
Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
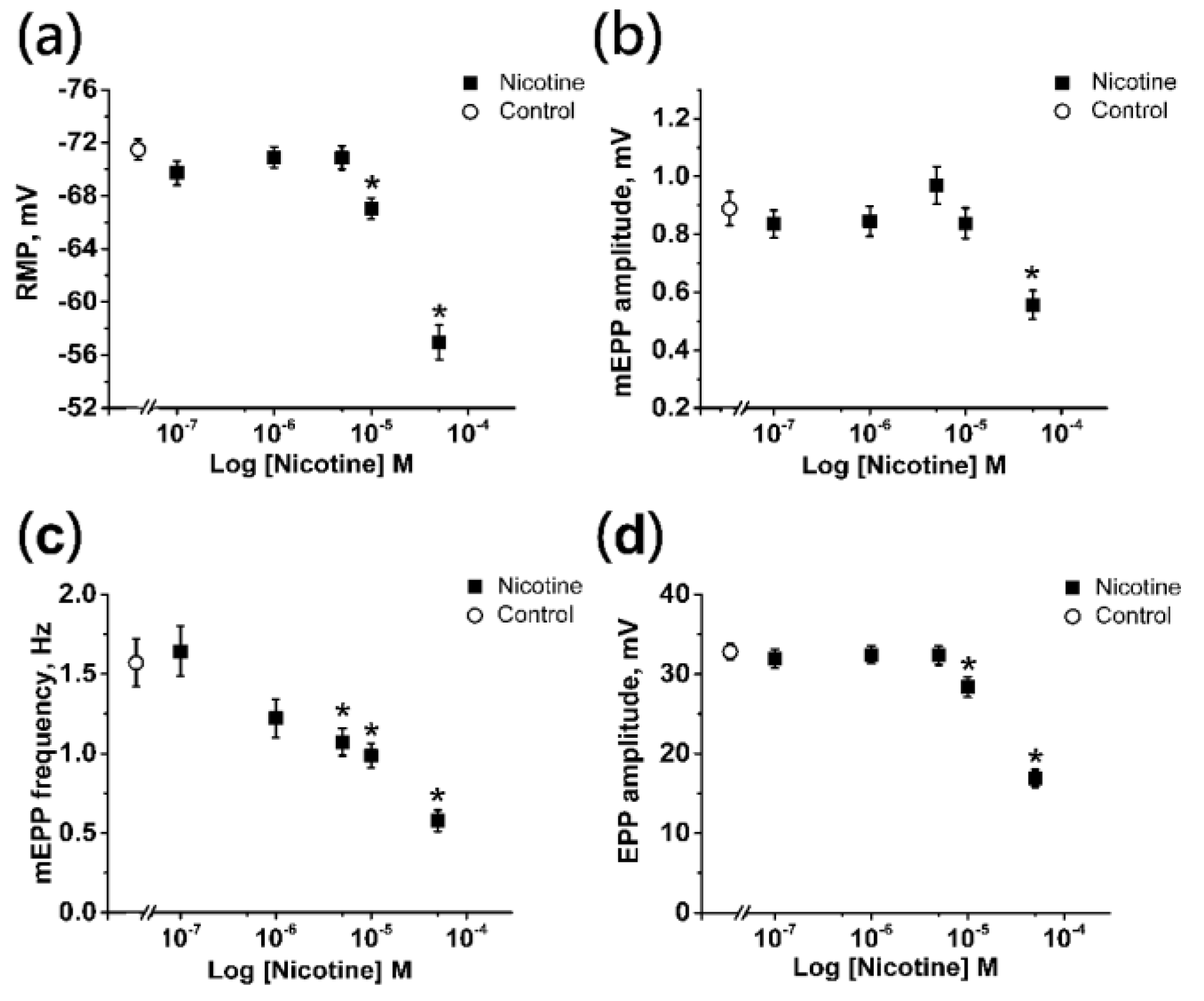
2.1. Effects of Nicotine on the Electrophysiological Parameters of the Neuromuscular Junction
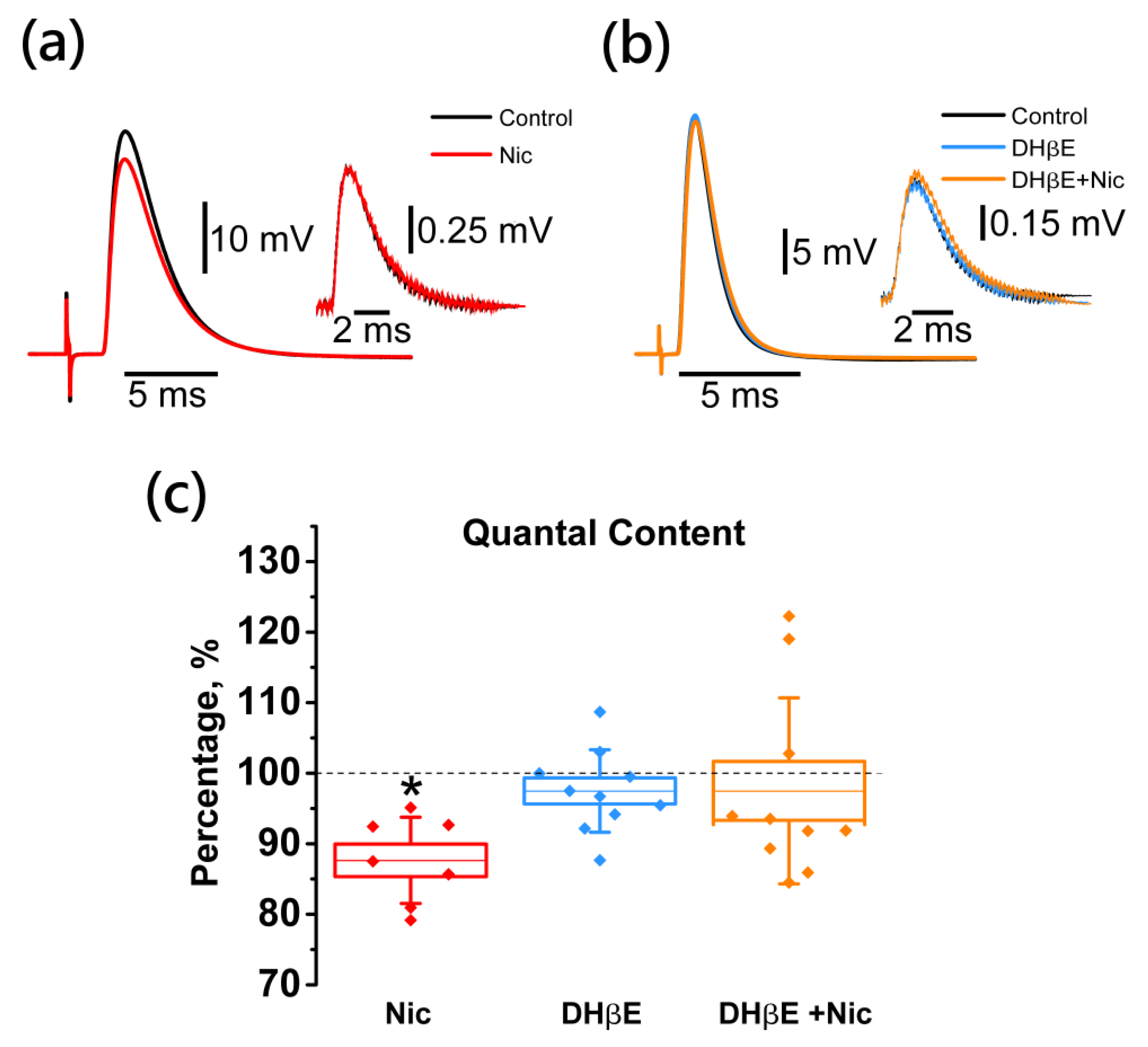
2.2. Activation of Neuronal Nicotinic Receptors Leads to Downregulation of the EPP Quantal Content
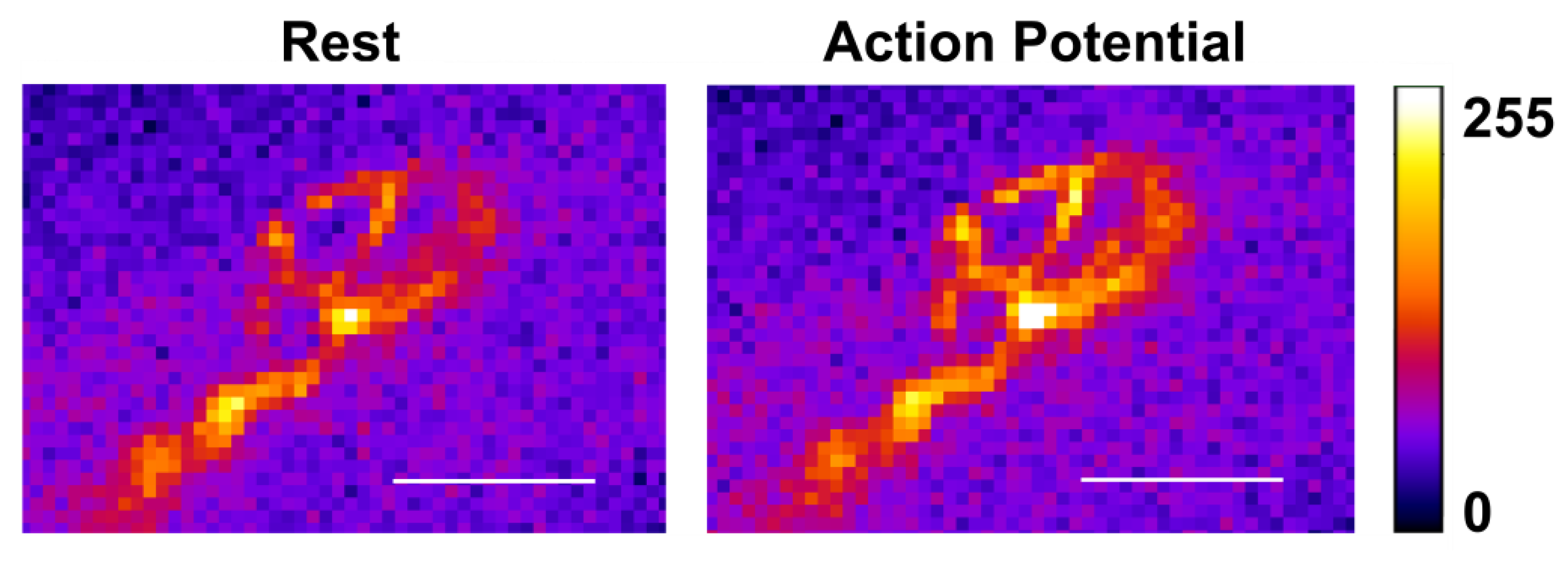
2.3. Activation of Neuronal Nicotinic Receptors Induces an Increase of the Calcium Level in the Motor Nerve Terminal
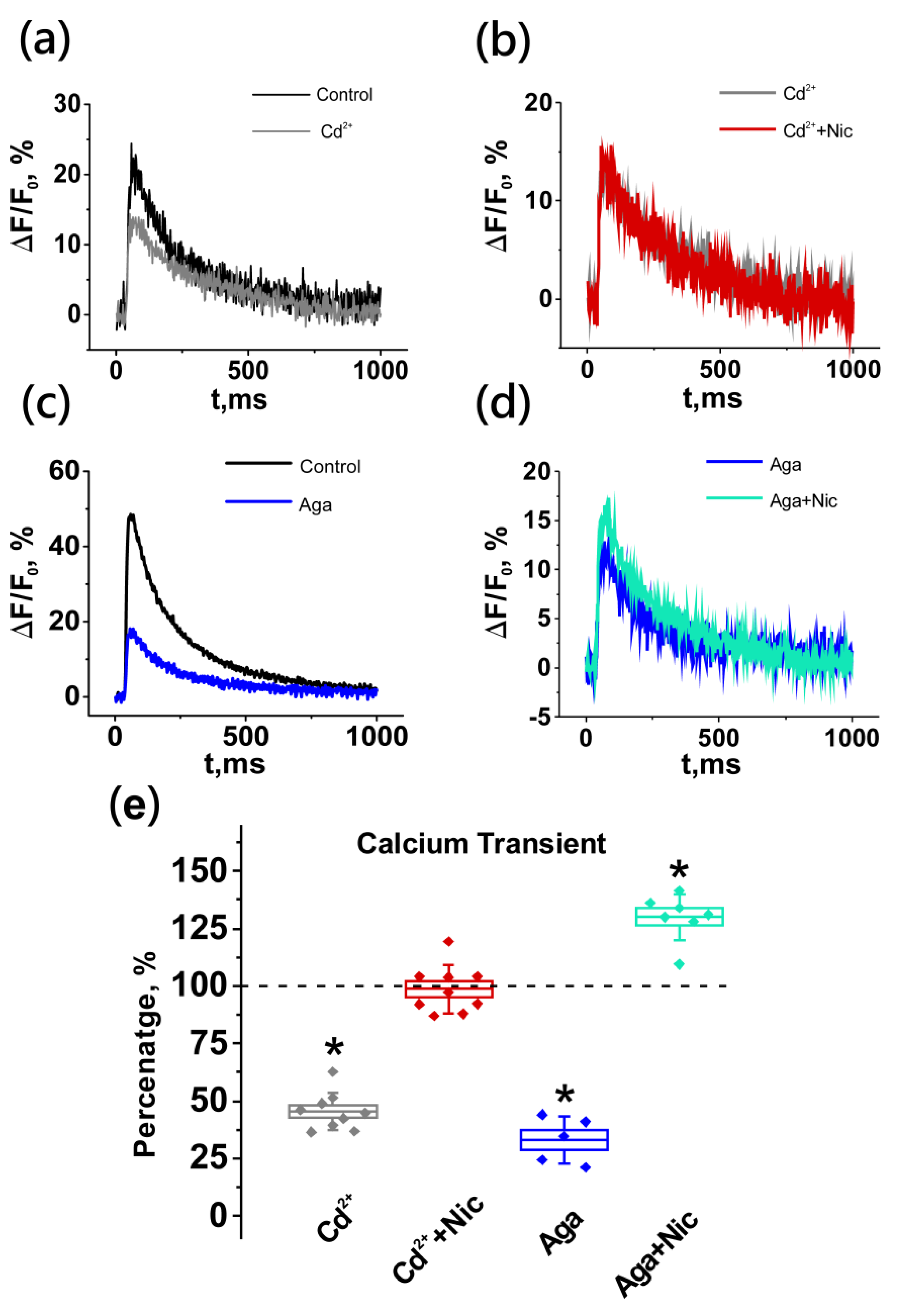
2.4. Neuronal Nicotinic Receptors Alter Calcium Level in Presynaptic Terminal by Gating L-Type (Cav1) Calcium Channels
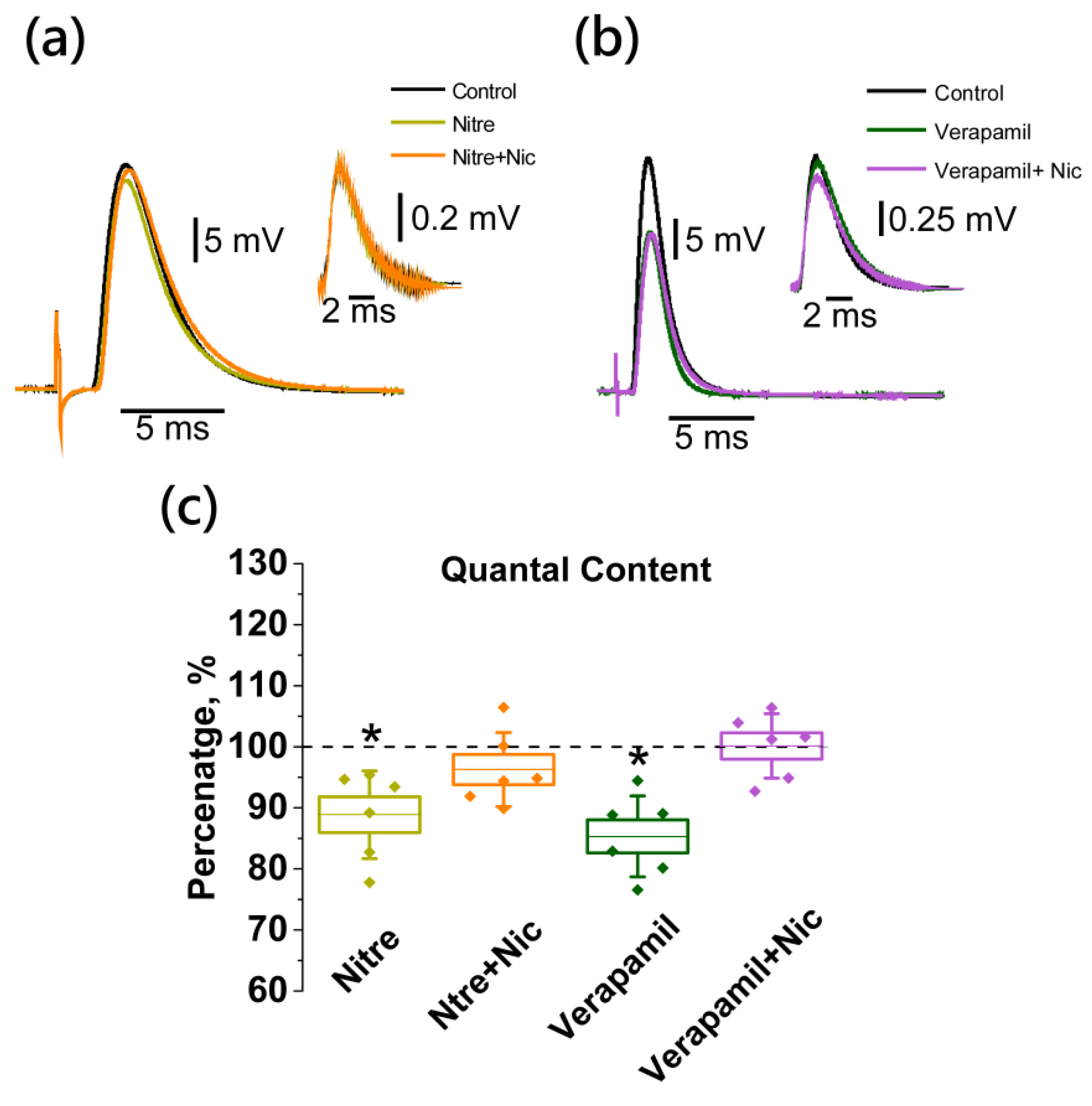
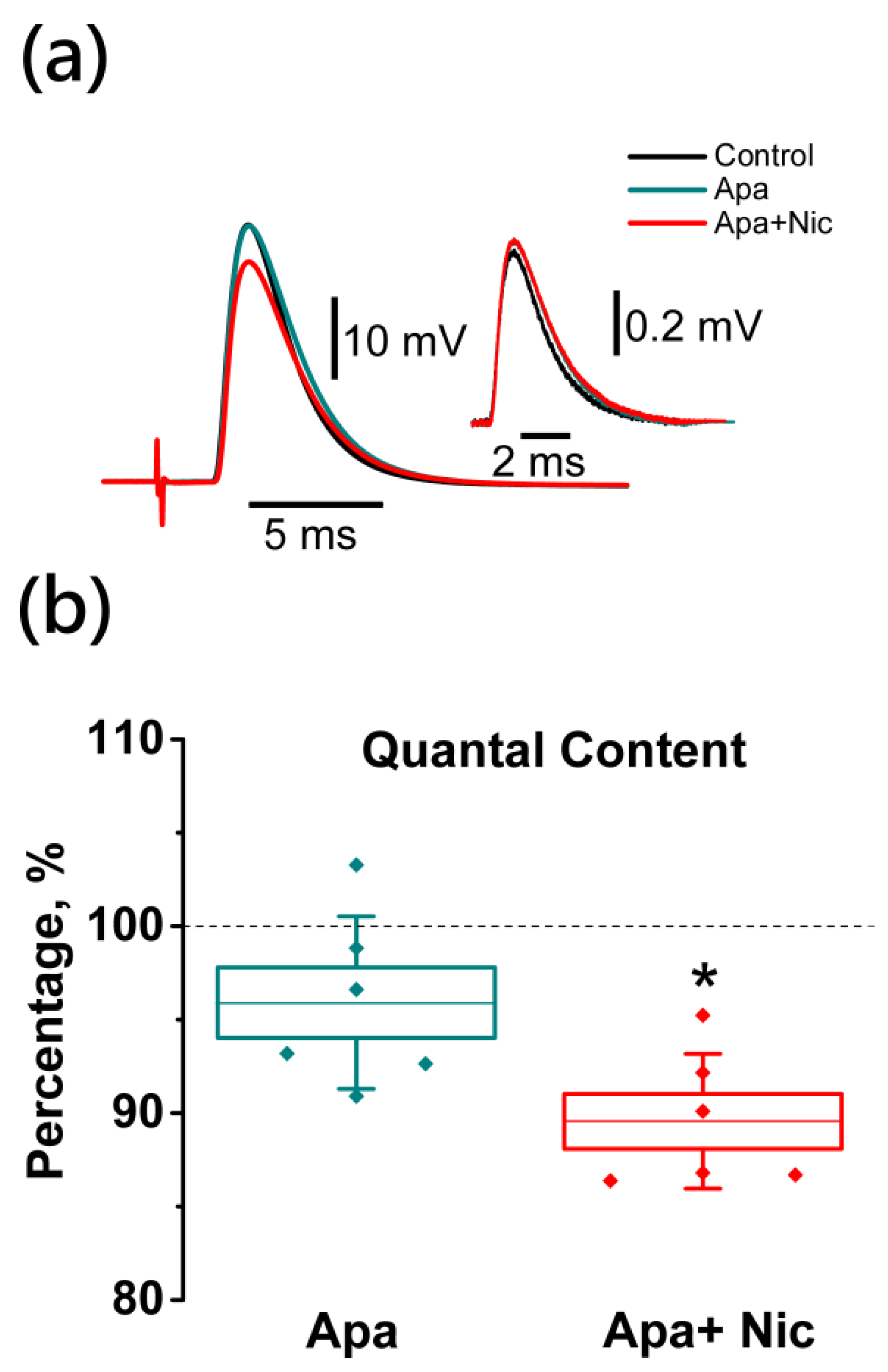
2.5. Nicotine-Induced Decrease in Acetylcholine Release Is Mediated by L-Type (Cav1) Calcium Channels and Not Coupled to Apamin-Sensitive KCa Channels
3. Discussion
3.1. Effects of Nicotine on ACh Release
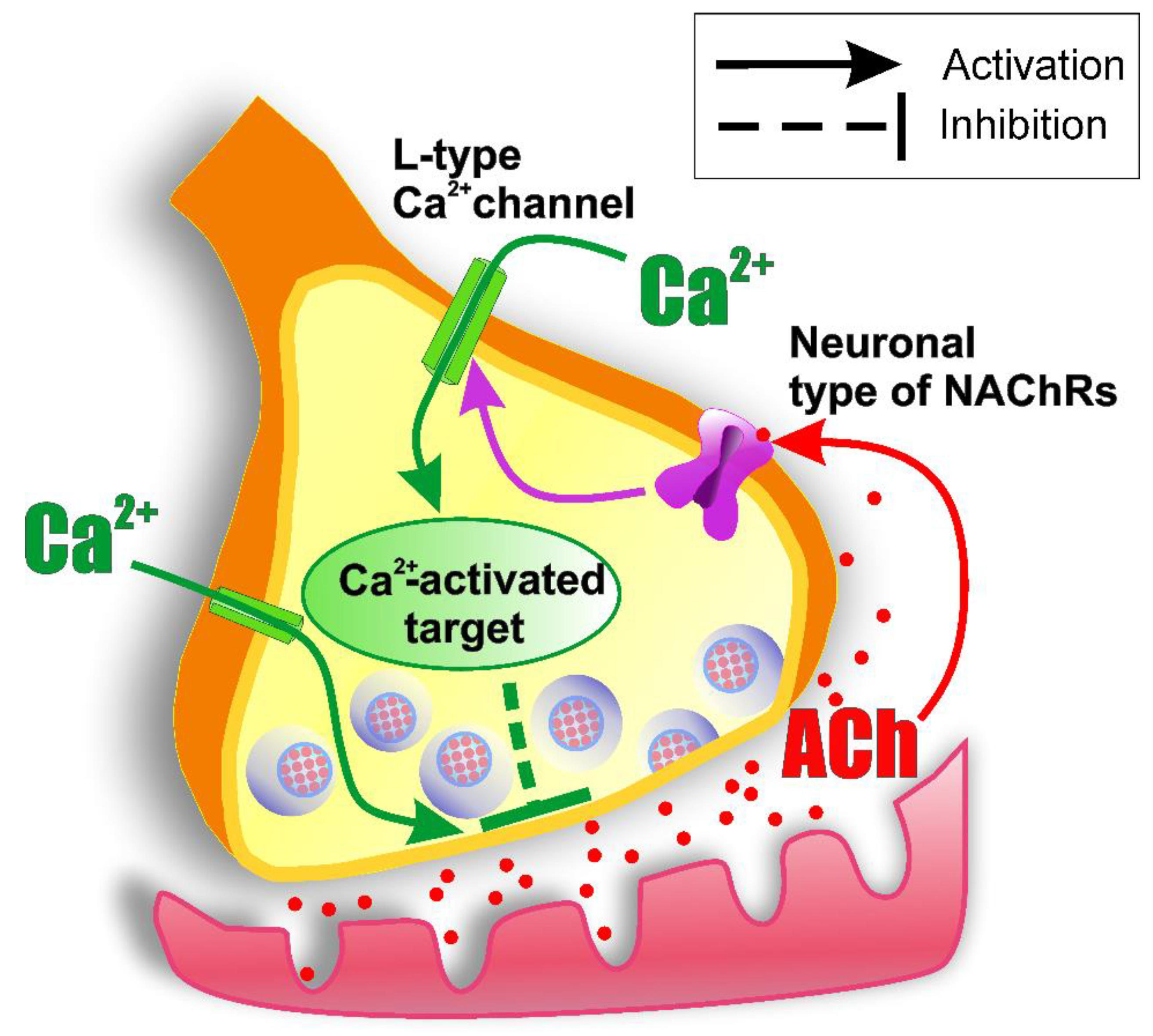
3.2. Presynaptic Cholinergic Receptors and the Role of Calcium Influx in the Mechanism of ACh Release Autoinhibition
3.3. Critical Issues in Establishing the Coupling between nNAChRs and L-Type (Cav1) Calcium Channels while Using a Pharmacological Approach
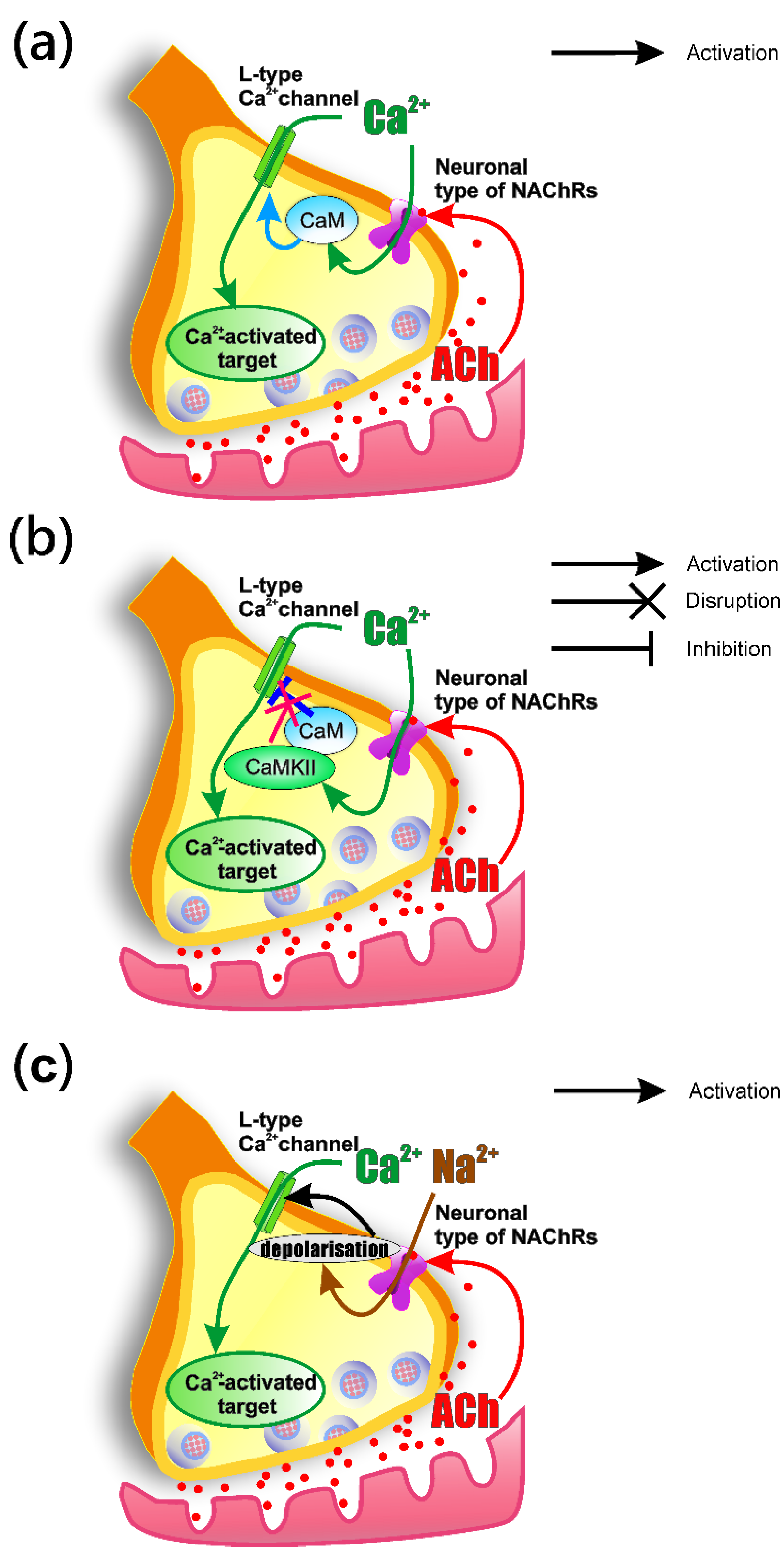
3.4. How the Activation of nNAChRs Modulates L-Type (Cav1) Channel Functioning
4. Materials and Methods
4.1. Animals
4.2. Tissue Preparations and Solutions
