
RT-qPCR Diagnostics: The “Drosten” SARS-CoV-2 Assay Paradigm


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
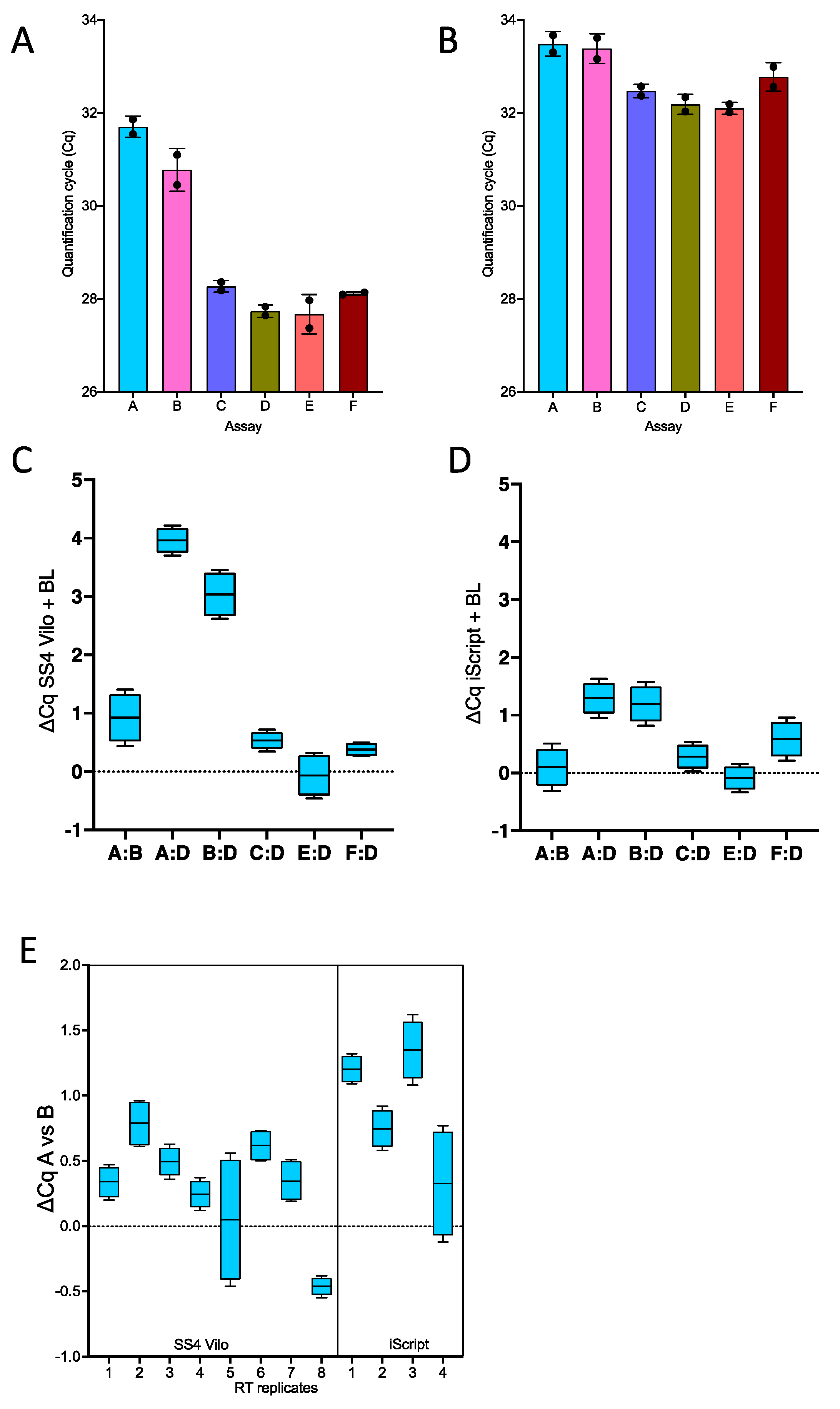
2.1. One-Step RT-qPCR
2.2. RT Temperature Effect
2.3. Separate RT Primed by Random Primers Followed by qPCR
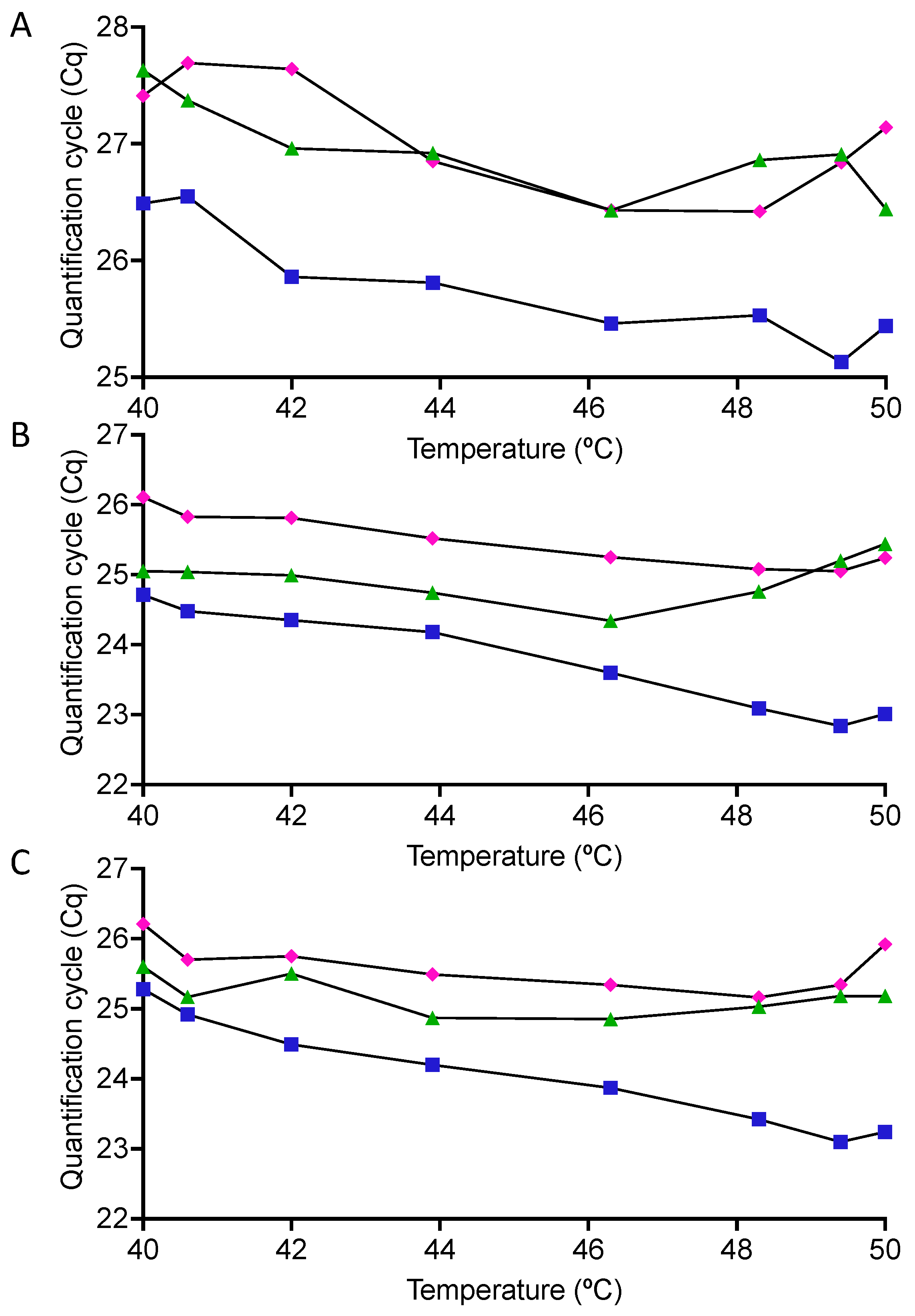
2.4. qPCR Temperature Effect
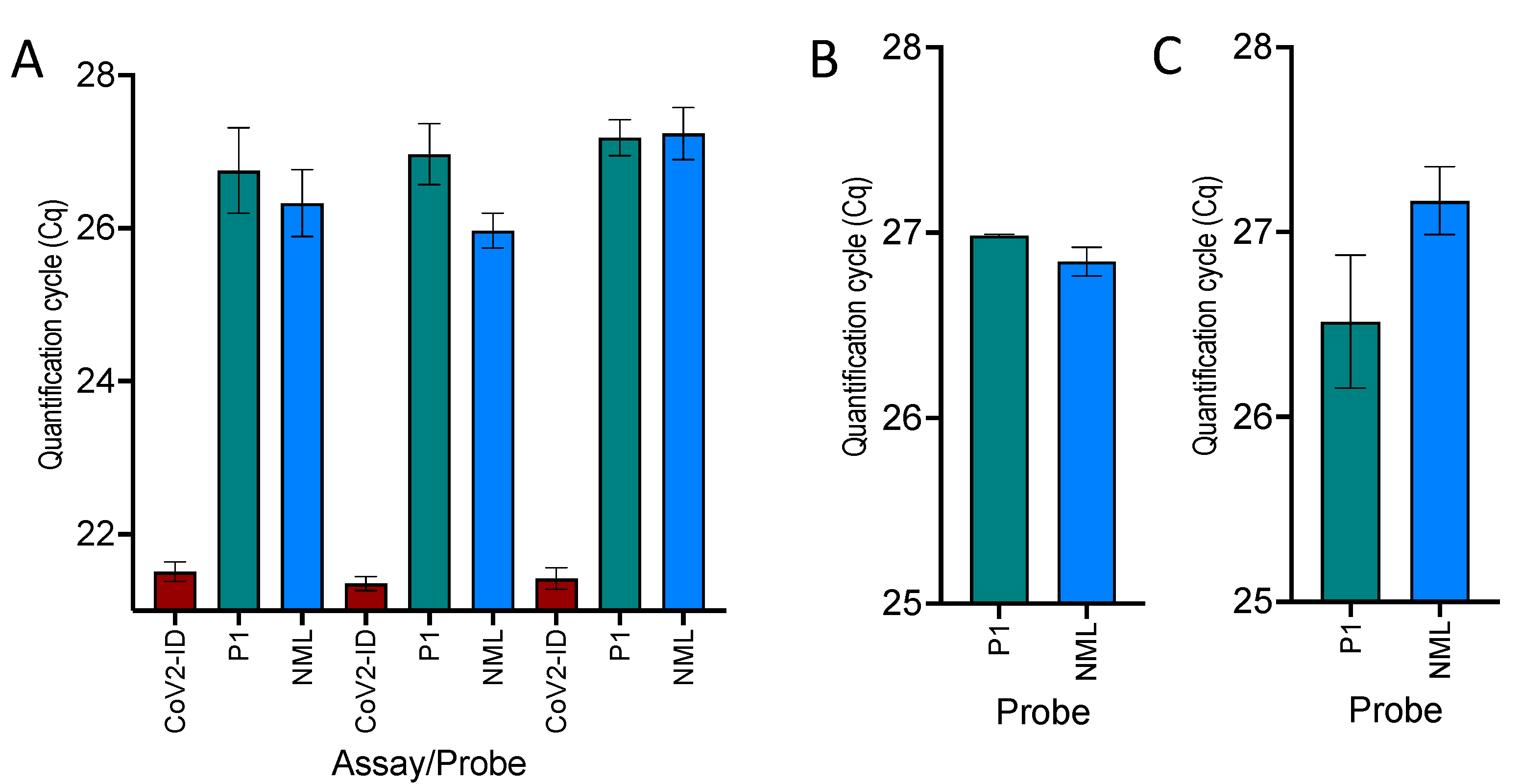
2.5. Comparison of Mismatched and Corrected RdRp Probes
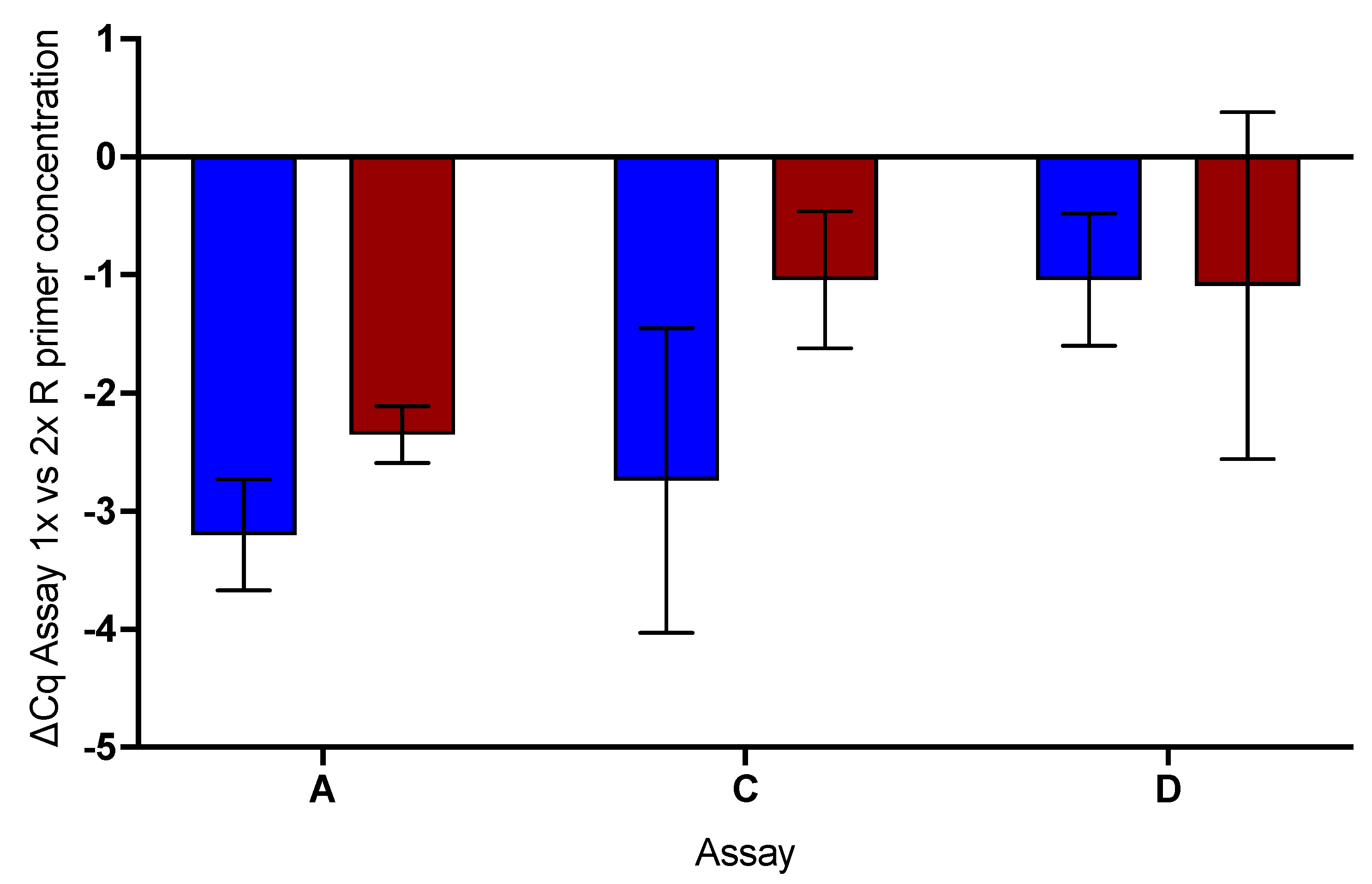
2.6. Effect of Increased Reverse Primer Concentration
3. Discussion
- The single base mismatch in the reverse primer reduces the sensitivity of the assay by affecting the RT step.
- The qPCR step is less affected by the primer mismatch than has been suggested.
- In one-step RT-qPCR reactions, specific primers perform better than those that incorporate wobble bases.
- The two mismatches in the RdRp_SARS-P1 probe do not affect the performance of the assay.
- Although there is a significant difference in the Tm between the forward and reverse primers, our data show that the RdRp assay performs reliably at a broad range of annealing temperatures and well above the calculated Tm for the R primer.
4. Methods and Materials
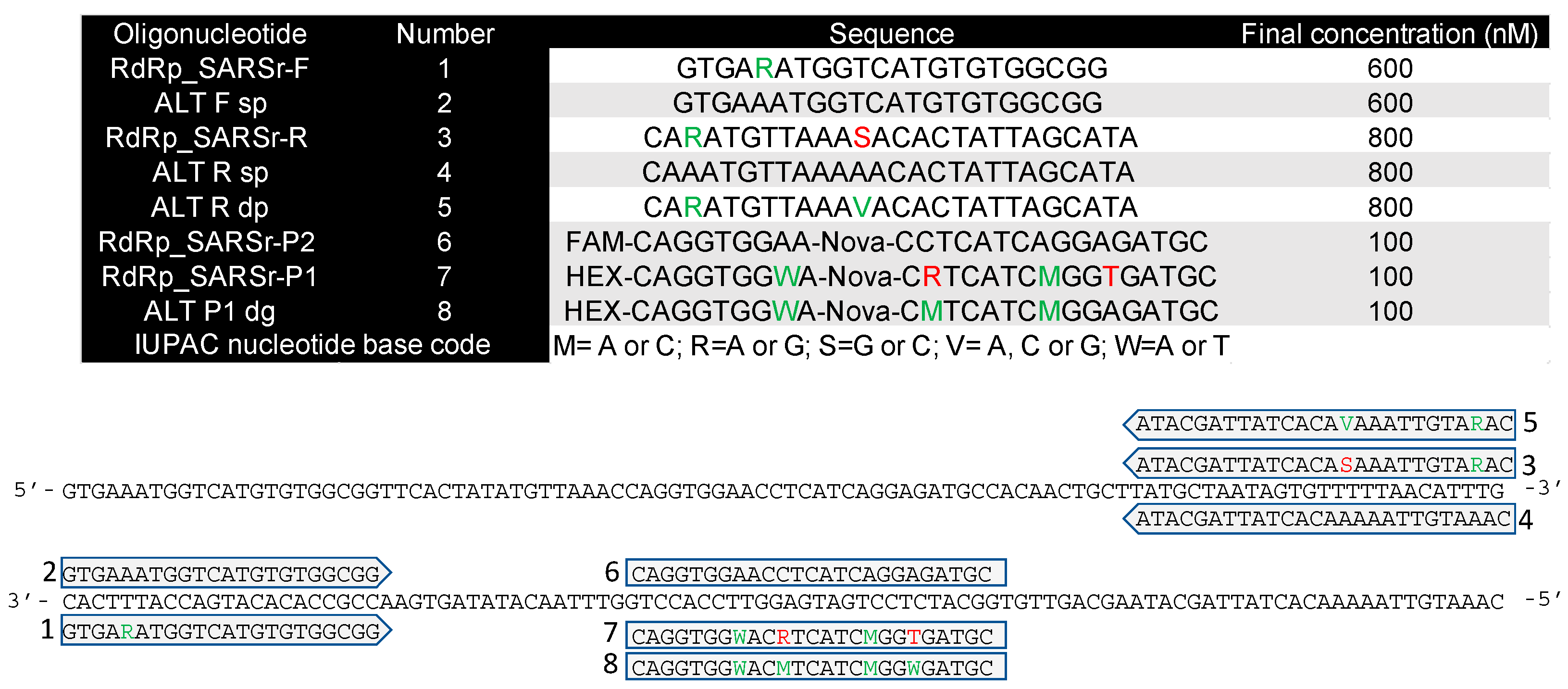
4.1. Primers and Probes
4.2. Instruments
4.3. RNA
4.4. RT-qPCR Reactions
4.4.1. RT Reactions
4.4.2. 1-Step RT-qPCR
4.4.3. 2-Step RT-qPCR
4.4.4. Gradient RT-qPCR
4.4.5. Gradient qPCR
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef]
- Bustin, S.A.; Dorudi, S. Molecular assessment of tumour stage and disease recurrence using PCR-based assays. Mol. Med. Today 1998, 4, 389–396. [Google Scholar] [CrossRef]
- Bustin, S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 2002, 29, 23–39. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef]
- Bustin, S.A.; Mueller, R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin. Sci. (Lond.) 2005, 109, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A. Real-time, fluorescence-based quantitative PCR: A snapshot of current procedures and preferences. Expert Rev. Mol. Diagn. 2005, 5, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Bustin, S.A. Real-time polymerase chain reaction—Towards a more reliable, accurate and relevant assay. Eur. Pharm. Rev. 2008, 6, 19–27. [Google Scholar]
