
A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis

 , and
, and
Abstract
:1. Introduction
2. Results
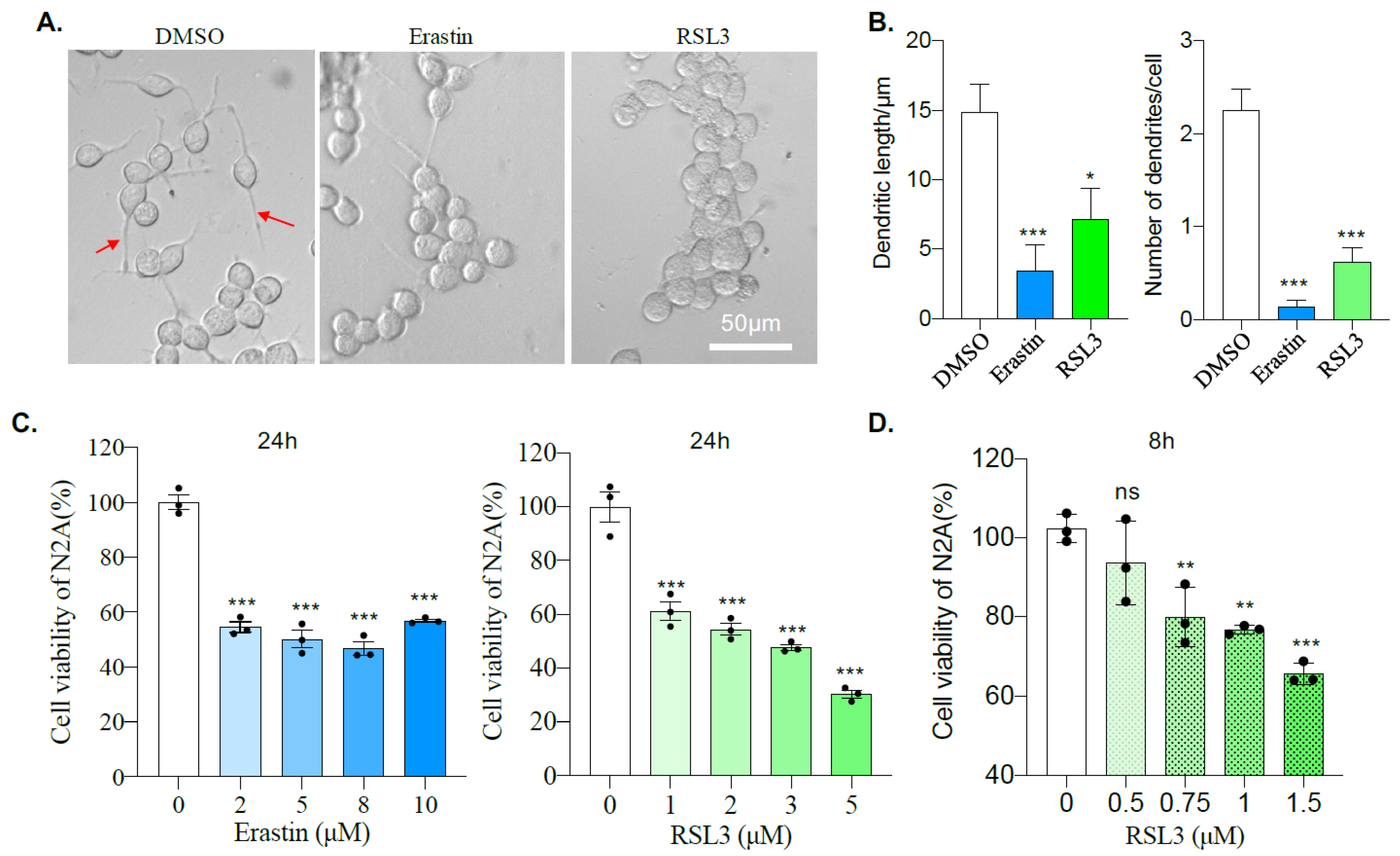
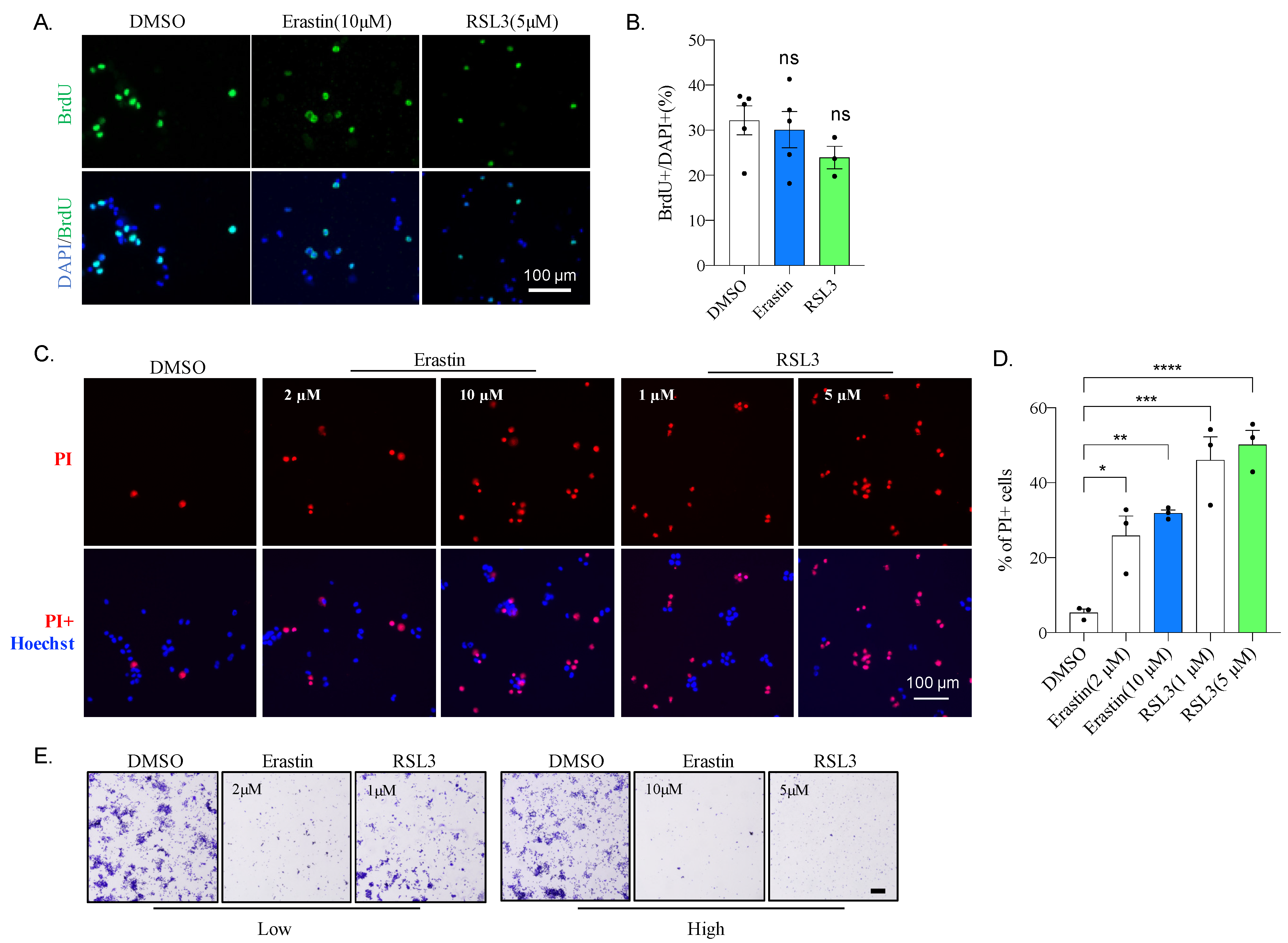
2.1. Mouse Neuroblastoma N2A Cells Are Sensitive to Erastin and RSL3
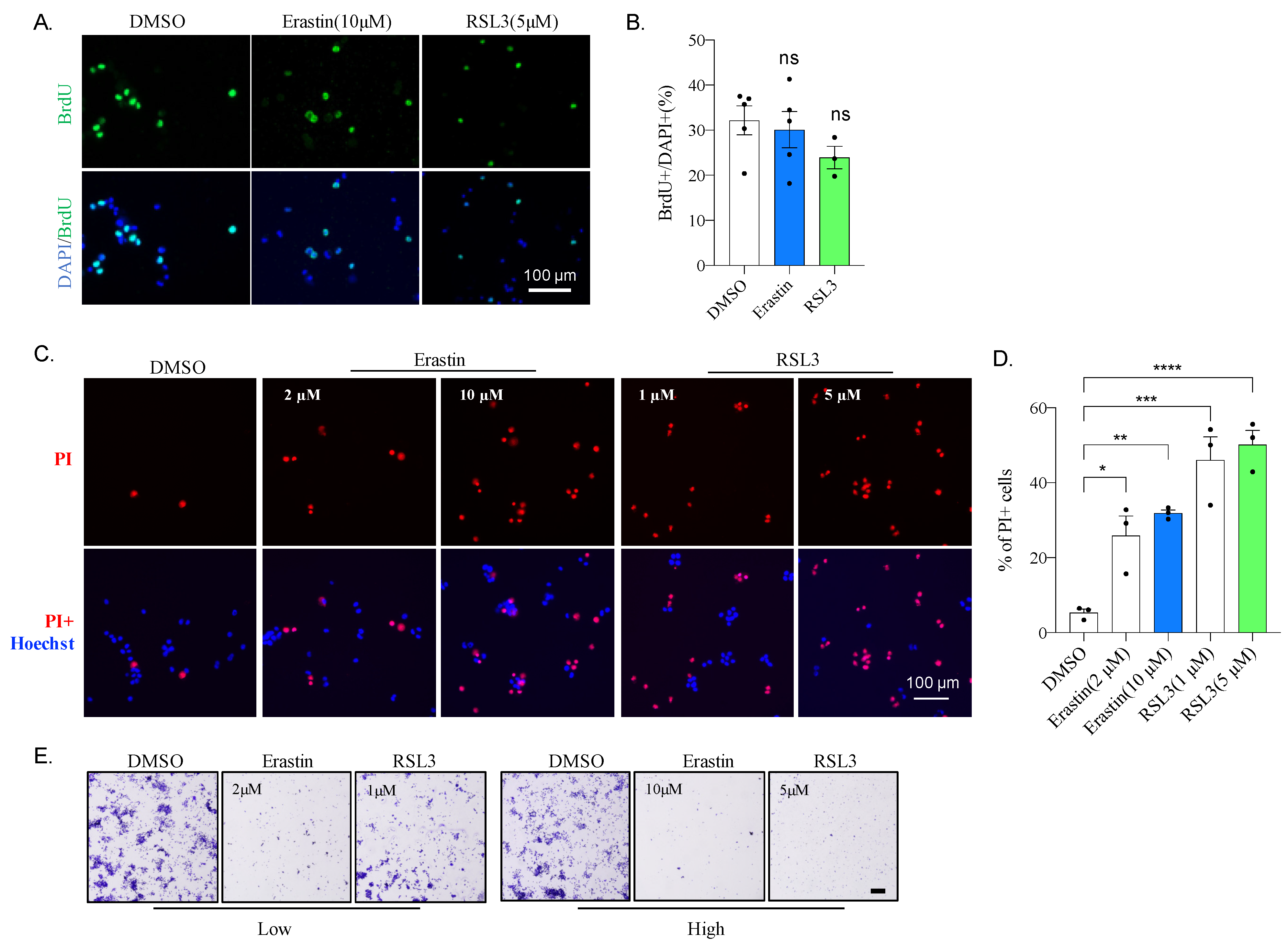
2.2. Erastin or RSL3 Treatment Leads to Cell Death without Affecting Proliferation
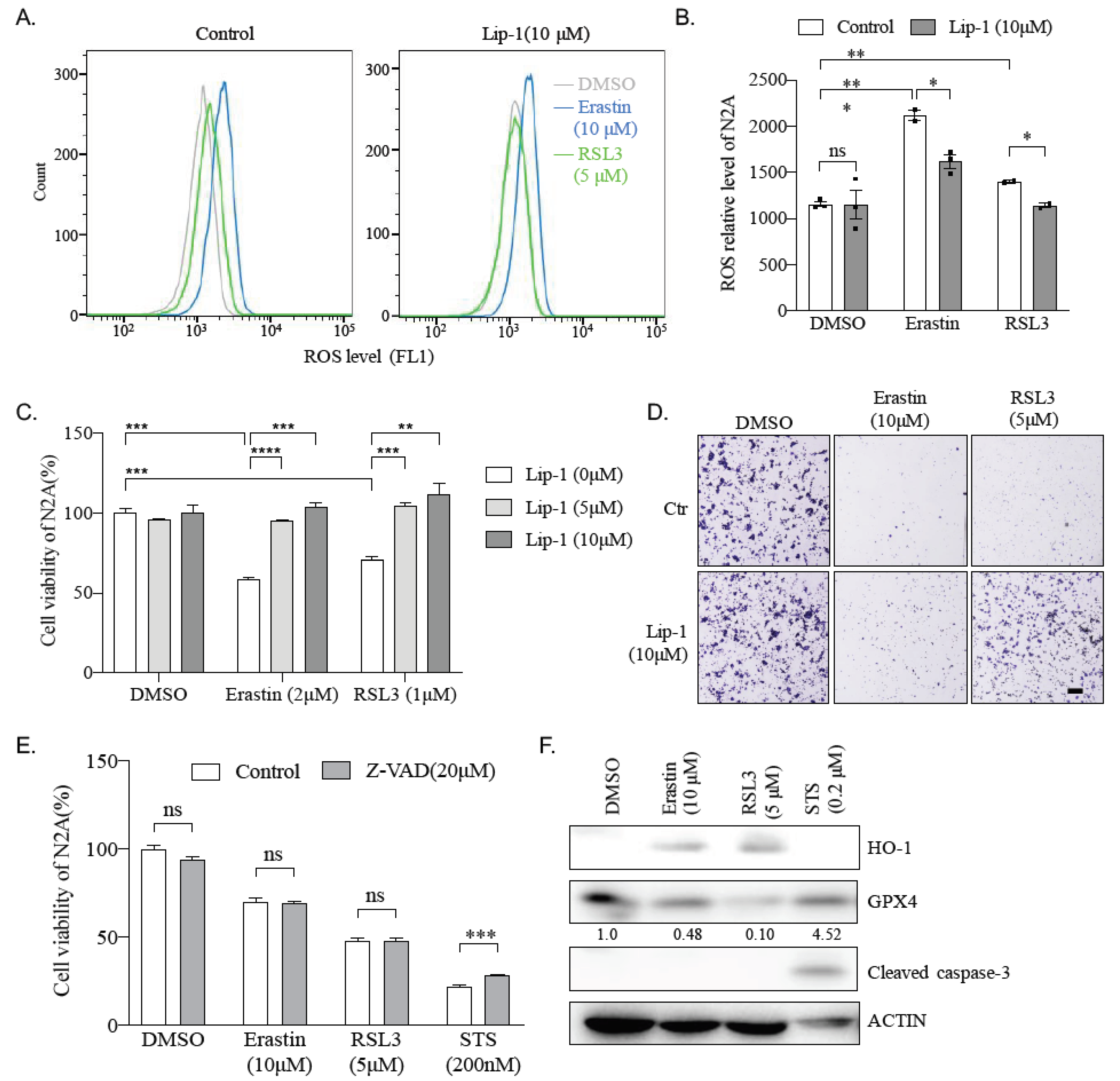
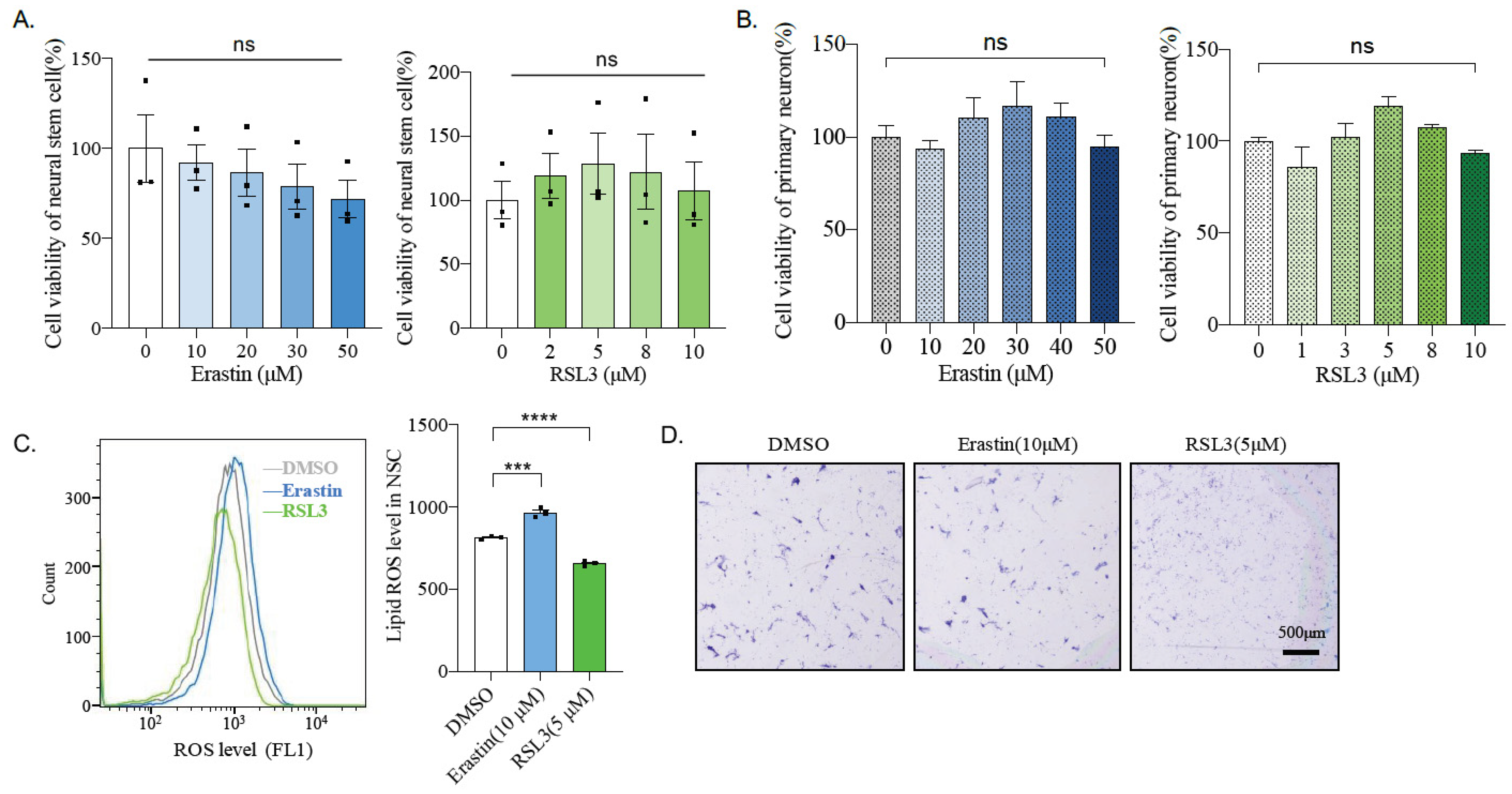
2.3. Erastin and RSL3 Induce ROS
2.4. Ferroptosis Inhibitor Liproxstatin-1 Blocks Effect Caused by Erastin and RSL3 in N2A Cells
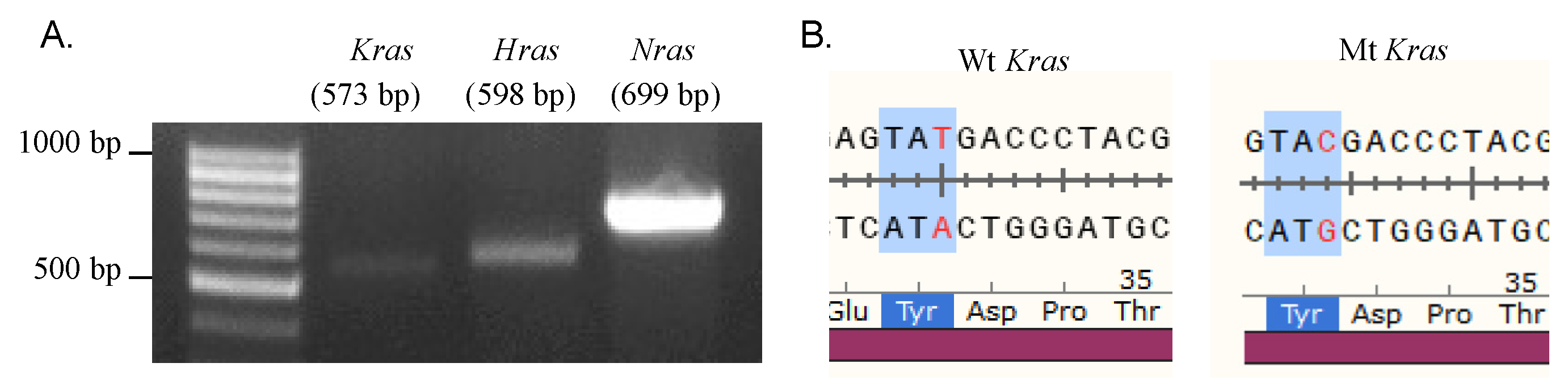
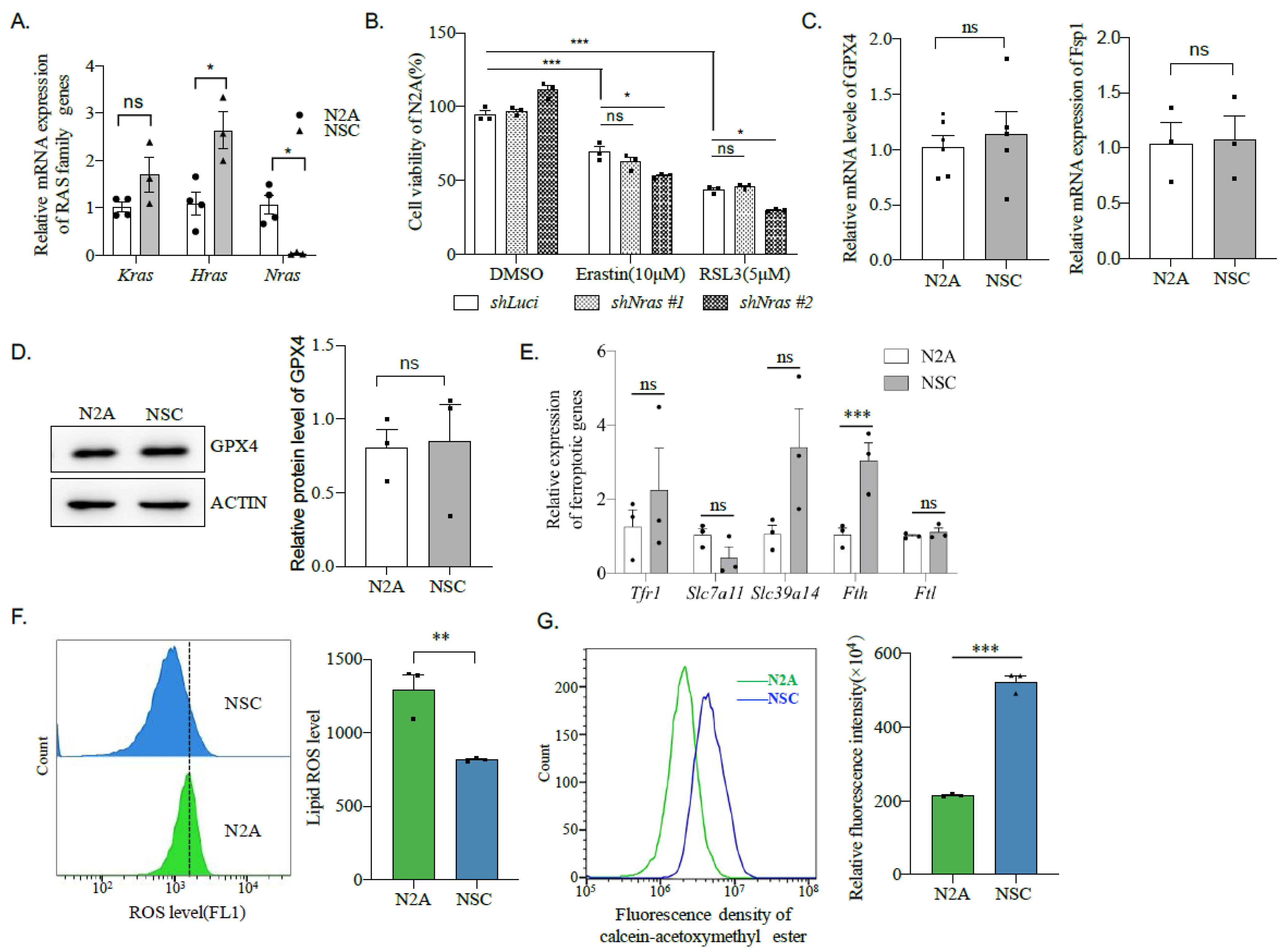
2.5. Erastin and RSL3 Induce Ferroptosis in N2A Cells Independent of RAS Mutations
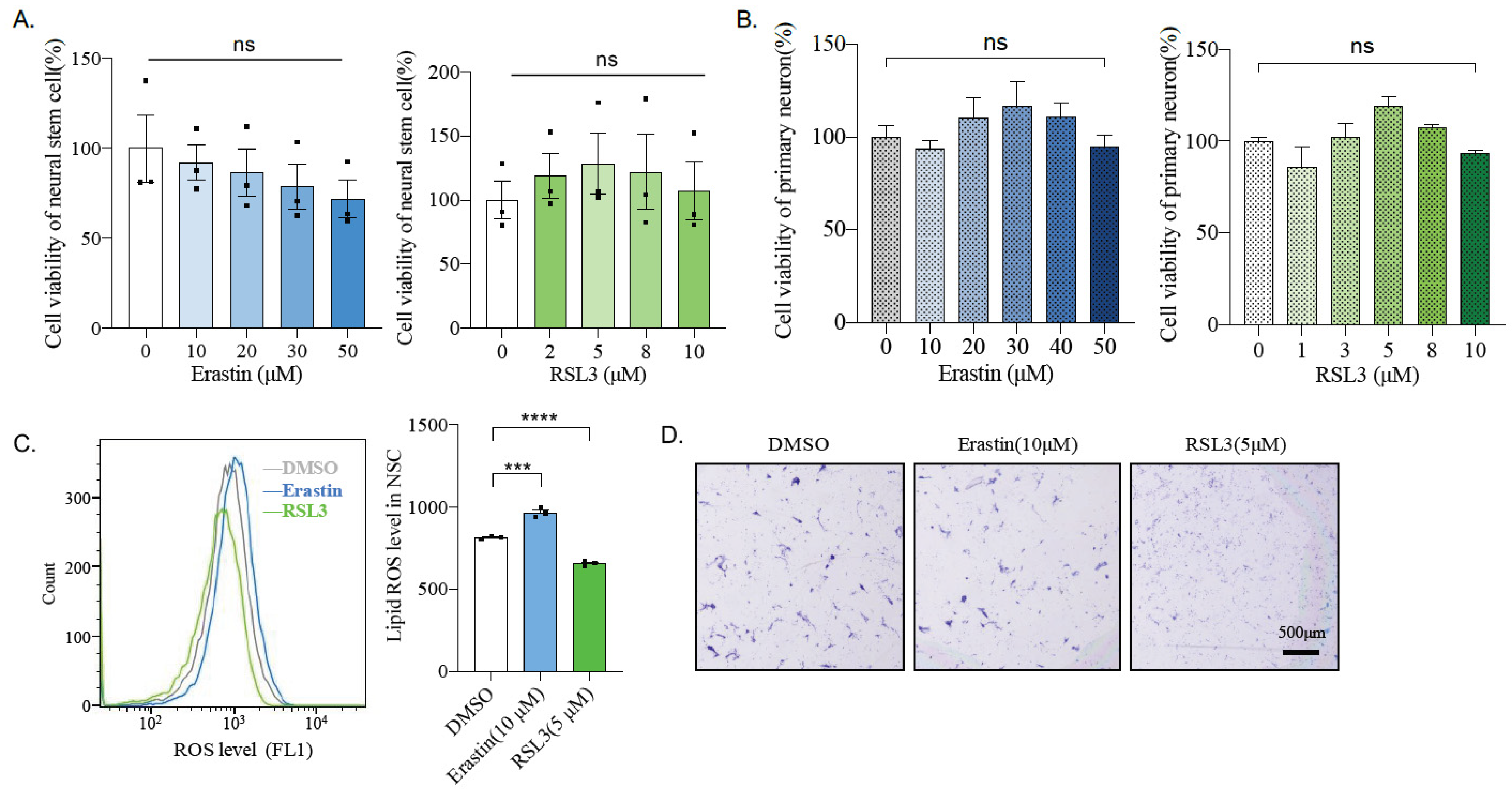
2.6. Mouse Primary Neural Stem Cells and Neurons Are Insensitive to Ferroptosis Inducers
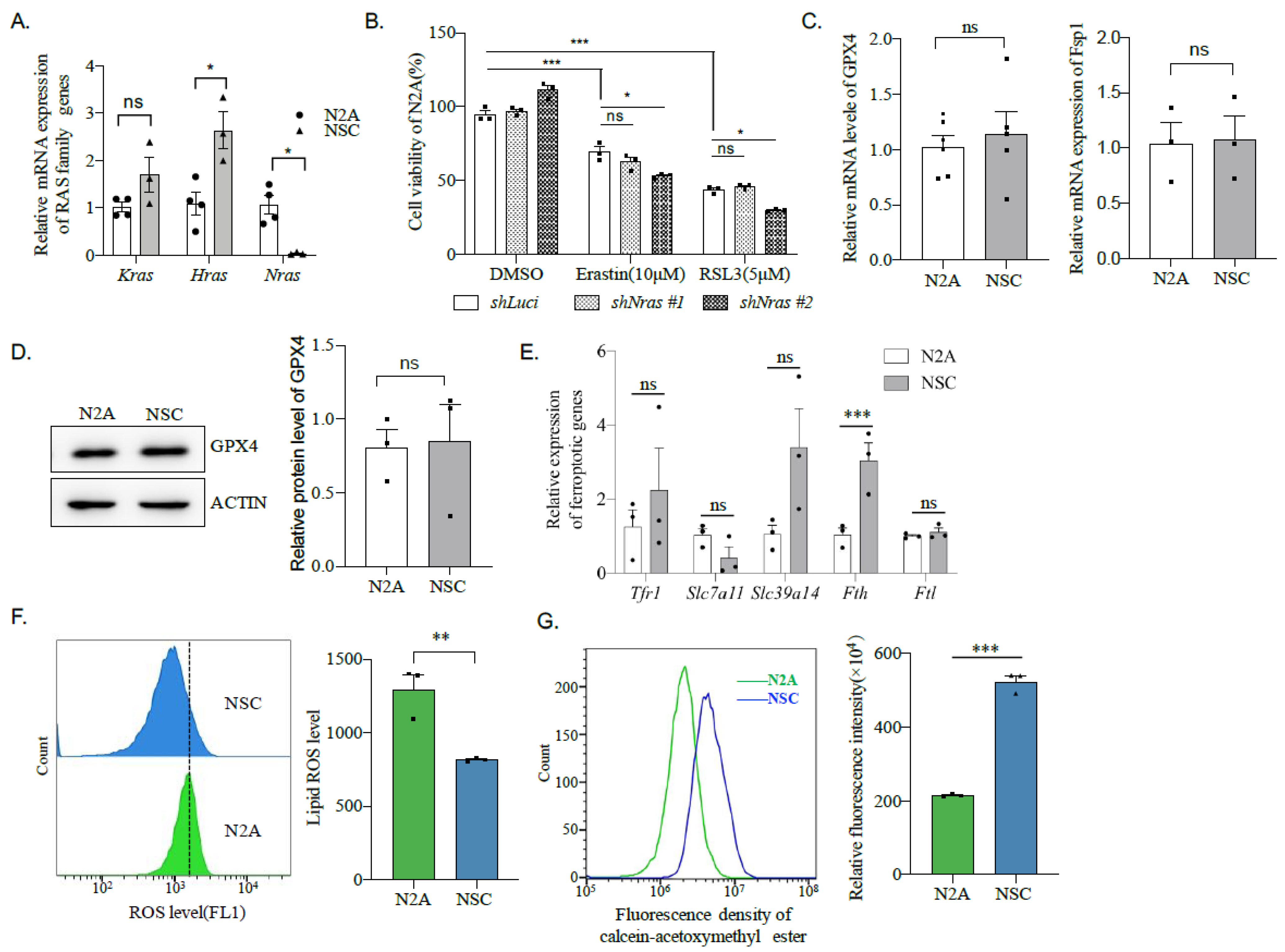