4.3. Electrophysiology
4.4. Calcium Transient Recording
4.5. Materials
4.6. Data and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| VGCCs | voltage-gated calcium channels |
| RMP | resting membrane potential |
| mEPPs | miniature endplate potentials |
| EPPs | end plate potentials |
| QC | quantal content |
| nNAChRs | neuronal nicotinic ACh receptors |
| DHβE | dihydro-β-erythroidine hydrobromide |
References
- Del Castillo, J.; Katz, B. Interaction at End-Plate Receptors between Different Choline Derivatives. Proc. R. Soc. Lond. B Biol. Sci. 1957, 146, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Malomouzh, A.I.; Nikolsky, E.E. Modern Concepts of Cholinergic Neurotransmission at the Motor Synapse. Biochem. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 209–222. [Google Scholar] [CrossRef]
- Starke, K.; Göthert, M.; Kilbinger, H. Modulation of Neurotransmitter Release by Presynaptic Autoreceptors. Physiol. Rev. 1989, 69, 864–989. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.C.; Prior, C.; Marshall, I.G. Presynaptic Receptors in the Neuromuscular Junction. Ann. N. Y. Acad. Sci. 1990, 604, 69–81. [Google Scholar] [CrossRef]
- Miller, R.J. Presynaptic Receptors. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 201–227. [Google Scholar] [CrossRef] [Green Version]
