
GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
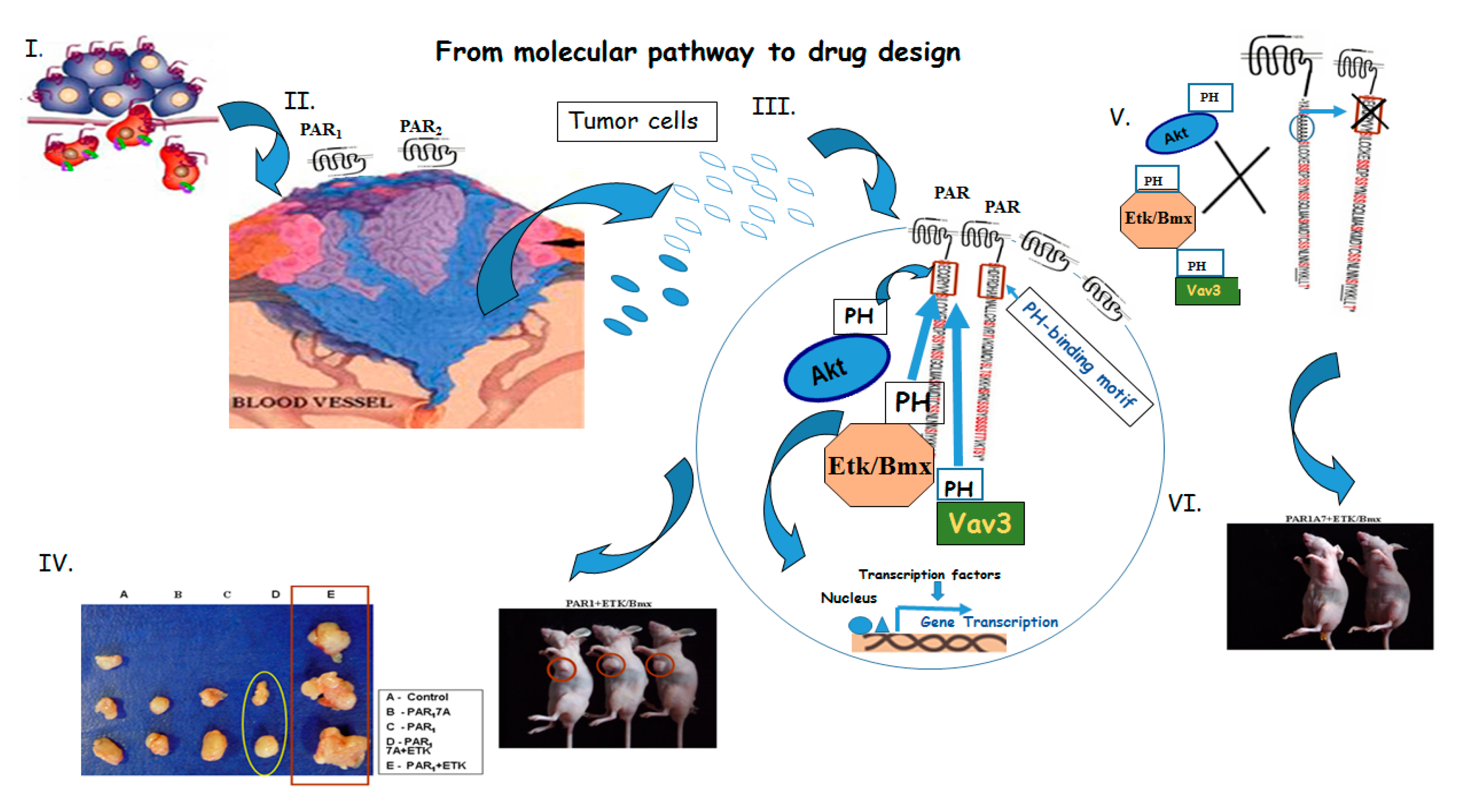
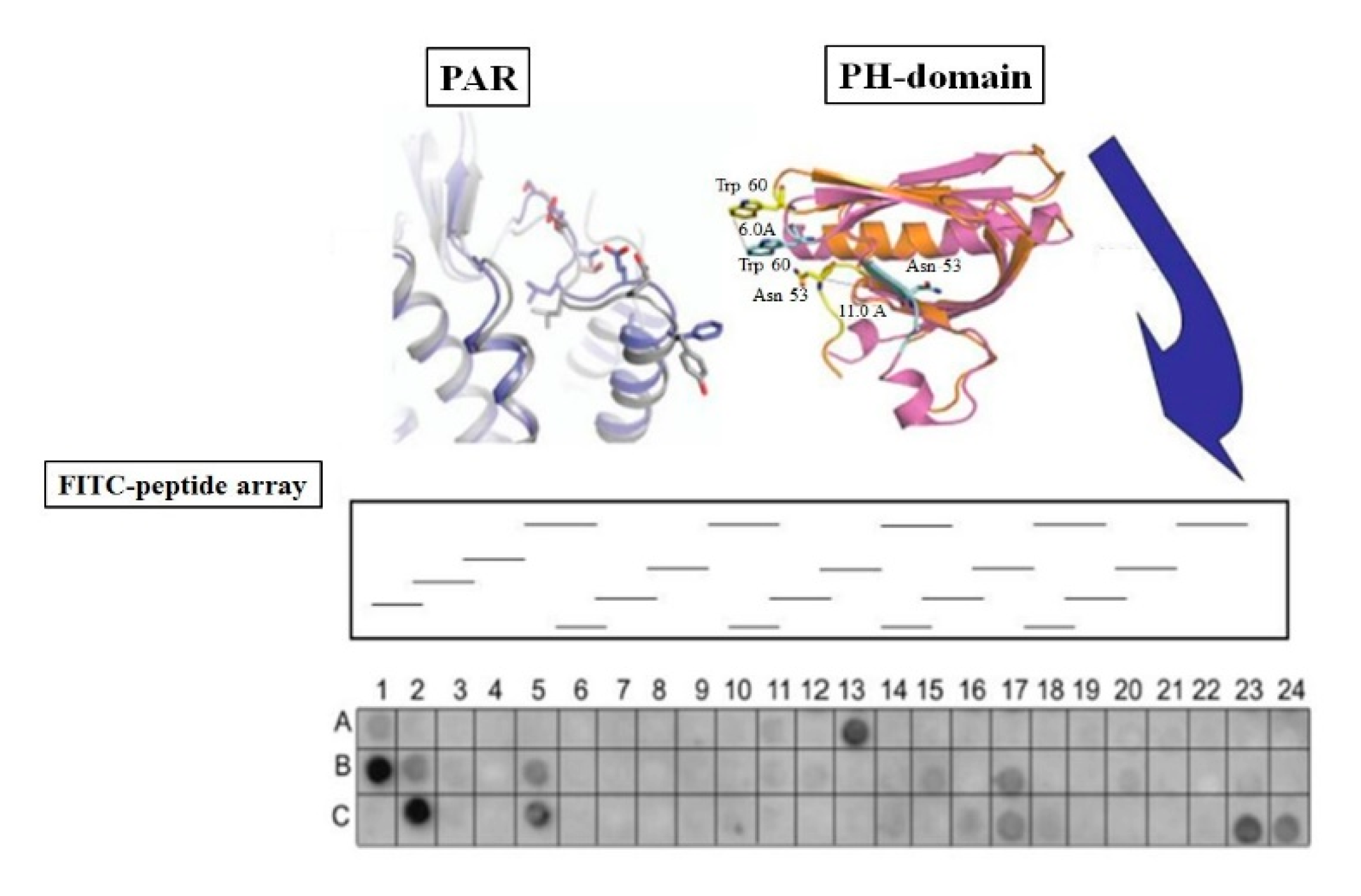
1.1. PAR1&2 Associate with PH-Signal Proteins via ‘Recognition Motifs’ That Endow Junctional Accesses to Cancer Growth
1.2. Tumor Growth, In Vivo
1.3. Placenta-EVT Invasion Is Enhanced via PAR2-PH Binding Domain
1.4. PAR2 Is Dominant over PAR1
1.5. A Lead Cyclic Peptide Directed toward the PAR2 PH-Binding Domain
1.6. Identification of Candidates PH-Domain Binding Motifs in a GPCR Array
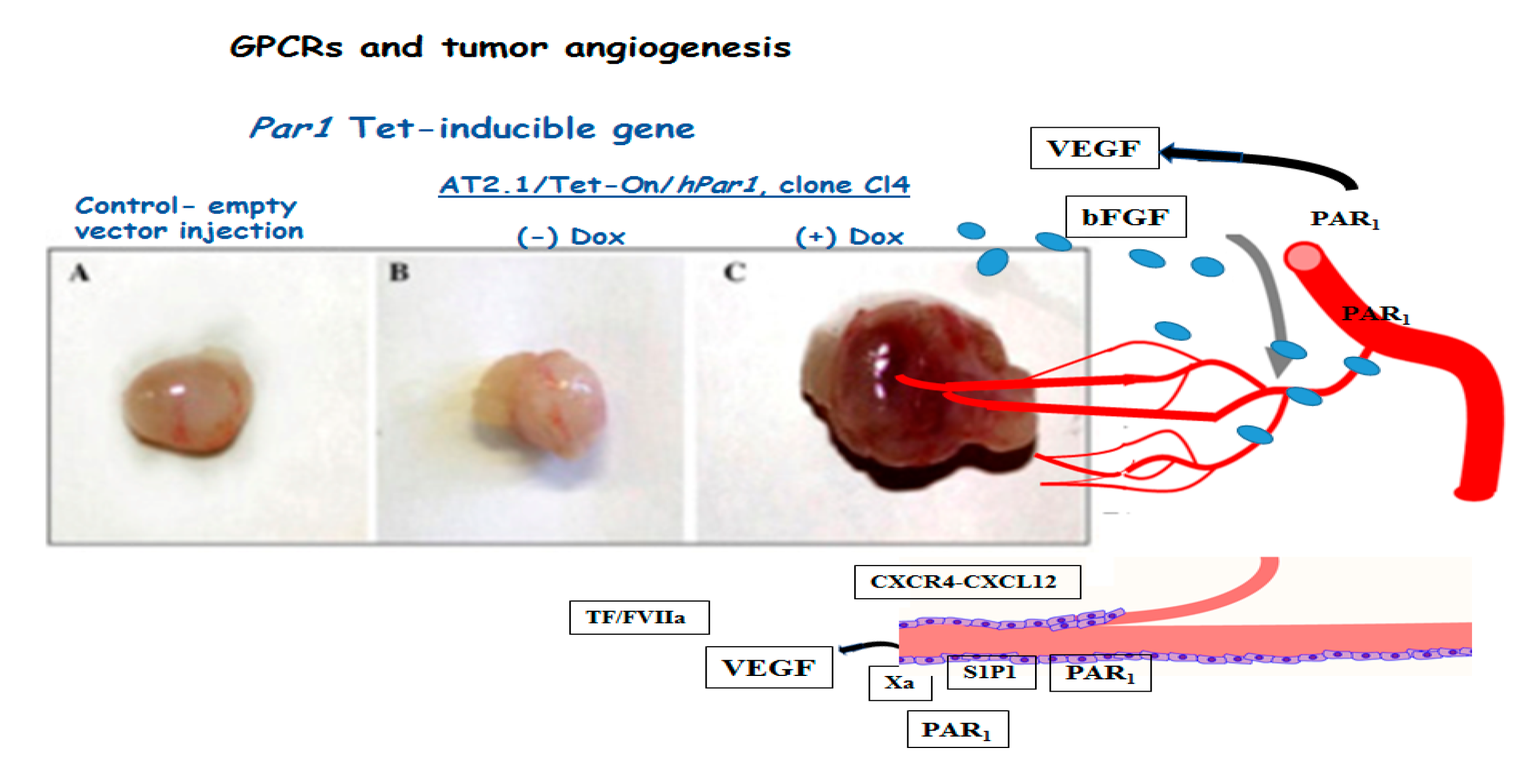
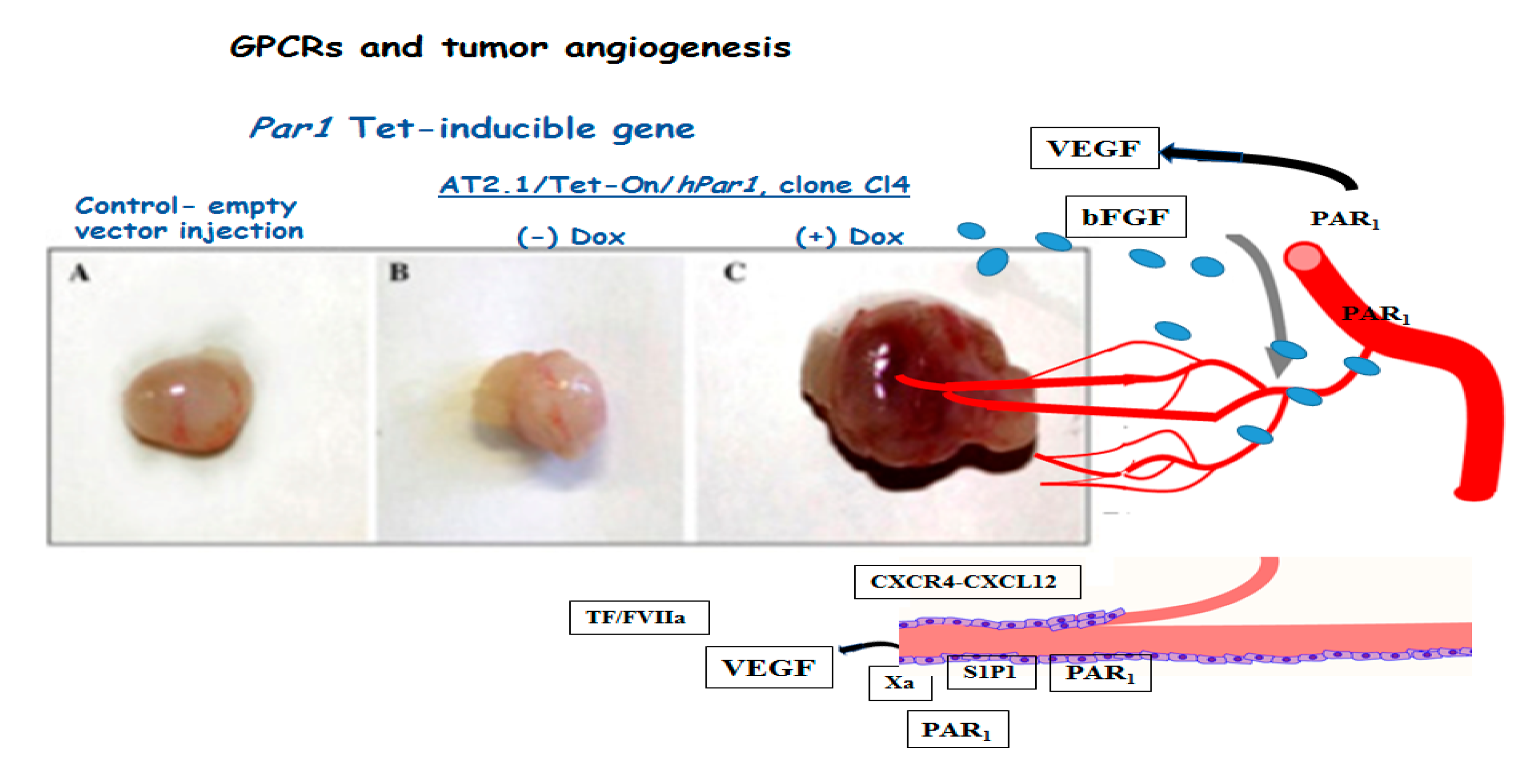
1.7. Tumor Angiogenesis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GPCRs | G-protein coupled receptors |
| PARs | protease-activated receptors |
| Znrf3 | zinc and ring finger 3 |
| Rnf43 | ring finger protein 43 |
| FZD | Frizzled |
| PH | domain-pleckstrin homology domain |
| FAK | focal adhesion kinase |
| Gab1-Grb2 | associated binder 1 |
| Sos | Son of Sevenless |
| EVT | extravillous trophoblasts |
| VEGF | vascular endothelial growth factor |
| bFGF | basic Fibroblast Growth Factor |
| mESC | mouse embryonic stem cell |
| S1P1 | Sphingosine-1-phosphate receptor 1 |
References
- López-Otín, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef]
- Dudani, J.S.; Warren, A.D.; Bhatia, S.N. Harnessing Protease Activity to Improve Cancer Care. Annu. Rev. Cancer Biol. 2017, 23, 353–376. [Google Scholar] [CrossRef]
- Hoeller, D.; Hecker, C.M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.-K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Gutkind, J.S.; Kostenis, E. Arrestins as rheostats of GPCR signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 615–616. [Google Scholar] [CrossRef]