2.7. N2A Cells Express a Lower Amount of Fth Compared to Neural Stem Cells
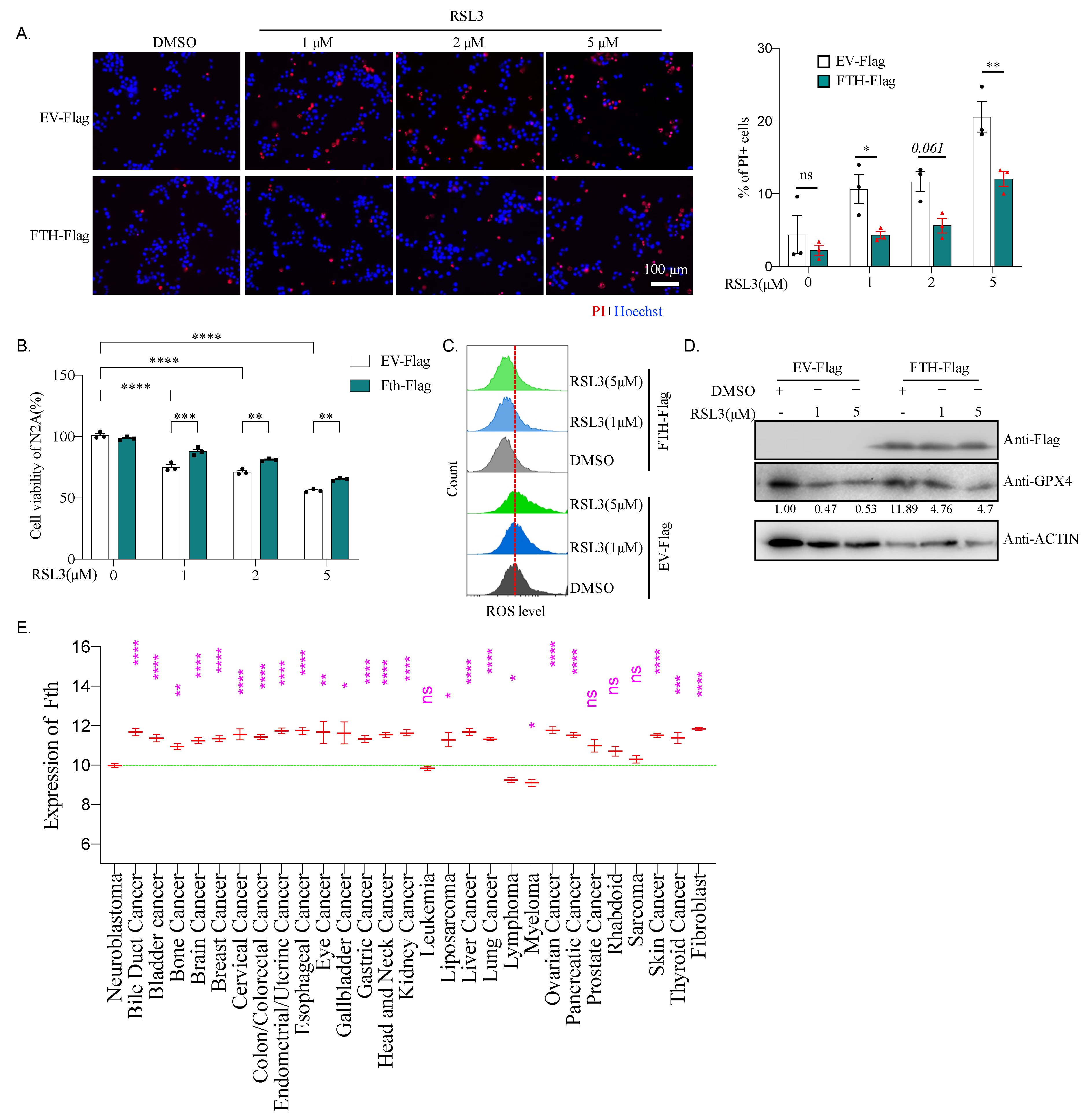
2.8. Fth May Be Responsible for the Sensitivity of Neuroblastoma to Ferroptosis
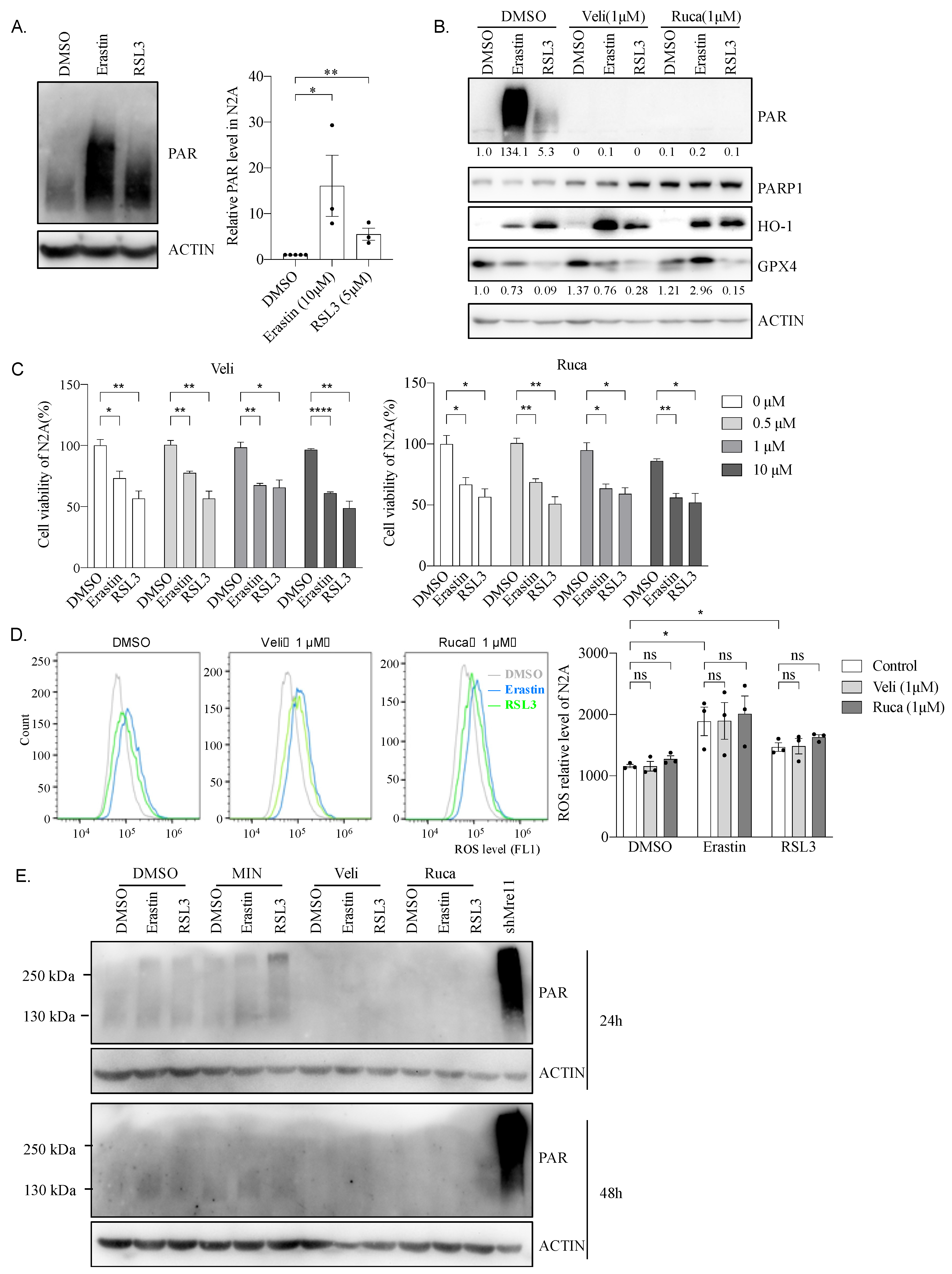
2.9. Hyper PARP Activity Induced by Erastin or RSL3 Is Independent of N2A Cell Viability
3. Discussion
4. Materials and Methods
4.1. The Mice
4.2. Plasmid Construction
4.3. Cell Culture
4.4. Cell Viability Measurement (CCK-8 Assay)
4.5. BrdU Labeling and Staining
4.6. Crystal Violet Staining
4.7. Propidium Iodide (PI) Staining
4.8. Western Blotting
4.9. Lipid ROS Detection
4.10. Measurement of Labile Iron Pool (LIP)
4.11. RNA Isolation and PCR Analysis
4.12. Analysis of Relative Expression of Fth, Gpx4, and Fsp1 in Different Cancer Cell Lines
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer. Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Wang, W.; Liu, G.; Wang, X.; Xian, W.; McKeon, F.; Foster, J.; Zhou, J.; Zhang, R. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med. Res. Rev. 2021, 41, 961–1021. [Google Scholar] [CrossRef]
