
Granuloma Formation in a Cyba-Deficient Model of Chronic Granulomatous Disease Is Associated with Myeloid Hyperplasia and the Exhaustion of B-Cell Lineage

 , ,
, ,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
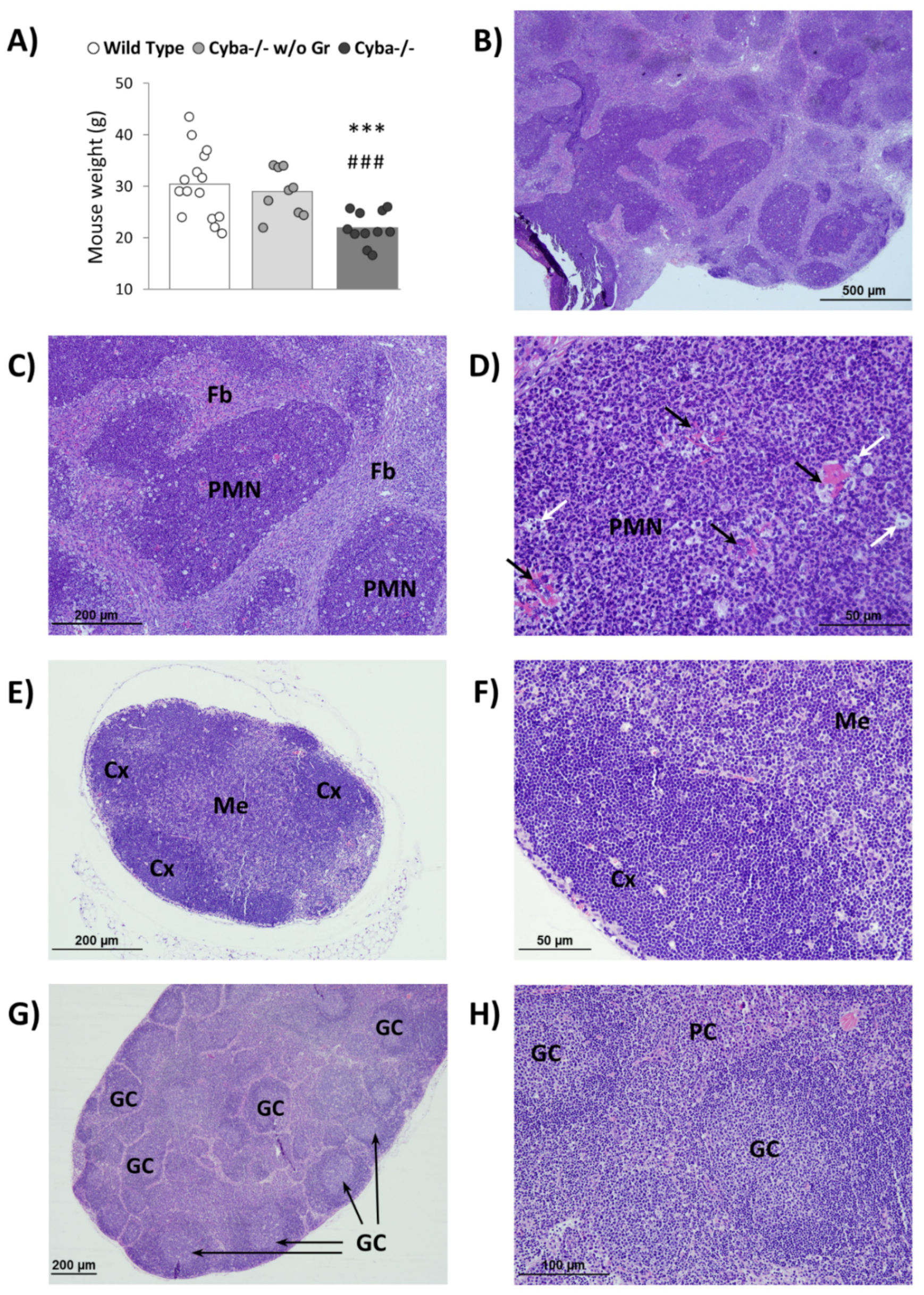