- Bustin, S.A. Real-time quantitative PCR-opportunities and pitfalls. Eur. Pharm. Rev. 2008, 4, 18–23. [Google Scholar]
- Murphy, J.; Bustin, S.A. Reliability of real-time reverse-transcription PCR in clinical diagnostics: Gold standard or substandard? Expert Rev. Mol. Diagn. 2009, 9, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Bustin, S.A.; Beaulieu, J.-F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.; Olsvik, P.A.; Penning, L.C.; Toegel, S. MIQE precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 2010, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. Primer sequence disclosure: A clarification of the MIQE guidelines. Clin. Chem. 2011, 57, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.; Nolan, T. Talking the talk, but not walking the walk: RT-qPCR as a paradigm for the lack of reproducibility in molecular research. Eur. J. Clin. Investig. 2017, 47, 756–774. [Google Scholar] [CrossRef]
- Sanders, R.; Bustin, S.; Huggett, J.; Mason, D. Improving the standardization of mRNA measurement by RT-qPCR. Biomol. Detect. Quantif. 2018, 15, 13–17. [Google Scholar] [CrossRef]
- Bustin, S. The continuing problem of poor transparency of reporting and use of inappropriate methods for RT-qPCR. Biomol. Detect. Quantif. 2017, 12, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Naber, S.P. Molecular pathology—Diagnosis of infectious disease. N. Engl. J. Med. 1994, 331, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Greub, G.; Sahli, R.; Brouillet, R.; Jaton, K. Ten years of R&D and full automation in molecular diagnosis. Future Microbiol. 2016, 11, 403–425. [Google Scholar]
- Read, S.J.; Burnett, D.; Fink, C.G. Molecular techniques for clinical diagnostic virology. J. Clin. Pathol. 2000, 53, 502–506. [Google Scholar] [CrossRef][Green Version]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Res. 2002, 30, 1292–1305. [Google Scholar] [CrossRef]