- Nikolsky, E.E.; Vyskocil, F.; Bukharaeva, E.A.; Samigullin, D.; Magazanik, L.G. Cholinergic Regulation of the Evoked Quantal Release at Frog Neuromuscular Junction. J. Physiol. 2004, 560, 77–88. [Google Scholar] [CrossRef]
- Prior, C.; Tian, L.; Dempster, J.; Marshall, I.G. Prejunctional Actions of Muscle Relaxants: Synaptic Vesicles and Transmitter Mobilization as Sites of Action. Gen. Pharmacol. 1995, 26, 659–666. [Google Scholar] [CrossRef]
- Santafé, M.M.; Salon, I.; Garcia, N.; Lanuza, M.A.; Uchitel, O.D.; Tomàs, J. Muscarinic Autoreceptors Related with Calcium Channels in the Strong and Weak Inputs at Polyinnervated Developing Rat Neuromuscular Junctions. Neuroscience 2004, 123, 61–73. [Google Scholar] [CrossRef]
- Tsentsevitsky, A.N.; Zakyrjanova, G.F.; Petrov, A.M.; Kovyazina, I.V. Breakdown of Phospholipids and the Elevated Nitric Oxide Are Involved in M3 Muscarinic Regulation of Acetylcholine Secretion in the Frog Motor Synapse. Biochem. Biophys. Res. Commun. 2020, 524, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Tsentsevitsky, A.N.; Kovyazina, I.V.; Nurullin, L.F.; Nikolsky, E.E. Muscarinic Cholinoreceptors (M1-, M2-, M3- and M4-Type) Modulate the Acetylcholine Secretion in the Frog Neuromuscular Junction. Neurosci. Lett. 2017, 649, 62–69. [Google Scholar] [CrossRef]
- Cilleros-Mañé, V.; Just-Borràs, L.; Polishchuk, A.; Durán, M.; Tomàs, M.; Garcia, N.; Tomàs, J.M.; Lanuza, M.A. M 1 and M 2 MAChRs Activate PDK1 and Regulate PKC ΒI and ε and the Exocytotic Apparatus at the NMJ. FASEB J. 2021, 35, e21724. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; Kimura, I.; Dezaki, K.; Kimura, M.; Sala, C.; Fumagalli, G. Immunohistochemical Localization of Neuronal Nicotinic Receptor Subtypes at the Pre- and Postjunctional Sites in Mouse Diaphragm Muscle. Neurosci. Lett. 1995, 196, 13–16. [Google Scholar] [CrossRef]
- Malomouzh, A.I.; Arkhipova, S.S.; Nikolsky, E.E.; Vyskočil, F. Immunocytochemical Demonstration of M1 Muscarinic Acetylcholine Receptors at the Presynaptic and Postsynaptic Membranes of Rat Diaphragm Endplates. Physiol. Res. 2011, 60, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Santafé, M.M.; Salon, I.; Garcia, N.; Lanuza, M.A.; Uchitel, O.D.; Tomàs, J. Modulation of ACh Release by Presynaptic Muscarinic Autoreceptors in the Neuromuscular Junction of the Newborn and Adult Rat. Eur. J. Neurosci. 2003, 17, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Timóteo, M.A.; Correia-de-Sá, P. Modulation by Adenosine of Both Muscarinic M1-Facilitation and M2-Inhibition of [3H]-Acetylcholine Release from the Rat Motor Nerve Terminals. Eur. J. Neurosci. 2002, 15, 1728–1736. [Google Scholar] [CrossRef]
- Zhilyakov, N.V.; Khaziev, E.F.; Latfullin, A.R.; Malomouzh, A.I.; Bukharaeva, E.A.; Nikolsky, E.E.; Samigullin, D.V. Changes in Calcium Levels in Motor Nerve Endings in Mice on Activation of Metabotropic Cholinoreceptors and GABA Receptors. Neurosci. Behav. Physiol. 2019, 49, 1092–1095. [Google Scholar] [CrossRef]