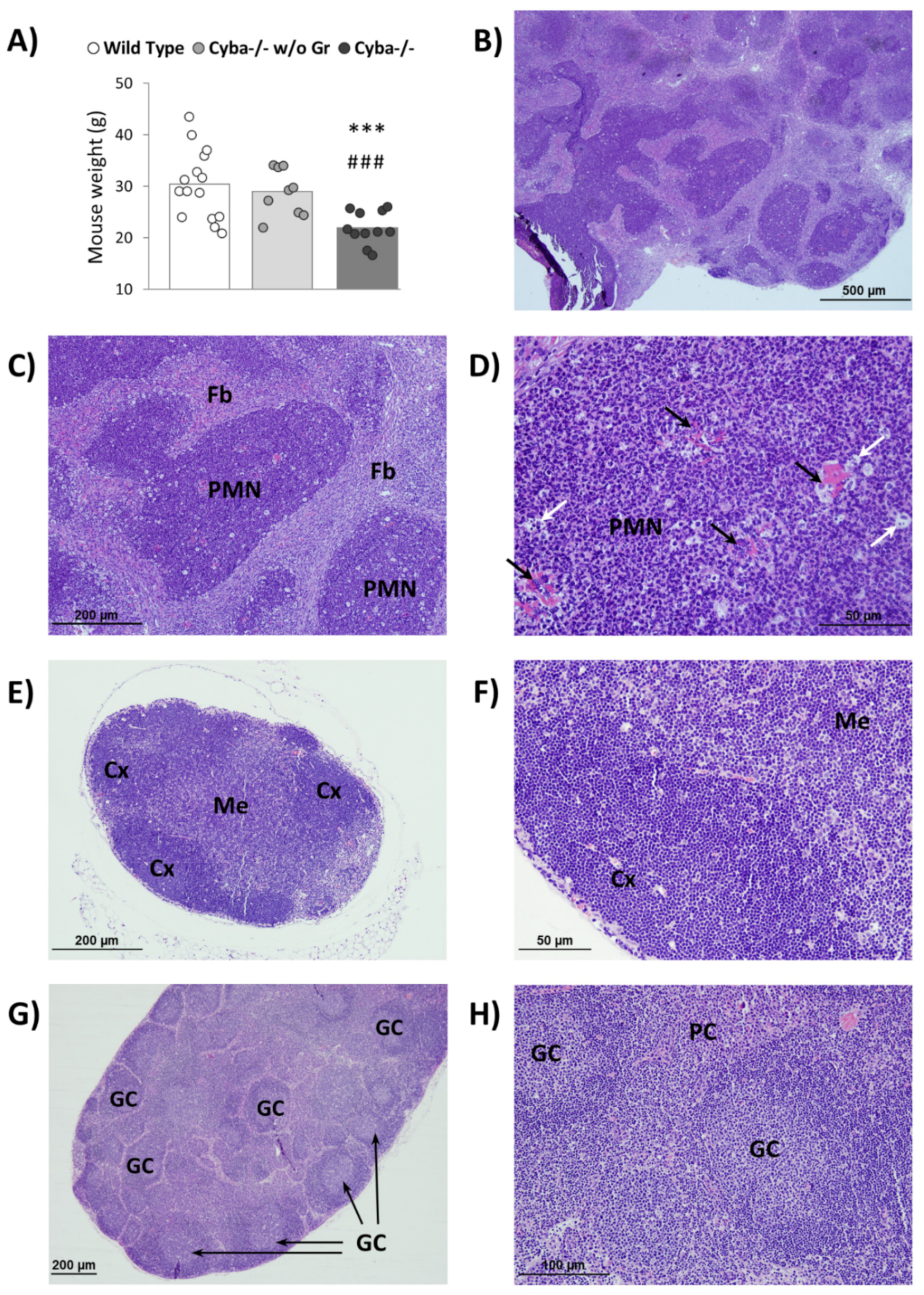
2.1. Cyba−/− Mice Spontaneously Develop Abscesses
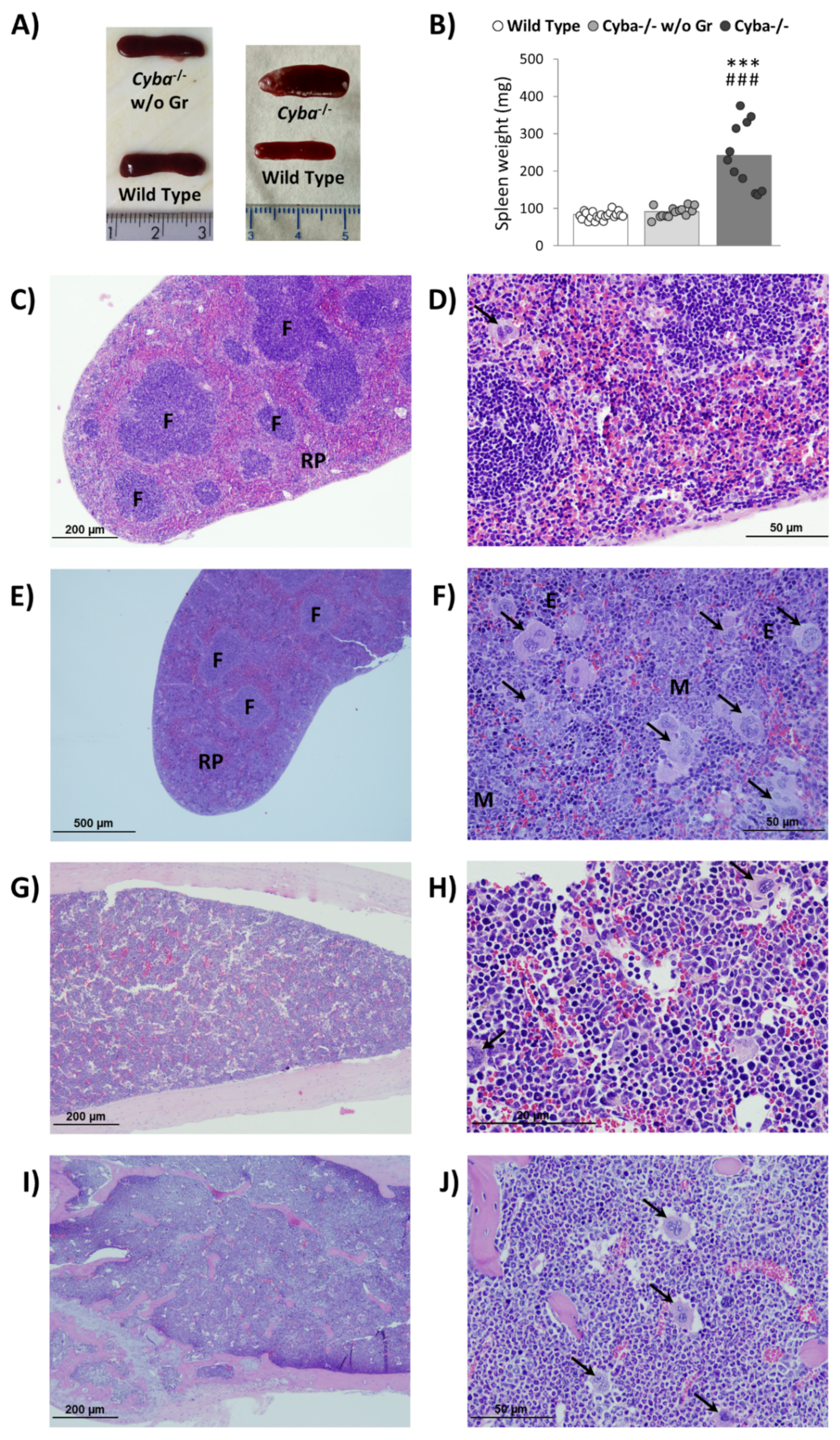