- Espy, M.; Uhl, J.; Sloan, L.; Buckwalter, S.; Jones, M.; Vetter, E.; Yao, J.; Wengenack, N.; Rosenblatt, J.; Cockerill, F., III; et al. Real-time PCR in clinical microbiology: Applications for routine laboratory testing. Clin. Microbiol. Rev. 2006, 19, 165–256. [Google Scholar] [CrossRef]
- Emmadi, R.; Boonyaratanakornkit, J.B.; Selvarangan, R.; Shyamala, V.; Zimmer, B.L.; Williams, L.; Bryant, B.; Schutzbank, T.; Schoonmaker, M.M.; Wilson, J.A.A.; et al. Molecular methods and platforms for infectious diseases testing a review of FDA-approved and cleared assays. J. Mol. Diagn. 2011, 13, 583–604. [Google Scholar] [CrossRef]
- Cobo, F. Application of molecular diagnostic techniques for viral testing. Open Virol. J. 2012, 6, 104–114. [Google Scholar] [CrossRef]
- Reta, D.H.; Tessema, T.S.; Ashenef, A.S.; Desta, A.F.; Labisso, W.L.; Gizaw, S.T.; Abay, S.M.; Melka, D.S.; Reta, F.A. Molecular and Immunological Diagnostic Techniques of Medical Viruses. Int. J. Microbiol. 2020, 2020, 8832728. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.; Mueller, R.; Shipley, G.; Nolan, T. COVID-19 and Diagnostic Testing for SARS-CoV-2 by RT-qPCR-Facts and Fallacies. Int. J. Mol. Sci. 2021, 22, 2459. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. RT-qPCR Testing of SARS-CoV-2: A Primer. Int. J. Mol. Sci. 2020, 21, 3004. [Google Scholar] [CrossRef]
- Cuong, H.Q.; Hai, N.D.; Linh, H.T.; Anh, N.H.; Hieu, N.T.; Thang, C.M.; Thao, N.T.T.; Lan, P.T. Comparison of Primer-Probe Sets among Different Master Mixes for Laboratory Screening of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). BioMed Res. Int. 2020, 2020, 7610678. [Google Scholar] [CrossRef] [PubMed]
- Bezier, C.; Anthoine, G.; Charki, A. Reliability of real-time RT-PCR tests to detect SARS-Cov-2: A literature review. Int. J. Metrol. Qual. Eng. 2020, 11, 13. [Google Scholar] [CrossRef]
- Fischer, C.; Mögling, R.; Melidou, A.; Kühne, A.; Oliveira-Filho, E.F.; Wolff, T.; Reiche, J.; Broberg, E.; Drosten, C.; Meijer, A.; et al. Variable sensitivity in molecular detection of SARS-CoV-2 in European Expert Laboratories: External Quality Assessment, June–July 2020. J. Clin. Microbiol. 2021, 59, e02676-20. [Google Scholar] [CrossRef]