- Khaziev, E.; Samigullin, D.; Zhilyakov, N.; Fatikhov, N.; Bukharaeva, E.; Verkhratsky, A.; Nikolsky, E. Acetylcholine-Induced Inhibition of Presynaptic Calcium Signals and Transmitter Release in the Frog Neuromuscular Junction. Front. Physiol. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slutsky, I.; Wess, J.; Gomeza, J.; Dudel, J.; Parnas, I.; Parnas, H. Use of Knockout Mice Reveals Involvement of M2-Muscarinic Receptors in Control of the Kinetics of Acetylcholine Release. J. Neurophysiol. 2003, 89, 1954–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kloot, W. Nicotinic Agonists Antagonize Quantal Size Increases and Evoked Release at Frog Neuromuscular Junction. J. Physiol. 1993, 468, 567–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, C.; Singh, S. Factors Influencing the Low-Frequency Associated Nicotinic ACh Autoreceptor-Mediated Depression of ACh Release from Rat Motor Nerve Terminals. Br. J. Pharmacol. 2000, 129, 1067–1074. [Google Scholar] [CrossRef]
- Balezina, O.P.; Fedorin, V.V.; Gaidukov, A.E. Effect of Nicotine on Neuromuscular Transmission in Mouse Motor Synapses. Bull. Exp. Biol. Med. 2006, 142, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Khaziev, E.F.; Samigullin, D.V.; Tsentsevitsky, A.N.; Bukharaeva, E.A.; Nikolsky, E.E. ATP Reduces the Entry of Calcium Ions into the Nerve Ending by Blocking L-Type Calcium Channels. Acta Naturae 