- Zhai, K.; Brockmuller, A.; Kubatka, P.; Shakibaei, M.; Busselberg, D. Curcumin’s Beneficial Effects on Neuroblastoma: Mechanisms, Challenges, and Potential Solutions. Biomolecules 2020, 10, 1469. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856. [Google Scholar] [CrossRef]
- Cartel, M.; Mouchel, P.L.; Gotanegre, M.; David, L.; Bertoli, S.; Mansat-De, M.V.; Besson, A.; Sarry, J.E.; Manenti, S.; Didier, C. Inhibition of ubiquitin-specific protease 7 sensitizes acute myeloid leukemia to chemotherapy. Leukemia 2021, 35, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int. J. Oncol. 2018, 52, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural Trans. 2011, 118, 301–314. [Google Scholar] [CrossRef]
- Thomas, G.; Leyland, L.A.; Schrag, A.E.; Lees, A.J.; Acosta-Cabronero, J.; Weil, R.S. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 418–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuss, J.; Jung, M.; Brune, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [Green Version]
- Blankenhaus, B.; Braza, F.; Martins, R.; Bastos-Amador, P.; Gonzalez-Garcia, I.; Carlos, A.R.; Mahu, I.; Faisca, P.; Nunes, J.M.; Ventura, P.; et al. Ferritin regulates organismal energy balance and thermogenesis. Mol. Metab. 2019, 24, 64–79. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da, S.M.; Ingold, I.; Goya, G.A.; Xavier, D.S.T.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Gene Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedűs, C.; Virág, L. Inputs and outputs of poly(ADP-ribosyl)ation: Relevance to oxidative stress. Redox Biol. 2014, 2, 978–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariani, K.; Ryu, D.; Menzies, K.; Yi, H.