- Roy, A.; Ansari, S.A.; Das, K.; Prasad, R.; Bhattacharya, A.; Mallik, S.; Mukherjee, A.; Sen, P. Coagulation factor VIIa-mediated protease-activated receptor 2 activation leads to β-catenin accumulation via the AKT/GSK3β pathway and contributes to breast cancer progression. J. Biol. Chem. 2017, 292, 13688–13701. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.R.; Ding, Y.; Ricks, T.K.; Gullapalli, A.; Wolfe, B.L.; Trejo, J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006, 66, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nierodzik, M.L.; Karpatkin, S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006, 10, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Even-Ram, S.; Uziely, B.; Cohen, P.; Grisaru-Granovsky, S.; Maoz, M.; Ginzburg, Y.; Reich, R.; Vlodavsky, I.; Bar-Shavit, R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nat. Med. 1998, 4, 909–914. [Google Scholar] [CrossRef]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerman, P.S.; Hayes, D.N.; Wilkerson, M.D.; Schultz, N.; Bose, R.; Chu, A.; Collisson, E.A.; Cope, L.; Creighton, C.J.; Getz, G.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- De Carvalho, D.D.; Sharma, S.; You, J.S.; Su, S.F.; Taberlay, P.C.; Kelly, T.K.; Yang, X.; Liang, G.; Jones, P.A. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012, 21, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G-proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M. Signal-dependent membrane targeting bypleckstrin homology (PH) domains. Biochem. J. 2000, 350, 1–18. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT-a major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Kancharla, A.; Maoz, M.; Jaber, M.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. PH motifs in PAR1&2 endow breast cancer growth. Nat. Commun. 2015, 6, 8853–8864. [Google Scholar]