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.-P.; Bours, V.; et al. A Recurrent Mutation at Position 26340 of SARS-CoV-2 Is Associated with Failure of the E Gene Quantitative Reverse Transcription-PCR Utilized in a Commercial Dual-Target Diagnostic Assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef]
- Pillonel, T.; Scherz, V.; Jaton, K.; Greub, G.; Bertelli, C. Letter to the editor: SARS-CoV-2 detection by real-time RT-PCR. Eurosurveillance 2020, 25, 2000880. [Google Scholar] [CrossRef]
- Jung, Y.; Park, G.-S.; Moon, J.H.; Ku, K.; Beak, S.-H.; Lee, C.-S.; Kim, S.; Park, E.C.; Park, D.; Lee, J.-H.; et al. Comparative Analysis of Primer-Probe Sets for RT-qPCR of COVID-19 Causative Virus (SARS-CoV-2). ACS Infect. Dis. 2020, 6, 2513–2523. [Google Scholar] [CrossRef]
- Nalla, A.K.; Casto, A.M.; Huang, M.-L.W.; Perchetti, G.A.; Sampoleo, R.; Shrestha, L.; Wei, Y.; Zhu, H.; Jerome, K.R.; Greninger, A.L. Comparative Performance of SARS-CoV-2 Detection Assays Using Seven Different Primer-Probe Sets and One Assay Kit. J. Clin. Microbiol. 2020, 58, e00557-20. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Etievant, S.; Bal, A.; Escuret, V.; Brengel-Pesce, K.; Bouscambert, M.; Cheynet, V.; Generenaz, L.; Oriol, G.; Destras, G.; Billaud, G.; et al. Performance Assessment of SARS-CoV-2 PCR Assays Developed by WHO Referral Laboratories. J. Clin. Med. 2020, 9, 1871. [Google Scholar] [CrossRef]
- Bustin, S.; Coward, A.; Sadler, G.; Teare, L.; Nolan, T. CoV2-ID, a MIQE-compliant sub-20-min 5-plex RT-PCR assay targeting SARS-CoV-2 for the diagnosis of COVID-19. Sci. Rep. 2020, 10, 22214. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, G.A.; Temin, H.M. High rate of mismatch extension during reverse transcription in a single round of retrovirus replication. Proc. Natl. Acad. Sci. USA 1994, 91, 9490–9494. [Google Scholar] [CrossRef]
- Perrino, F.W.; Preston, B.D.; Sandell, L.L.; Loeb, L.A. Extension of mismatched 3′ termini of DNA is a major determinant of the infidelity of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1989, 86, 8343–8347. [Google Scholar] [CrossRef]