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly-ADP ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2016, 66, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czapski, G.A.; Cieślik, M.; Wencel, P.L.; Wójtowicz, S.; Strosznajder, R.P.; Strosznajder, J.B. Inhibition of poly(ADP-ribose) polymerase-1 alters expression of mitochondria-related genes in PC12 cells: Relevance to mitochondrial homeostasis in neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, X.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radical. Biol. Med. 2014, 67, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354. [Google Scholar] [CrossRef] [Green Version]
- Hadian, K.; Stockwell, B.R. SnapShot: Ferroptosis. Cell 2020, 181, 1188. [Google Scholar] [CrossRef] [PubMed]
- Schuhwerk, H.; Atteya, R.; Siniuk, K.; Wang, Z.Q. PARPing for balance in the homeostasis of poly(ADP-ribosyl)ation. Semin. Cell Dev. Biol. 2017, 63, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021, 42, 101928. [Google Scholar] [CrossRef]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, T.; Wang, H.; Li, Q.; Cheng, Y.; Gao, Y.; Fang, X.; Ma, X.; Chen, G.; Gao, C.; Gu, Z.; et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 2021, 70, e12704. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [Green Version]
- Moroz, V.; Machin, D.; Hero, B.; Ladenstein, R.; Berthold, F.; Kao, P.; Obeng, Y.; Pearson, A.; Cohn, S.L.; London, W.B. The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr. Blood Cancer 2020, 67, e28359. [Google Scholar] [CrossRef]
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Garringer, H.J.; Goodwin, C.B.; Richine, B.; Acton, A.; VanDuyn, N.; Muhoberac, B.B.; Irimia-Dominguez, J.; Chan, R.J.; Peacock, M.; et al. Systemic and cerebral iron homeostasis in ferritin knock-out mice. PLoS ONE 2015, 10, e117435. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Yen, J.; Kou, M.; Wu, M. Luteolin and Apigenin Attenuate 4-Hydroxy-2-Nonenal-Mediated Cell Death through Modulation of UPR, Nrf2-ARE and MAPK Pathways in PC12 Cells. PLoS ONE 2015, 10, e130599. [Google Scholar] [CrossRef] [Green Version]