- Chen, R.; Kim, O.; Li, M.; Xiong, X.; Guan, J.L.; Kung, H.J.; Chen, H.; Shimizu, Y.; Qiu, Y. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 2001, 3, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2004, 2, 395–402. [Google Scholar] [PubMed]
- Salah, Z.; Maoz, M.; Cohen, I.; Pizov, G.; Pode, D.; Runge, M.S.; Bar-Shavit, R. Identification of a novel functional androgen response element within hPar1 promoter: Implications to prostate cancer progression. FASEB J. 2005, 19, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, L.M.; Austin, K.M.; Zhang, P.; Kasuda, S.; Koukos, G.; Sharifi, S.; Covic, L.; Kuliopulos, A. Protease-activated receptor-2 modulates protease-activated receptor-1-driven neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e100–e106. [Google Scholar] [CrossRef] [Green Version]
- Jaber, M.; Maoz, M.; Kancharla, A.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. Protease-activated-receptor-2 affects protease-activatedreceptor-1-driven breast cancer. Cell Mol. Life Sci. 2014, 71, 2517–2533. [Google Scholar] [CrossRef]
- Modlin, I.M.; Pavel, M.; Kidd, M.; Gustafsson, B.I. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrin (carcinoid) tumours. Aliment. Pharmacol. Ther. 2010, 31, 169–188. [Google Scholar]
- Jain, S.; Zain, J. Romidepsin in the treatment of cutaneous T-cell lymphoma. J. Blood Med. 2011, 2, 37–47. [Google Scholar]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Javaherian, K.; Lo, K.M.; Folkman, J.; Hanahan, D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Alitalo, K. Clinical applications of angiogenic growth factors and their inhibitors. Nat. Med. 1999, 5, 1359–1364. [Google Scholar] [CrossRef]
