International Journal of Molecular Sciences, 22(16), 8772. https://doi.org/10.3390/ijms22168772