- Bakhanashvili, M.; Hizi, A. The fidelity of the reverse transcriptases of human immunodeficiency viruses and murine leukemia virus, exhibited by the mispair extension frequencies, is sequence dependent and enzyme related. FEBS Lett. 1993, 319, 201–205. [Google Scholar] [CrossRef]
- Yu, H.; Goodman, M.F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 1992, 267, 10888–10896. [Google Scholar] [CrossRef]
- Persson, S.; Karlsson, M.; Borsch-Reniers, H.; Ellström, P.; Eriksson, R.; Simonsson, M. Missing the Match Might Not Cost You the Game: Primer-Template Mismatches Studied in Different Hepatitis A Virus Variants. Food Environ. Virol. 2019, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, C.; Sninsky, J.; Kwok, S. The effects of internal primer-template mismatches on RT-PCR: HIV-1 model studies. Nucleic Acids Res. 1997, 25, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Mendelman, L.V.; Petruska, J.; Goodman, M.F. Base mispair extension kinetics. Comparison of DNA polymerase alpha and reverse transcriptase. J. Biol. Chem. 1990, 265, 2338–2346. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- SantaLucia, J.J.; Hicks, D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [Google Scholar] [CrossRef]
- Huang, M.M.; Arnheim, N.; Goodman, M.F. Extension of base mispairs by Taq DNA polymerase: Implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992, 20, 4567–4573. [Google Scholar] [CrossRef]
- Creighton, S.; Huang, M.M.; Cai, H.; Arnheim, N.; Goodman, M.F. Base mispair extension kinetics. Binding of avian myeloblastosis reverse transcriptase to matched and mismatched base pair termini. J. Biol. Chem. 1992, 267, 2633–2639. [Google Scholar] [CrossRef]
- Mendelman, L.V.; Boosalis, M.S.; Petruska, J.; Goodman, M.F. Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem. 1989, 264, 14415–14423. [Google Scholar] [CrossRef]
- Newton, C.; Graham, A.; Heptinstall, L.; Powell, S.; Summers, C.; Kalsheker, N.; Smith, J.; Markham, A. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef]