- Wong, P.-P.; Demircioglu, F.; Ghazaly, E.; Alrawashdeh, W.; Stratford, M.R.L.; Scudamore, C.L.; Cereser, B.; Crnogorac, J.T.; McDonald, S.; Elia, G.; et al. Dual-Action Combination Therapy Enhances Angiogenesis while Reducing Tumor Growth and Spread. Cancer Cell 2015, 27, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, R.O.; Bader, B.L.; Hodivala-Dilke, K. Integrins in vascular development. Braz. J. Med. Biol. Res. 1999, 32, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Pattern and emerging mechanisms of angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S.; Mancino, V.; Revel, J.P.; Simon, M.I. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 1997, 275, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Vouret-Craviari, V.; Pouysségur, J. Angiogenesis and G-protein-coupled receptors: Signals that bridge the gap. Oncogene 2001, 20, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Clarke, R.B.; Lisanti, M.P.; Maggiolini, M.G. Protein-Coupled Receptors at the Crossroad between Physiologic and Pathologic Angiogenesis: Old Paradigms and Emerging Concepts. Int. J. Mol. Sci. 2017, 18, 2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.; Demory, A.; Peyre, F.; Kroll, J.; Augustin, H.G.; Helfrich, W.; Kzhyshkowska, J.; Schledzewski, K.; Arnold, B.; Goerdt, S. Wnt2 acts as a cell type-specific, autocrine growth factor in rat hepatic sinusoidal endothelial cells cross-stimulating the VEGF pathway. Hepatology 2008, 47, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Barcelos, L.S.; Duplaa, C.; Kränkel, N.; Graiani, G.; Invernici, G.; Katare, R.; Siragusa, M.; Meloni, M.; Campesi, I.; Monica, M.; et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ. Res. 2009, 104, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Allende, M.L.; Yamashita, T.; Proia, R.L. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [Google Scholar] [CrossRef]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.J.; Mao, X.B.; Ren, L.L.; Chum, X.Y. Inhibition of CXCL12/CXCR4 axis as a potential targeted therapy of advanced gastric carcinoma. Cancer Med. 2017, 6, 1424–1436. [Google Scholar] [CrossRef]