- Canli, O.; Alankus, Y.B.; Grootjans, S.; Vegi, N.; Hultner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gani, O.A.; Engh, R.A. Protein kinase inhibition of clinically important staurosporine analogues. Nat. Prod. Rep. 2010, 27, 489–498. [Google Scholar] [CrossRef]
- Jantas, D.; Szymanska, M.; Budziszewska, B.; Lason, W. An involvement of BDNF and PI3-K/Akt in the anti-apoptotic effect of memantine on staurosporine-evoked cell death in primary cortical neurons. Apoptosis 2009, 14, 900–912. [Google Scholar] [CrossRef]
- Zhou, Z.; Kirtay, M.; Schneble, N.; Yakoub, G.; Ding, M.; Rüdiger, T.; Siniuk, K.; Lu, R.; Jiang, Y.; Li, T.; et al. NBS1 interacts with Notch signaling in neuronal homeostasis. Nucleic Acids Res. 2020, 48, 10924–10939. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequencing Results |
|---|---|
| Kras | Synonymous mutations, c.T96>C |
| Hras | No mutation |
| Nras | No mutation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, R.; Jiang, Y.; Lai, X.; Liu, S.; Sun, L.; Zhou, Z.-W. A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis. Int. J. Mol. Sci. 2021, 22, 8898. https://doi.org/10.3390/ijms22168898
Lu R, Jiang Y, Lai X, Liu S, Sun L, Zhou Z-W. A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis. International Journal of Molecular Sciences. 2021; 22(16):8898. https://doi.org/10.3390/ijms22168898
Chicago/Turabian StyleLu, Ruiqing, Yinan Jiang, Xianxin Lai, Shujie Liu, Litao Sun, and Zhong-Wei Zhou. 2021. "A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis" International Journal of Molecular Sciences 22, no. 16: 8898. https://doi.org/10.3390/ijms22168898
APA StyleLu, R., Jiang, Y., Lai, X., Liu, S., Sun, L., & Zhou, Z.-W. (2021). A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis. International Journal of Molecular Sciences, 22(16), 8898. https://doi.org/10.3390/ijms22168898