- Ehlen, T.; Dubeau, L. Detection of ras point mutations by polymerase chain reaction using mutation-specific, inosine-containing oligonucleotide primers. Biochem. Biophys. Res. Commun. 1989, 160, 441–447. [Google Scholar] [CrossRef]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar] [CrossRef]
- Rejali, N.A.; Moric, E.; Wittwer, C.T. The Effect of Single Mismatches on Primer Extension. Clin. Chem. 2018, 64, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Okayama, H.; Curiel, D.T.; Brantly, M.L.; Holmes, M.D.; Crystal, R.G. Rapid, nonradioactive detection of mutations in the human genome by allele-specific amplification. J. Lab. Clin. Med. 1989, 114, 105–113. [Google Scholar] [PubMed]
- Kwok, S.; Kellogg, D.; McKinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J. Effects of primer-template mismatches on the polymerase chain reaction: Human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990, 18, 999–1005. [Google Scholar] [CrossRef]
- Bru, D.; Martin-Laurent, F.; Philippot, L. Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl. Environ. Microbiol. 2008, 74, 1660–1663. [Google Scholar] [CrossRef]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef]
- Wu, J.H.; Hong, P.Y.; Liu, W.T. Quantitative effects of position and type of single mismatch on single base primer extension. J. Microbiol. Methods 2009, 77, 267–275. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Pierce, K.E.; Rice, J.E.; Wangh, L.J. Linear-after-the-exponential (LATE)-PCR: An advanced method of asymmetric PCR and its uses in quantitative real-time analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 1933–1938. [Google Scholar] [CrossRef]
- Pierce, K.E.; Sanchez, J.A.; Rice, J.E.; Wangh, L.J. Linear-After-The-Exponential (LATE)-PCR: Primer design criteria for high yields of specific single-stranded DNA and improved real-time detection. Proc. Natl. Acad. Sci. USA 2005, 102, 8609–8614. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, R.; Aherfi, S.; Wurtz, N.; Grimaldier, C.; Hoang, V.; Colson, P.; Raoult, D.; La Scola, B. Correlation between 3790 qPCR positives samples and positive cell cultures including 1941 SARS-CoV-2 isolates. Clin. Infect. Dis. 2021, 72, e921. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.; Peaper, D.R.; She, R.C.; Nolte, F.S.; Wojewoda, C.M.; Anderson, N.W.; Pritt, B.S. College of American Pathologists (CAP) Microbiology Committee Perspective: Caution must be used in interpreting the Cycle Threshold (Ct) value. Clin. Infect. Dis. 2021, 72, e685–e686. [Google Scholar] [CrossRef]
- Bullard, J.; Dust, K.; Funk, D.; Strong, J.E.; Alexander, D.; Garnett, L.; Boodman, C.; Bello, A.; Hedley, A.; Schiffman, Z.; et al. Predicting infectious severe acute respiratory syndrome coronavirus 2 from diagnostic samples. Clin. Infect. Dis. 2020, 71, 2663–2666. [Google Scholar] [CrossRef] [PubMed]
- Arons, M.M.; Hatfield, K.M.; Reddy, S.C.; Kimball, A.; James, A.; Jacobs, J.R.; Taylor, J.; Spicer, K.; Bardossy, A.C.; Oakley, L.P.; et al. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N. Engl. J. Med. 2020, 382, 2081–2090. [Google Scholar] [CrossRef]
- La Scola, B.; Le Bideau, M.; Andreani, J.; Grimaldier, C.; Colson, P.; Gautret, P.; Raoult, D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 patients from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Dahdouh, E.; Lázaro-Perona, F.; Romero-Gomez, M.P.; Mingorance, J.; Garcia-Rodriguez, J. Ct values from SARS-CoV-2 diagnostic PCR assays should not be used as direct estimates of viral load. J. Infect. 2021, 82, 414–451. [Google Scholar] [CrossRef]
- Tom, M.R.; Mina, M.J. To interpret the SARS-CoV-2 test, consider the cycle threshold value. Clin. Infect. Dis. 2020, 71, 2252–2254. [Google Scholar] [CrossRef]
- Shah, S.; Singhal, T.; Davar, N.; Thakkar, P. No correlation between Ct values and severity of disease or mortality in patients with COVID 19 disease. Indian J. Med. Microbiol. 2021, 39, 116–117. [Google Scholar] [CrossRef]