2018, 10, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Atchison, W.D. Dihydropyridine-Sensitive and -Insensitive Components of Acetylcholine Release from Rat Motor Nerve Terminals. J. Pharmacol. Exp. Ther. 1989, 251, 672–678. [Google Scholar] [PubMed]
- Protti, D.A.; Szczupak, L.; Scornik, F.S.; Uchitel, O.D. Effect of ω-Conotoxin GVIA on Neurotransmitter Release at the Mouse Neuromuscular Junction. Brain Res. 1991, 557, 336–339. [Google Scholar] [CrossRef]
- Penner, R.; Dreyer, F. Two Different Presynaptic Calcium Currents in Mouse Motor Nerve Terminals. Pflügers Arch. Eur. J. Physiol. 1986, 406, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Bowersox, S.S.; Miljanich, G.P.; Sugiura, Y.; Li, C.; Nadasdi, L.; Hoffman, B.B.; Ramachandran, J.; Ko, C.P. Differential Blockade of Voltage-Sensitive Calcium Channels at the Mouse Neuromuscular Junction by Novel ω-Conopeptides and ω-Agatoxin-IVA. J. Pharmacol. Exp. Ther. 1995, 273, 248–256. [Google Scholar] [PubMed]
- Moroni, M.; Zwart, R.; Sher, E.; Cassels, B.K.; Bermudez, I. A4β2 Nicotinic Receptors with High and Low Acetylcholine Sensitivity: Pharmacology, Stoichiometry, and Sensitivity to Long-Term Exposure to Nicotine. Mol. Pharmacol. 2006, 70, 755–768. [Google Scholar] [CrossRef]
- Wonnacott, S. Nicotinic ACh Receptors. Available online: https://www.tocris.com/literature/scientific-reviews/nicotinic-ach-receptors (accessed on 27 August 2021).