- Chen, Q.; Zhong, T. The association of CXCR4 expression with clinicopathological significance and potential drug target in prostate cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 5115–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupertus, K.; Sinistra, J.; Scheuer, C.; Nickels, R.M.; Schilling, M.K.; Menger, M.D.; Kollmar, O. Interaction of the chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the regulation of tumor angiogenesis of colorectal cancer. Clin. Exp. Metastasis 2014, 31, 447–459. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.; Chen, J.H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Salah, Z.; Maoz, M.; Even Ram, S.C.; Ochayon, S.; Neufeld, G.; Katzav, S.; Bar-Shavit, R. Oncogenic transformation induces tumor angiogenesis: A role for PAR1 activation. FASEB J. 2005, 17, 163–174. [Google Scholar] [CrossRef]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Van Brocklyn, J.R.; Hobson, J.P.; Movafagh, S.; Zukowska-Grojec, Z.; Milstien, S.; Spiegel, S. Sphingosine 1-phosphate stimulates cell migration through a G(i)-coupled cell surface receptor. Potential involvement in angiogenesis. J. Biol. Chem. 1999, 274, 35343–35350. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; An, S.; Sassmann, A.; Liu, M.; Mastej, V.; Mittal, M.; Zhang, W.; Hong, Z.; Offermanns, S.; Rehman, J.; et al. PAR1 Scaffolds TGFbRII to Downregulate TGF-b Signaling and Activate ESC Differentiation to Endothelial Cells. Stem Cell Rep. 2016, 7, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Griffin, C.T.; Srinivasan, Y.; Zheng, Y.W.; Huang, W.; Coughlin, S.R. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science 2001, 293, 1666–1670. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Kim, H.Y.; Song, I.-C.; Yun, H.-J.; Jo, D.-Y.; Kim, S.; Lee, H.J. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol. Rep. 2012, 28, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.; Cohen, K.S.; Klein, R.J.; Scadden, D.T.; Chiocca, E.A. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006, 66, 9054–9064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.Y.; Soloviev, I.; Chang, P.; Lee, J.; Huang, X.D.; Zhong, C.; Ferrara, N.; Polakis, P.; Sakanaka, C. Stromal cell-derived factor-1/CXCL12 contributes to MMTV-Wnt1 tumor growth involving Gr1+CD11b+ cells. PLoS ONE 2010, 5, e8611. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nag, J.K.; Malka, H.; Appasamy, P.; Sedley, S.; Bar-Shavit, R. GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network. Int. J. Mol. Sci. 2021, 22, 8985. https://doi.org/10.3390/ijms22168985
Nag JK, Malka H, Appasamy P, Sedley S, Bar-Shavit R. GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network. International Journal of Molecular Sciences. 2021; 22(16):8985. https://doi.org/10.3390/ijms22168985
Chicago/Turabian StyleNag, Jeetendra Kumar, Hodaya Malka, Priyanga Appasamy, Shoshana Sedley, and Rachel Bar-Shavit. 2021. "GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network" International Journal of Molecular Sciences 22, no. 16: 8985. https://doi.org/10.3390/ijms22168985
APA StyleNag, J. K., Malka, H., Appasamy, P., Sedley, S., & Bar-Shavit, R. (2021). GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network. International Journal of Molecular Sciences, 22(16), 8985. https://doi.org/10.3390/ijms22168985