- Trunfio, M.; Venuti, F.; Alladio, F.; Longo, B.M.; Burdino, E.; Cerutti, F.; Ghisetti, V.; Bertucci, R.; Picco, C.; Bonora, S.; et al. Diagnostic SARS-CoV-2 Cycle Threshold Value Predicts Disease Severity, Survival, and Six-Month Sequelae in COVID-19 Symptomatic Patients. Viruses 2021, 13, 281. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Aita, A.; Navaglia, F.; Franchin, E.; Fioretto, P.; Moz, S.; Bozzato, D.; Zambon, C.-F.; Martin, B.; Dal Prà, C.; et al. SARS-CoV-2 RNA identification in nasopharyngeal swabs: Issues in pre-analytics. Clin. Chem. Lab. Med. 2020, 58, 1579–1586. [Google Scholar] [CrossRef]
- Binnicker, M.J. Challenges and Controversies to Testing for COVID-19. J. Clin. Microbiol. 2020, 58, e01695-20. [Google Scholar] [CrossRef]
- Carroll, A.; McNamara, E. Comparison and correlation of commercial SARS-CoV-2 real-time-PCR assays, Ireland, June 2020. Eurosurveillance 2021, 26, 2002079. [Google Scholar] [CrossRef] [PubMed]
- Aquino-Jarquin, G. The Raw Cycle Threshold Values from Reverse-transcription Polymerase Chain Reaction Detection Are Not Viral Load Quantitation Units. Clin. Infect. Dis. 2021, 72, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gómez, M.P.; Gómez-Sebastian, S.; Cendejas-Bueno, E.; Montero-Vega, M.D.; Mingorance, J.; García-Rodríguez, J.; Group, S.-C.-W. Ct value is not enough to discriminate patients harbouring infective virus. J. Infect. 2021, 82, e35–e37. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bustin, S.; Kirvell, S.; Huggett, J.F.; Nolan, T. RT-qPCR Diagnostics: The “Drosten” SARS-CoV-2 Assay Paradigm. Int. J. Mol. Sci. 2021, 22, 8702. https://doi.org/10.3390/ijms22168702
Bustin S, Kirvell S, Huggett JF, Nolan T. RT-qPCR Diagnostics: The “Drosten” SARS-CoV-2 Assay Paradigm. International Journal of Molecular Sciences. 2021; 22(16):8702. https://doi.org/10.3390/ijms22168702
Chicago/Turabian StyleBustin, Stephen, Sara Kirvell, Jim F. Huggett, and Tania Nolan. 2021. "RT-qPCR Diagnostics: The “Drosten” SARS-CoV-2 Assay Paradigm" International Journal of Molecular Sciences 22, no. 16: 8702. https://doi.org/10.3390/ijms22168702
APA StyleBustin, S., Kirvell, S., Huggett, J. F., & Nolan, T. (2021). RT-qPCR Diagnostics: The “Drosten” SARS-CoV-2 Assay Paradigm. International Journal of Molecular Sciences, 22(16), 8702. https://doi.org/10.3390/ijms22168702