- Katz, B. The Release of Neural Transmitter Substances; Liverpool University Press: Liverpool, UK, 1969; pp. 5–39. [Google Scholar]
- Crawford, A.C. The Dependence of Evoked Transmitter Release on External Calcium Ions at Very Low Mean Quantal Contents. J. Physiol. 1974, 240, 255–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protti, D.A.; Uchitel, O.D. Transmitter Release and Presynaptic Ca2+ Currents Blocked by the Spider Toxin ω-Aga-IVA. Neuroreport 1993, 5, 333–336. [Google Scholar] [CrossRef]
- Tian, L.; Prior, C.; Dempster, J.; Marshall, I.G. Nicotinic Antagonist-produced Frequency-dependent Changes in Acetylcholine Release from Rat Motor Nerve Terminals. J. Physiol. 1994, 476, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez-Noriega, L.E.; Gillespie, A.; Stauderman, K.A.; Crona, J.H.; Claeps, B.O.; Elliott, K.J.; Reid, R.T.; Rao, T.S.; Veliçelebi, G.; Harpold, M.M.; et al. Characterization of the Recombinant Human Neuronal Nicotinic Acetylcholine Receptors A3β2 and A4β2 Stably Expressed in HEK293 Cells. Neuropharmacology 2000, 39, 2543–2560. [Google Scholar] [CrossRef]
- Papke, R.L.; Wecker, L.; Stitzel, J.A. Activation and Inhibition of Mouse Muscle and Neuronal Nicotinic Acetylcholine Receptors Expressed in Xenopus Oocytes. J. Pharmacol. Exp. Ther. 2010, 333, 501–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karadsheh, M.S.; Shah, M.S.; Tang, X.; Macdonald, R.L.; Stitzel, J.A. Functional Characterization of Mouse A4β2 Nicotinic Acetylcholine Receptors Stably Expressed in HEK293T Cells. J. Neurochem. 2004, 91, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michael McIntosh, J.; Rich, M.M. Muscle Nicotinic Acetylcholine Receptors May Mediate Trans-Synaptic Signaling at the Mouse Neuromuscular Junction. J. Neurosci. 2018, 38, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Vyskocil, F.; Malomouzh, A.; Nikolsky, E. Non-Quantal Acetylcholine Release at the Neuromuscular Junction. Physiol. Res. 2009, 58, 763–784. [Google Scholar] [CrossRef]
- Malomouzh, A.; Mukhtarov, M.; Nikolsky, E.; Vyskočil, F. Muscarinic M1 Acetylcholine Receptors Regulate the Non-Quantal Release of Acetylcholine in the Rat Neuromuscular Junction via NO-Dependent Mechanism. J. Neurochem. 2007, 102, 2110–2117. [Google Scholar] [CrossRef]
- Hess, P.; Lansman, J.B.; Tsien, R.W. Different Modes of Ca Channel Gating Behaviour Favoured by Dihydropyridine Ca Agonists and Antagonists. Nature 1984, 311, 538–544. [Google Scholar] [CrossRef]
- Radford Deckera, E.; Dani, J.A. Calcium Permeability of the Nicotinic Acetylcholine Receptor: The Single-Channel Calcium Influx Is Significant. J. Neurosci. 1990, 10, 3413–3420. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F. Neuronal Nicotinic Receptors: From Structure to Pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.G.; Barrett, C.F.; Tsien, R.W. L-Type Calcium Channel Ligands Block Nicotine-Induced Signaling to CREB by Inhibiting Nicotinic Receptors. Neuropharmacology 2006, 51, 27–36. [Google Scholar] [CrossRef]
- Lansman, J.B.; Hess, P.; Tsien, R.W. Blockade of Current through Single Calcium Channels by Cd2+, Mg2+, and Ca2+s: Voltage and Concentration Dependence of Calcium Entry into the Pore. J. Gen. Physiol. 1986, 88, 321–347. [Google Scholar] [CrossRef] [Green Version]
- Samigullin, D.V.; Khaziev, E.F.; Zhilyakov, N.V.; Sudakov, I.A.; Bukharaeva, E.A.; Nikolsky, E.E. Calcium Transient Registration in Response to Single Stimulation and during Train of Pulses in Mouse Neuromuscular Junction. Bionanoscience 2017, 7, 162–166. [Google Scholar] [CrossRef]
- Nachshen, D.A.; Blaustein, M.P. The Effects of Some Organic “Calcium Antagonists” on Calcium Influx in Presynaptic Nerve Terminals. Mol. Pharmacol. 1979, 16, 576–586. [Google Scholar] [PubMed]
- Katz, E.; Ferro, P.A.; Weisz, G.; Uchitel, O.D. Calcium Channels Involved in Synaptic Transmission at the Mature and Regenerating Mouse Neuromuscular Junction. J. Physiol. 1996, 497, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Pagani, R.; Song, M.; Mcenery, M.; Qin, N.; Tsien, R.W.; Toro, L.; Stefani, E.; Uchitel, O.D. Differential Expression of A1 and β Subunits of Voltage Dependent Ca2+ Channel at the Neuromuscular Junction of Normal and P/Q Ca2+ Channel Knockout Mouse. Neuroscience 2004, 123, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Perissinotti, P.P.; Tropper, B.G.; Uchitel, O.D. L-Type Calcium Channels Are Involved in Fast Endocytosis at the Mouse Neuromuscular Junction. Eur. J. Neurosci. 2008, 27, 1333–1344. [Google Scholar] [CrossRef]
- Urbano, F.J.; Rosato-Siri, M.D.; Uchitel, O.D. Calcium Channels Involved in Neurotransmitter Release at Adult, Neonatal and P/Q-Type Deficient Neuromuscular Junctions. Mol. Membr. Biol. 2002, 19, 293–300. [Google Scholar] [CrossRef]
- Houlihan, L.M.; Slater, E.Y.; Beadle, D.J.; Lukas, R.J.; Bermudez, I. Effects of Diltiazem on Human Nicotinic Acetylcholine and GABA(A) Receptors. Neuropharmacology 2000, 39, 2533–2542. [Google Scholar] [CrossRef]
- Garduño, J.; Galindo-Charles, L.; Jiménez-Rodríguez, J.; Galarraga, E.; Tapia, D.; Mihailescu, S.; Hernandez-Lopez, S. Presynaptic A4β2 Nicotinic Acetylcholine Receptors Increase Glutamate Release and Serotonin Neuron Excitability in the Dorsal Raphe Nucleus. J. Neurosci. 2012, 32, 15148–15157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Rumpf, C.H.; Fujiwara, Y.; Cooley, E.S.; Van Petegem, F.; Minor, D.L. Structures of CaV2 Ca2+/CaM-IQ Domain Complexes Reveal Binding Modes That Underlie Calcium-Dependent Inactivation and Facilitation. Structure 2008, 16, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Abiria, S.A.; Colbran, R.J. CaMKII Associates with Ca V 1.2 L-Type Calcium Channels via Selected β Subunits to Enhance Regulatory Phosphorylation. J. Neurochem. 2010, 112, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsura, M.; Mohri, Y.; Shuto, K.; Hai-Du, Y.; Amano, T.; Tsujimura, A.; Sasa, M.; Ohkuma, S. Up-Regulation of L-Type Voltage-Dependent Calcium Channels after Long Term Exposure to Nicotine in Cerebral Cortical Neurons. J. Biol. Chem. 2002, 277, 7979–7988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo-Parada, L.; Bose, C.M.; Landmesser, L.T. Alterations in Transmission, Vesicle Dynamics, and Transmitter Release Machinery at NCAM-Deficient Neuromuscular Junctions. Neuron 2001, 32, 815–828. [Google Scholar] [CrossRef] [Green Version]
- De Lorenzo, S.; Veggetti, M.; Muchnik, S.; Losavio, A. Presynaptic Inhibition of Spontaneous Acetylcholine Release Mediated by P2Y Receptors at the Mouse Neuromuscular Junction. Neuroscience 2006, 142, 71–85. [Google Scholar] [CrossRef]
- Tarasova, E.; Gaydukov, A.E.; Balezina, O.P. Calcineurin and Its Role in Synaptic Transmission. Biochemistry 2018, 83, 674–689. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The Arrive Guidelines 2.0: Updated Guidelines for Reporting Animal Research. PLoS Biol. 2020, 18, 1769–1777. [Google Scholar] [CrossRef]
- Lilley, E.; Stanford, S.C.; Kendall, D.E.; Alexander, S.P.H.; Cirino, G.; Docherty, J.R.; George, C.H.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. ARRIVE 2.0 and the British Journal of Pharmacology: Updated Guidance for 2020. Br. J. Pharmacol. 2020, 177, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Angaut-Petit, D.; Molgo, J.; Connold, A.L.; Faille, L. The Levator Auris Longus Muscle of the Mouse: A Convenient Preparation for Studies of Short- and Long-Term Presynaptic Effects of Drugs or Toxins. Neurosci. Lett. 1987, 82, 83–88. [Google Scholar] [CrossRef]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Three-Dimensional Solution Structure of μ-Conotoxin GIIIB, a Specific Blocker of Skeletal Muscle Sodium Channels. Biochemistry 1996, 35, 8824–8835. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, S. A Study of the Interaction between Neuromuscular Blocking Agents and Acetylcholine at the Mammalian Motor End-Plate. Acta Anaesthesiol. Scand. 1958, 2, 69–79. [Google Scholar] [CrossRef]
- Samigullin, D.V.; Khaziev, E.F.; Zhilyakov, N.V.; Bukharaeva, E.A.; Nikolsky, E.E. Loading a Calcium Dye into Frog Nerve Endings through the Nerve Stump: Calcium Transient Registration in the Frog Neuromuscular Junction. J. Vis. Exp. 2017, 125, e55122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental Design and Analysis and Their Reporting II: Updated and Simplified Guidance for Authors and Peer Reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, P.; Cheeta, S.; Tucci, S.; File, S.E. Nicotinic–Serotonergic Interactions in Brain and Behaviour. Pharmacol. Biochem. Behav. 2002, 71, 795–805. [Google Scholar] [CrossRef]
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2003; Volume 147, pp. 1–46. [Google Scholar]
- Picciotto, M.R. Nicotine as a Modulator of Behavior: Beyond the Inverted U. Trends Pharmacol. Sci. 2003, 24, 493–499. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhilyakov, N.; Arkhipov, A.; Malomouzh, A.; Samigullin, D. Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels. Int. J. Mol. Sci. 2021, 22, 9031. https://doi.org/10.3390/ijms22169031
Zhilyakov N, Arkhipov A, Malomouzh A, Samigullin D. Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels. International Journal of Molecular Sciences. 2021; 22(16):9031. https://doi.org/10.3390/ijms22169031
Chicago/Turabian StyleZhilyakov, Nikita, Arsenii Arkhipov, Artem Malomouzh, and Dmitry Samigullin. 2021. "Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels" International Journal of Molecular Sciences 22, no. 16: 9031. https://doi.org/10.3390/ijms22169031
APA StyleZhilyakov, N., Arkhipov, A., Malomouzh, A., & Samigullin, D. (2021). Activation of Neuronal Nicotinic Receptors Inhibits Acetylcholine Release in the Neuromuscular Junction by Increasing Ca2+ Flux through Cav1 Channels. International Journal of Molecular Sciences, 22(16), 9031. https://doi.org/10.3390/ijms22169031