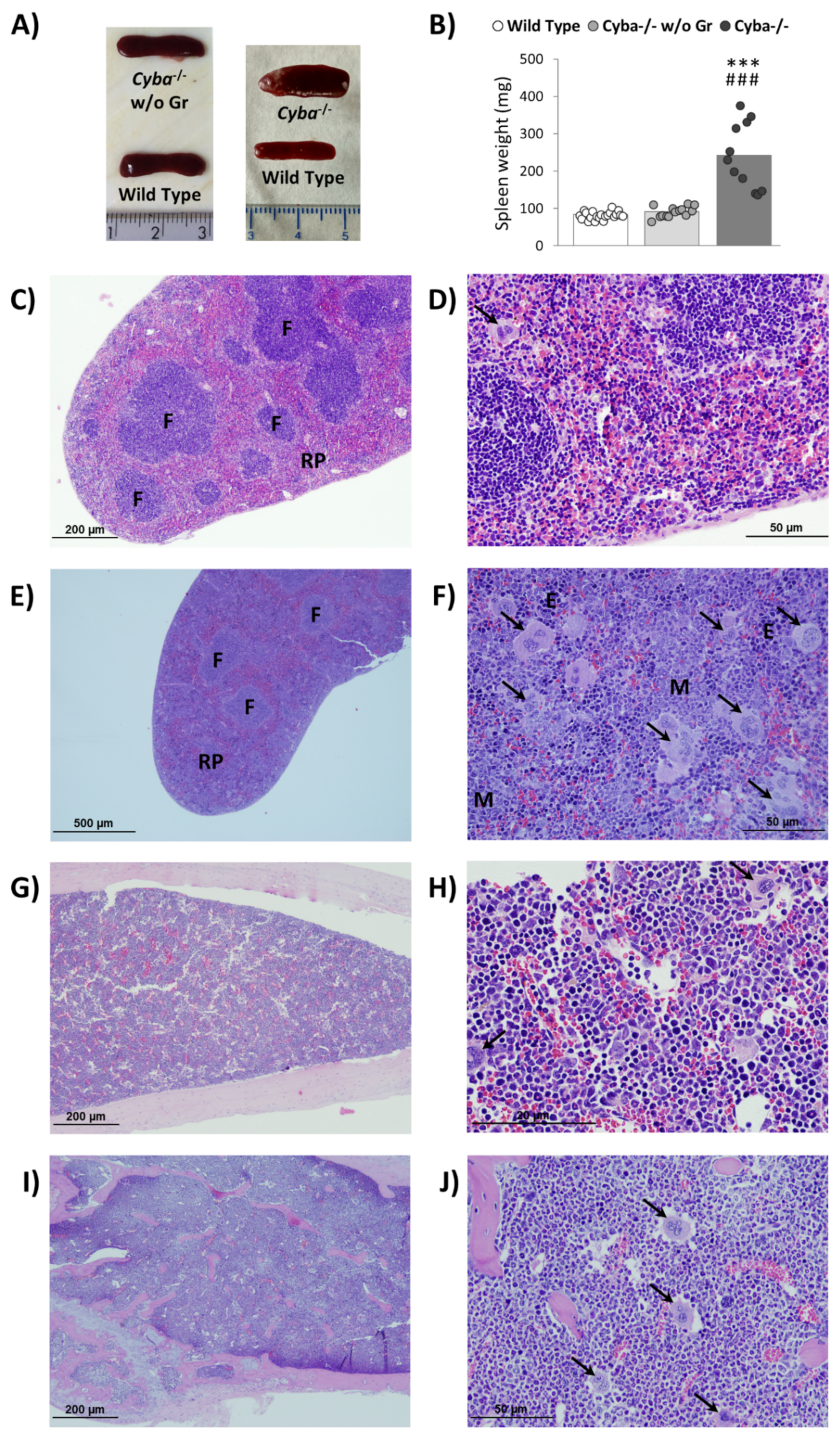
2.2. Cyba−/− Mice Bearing Abscesses Display Splenomegaly and Bone Marrow Myeloid Hyperplasia
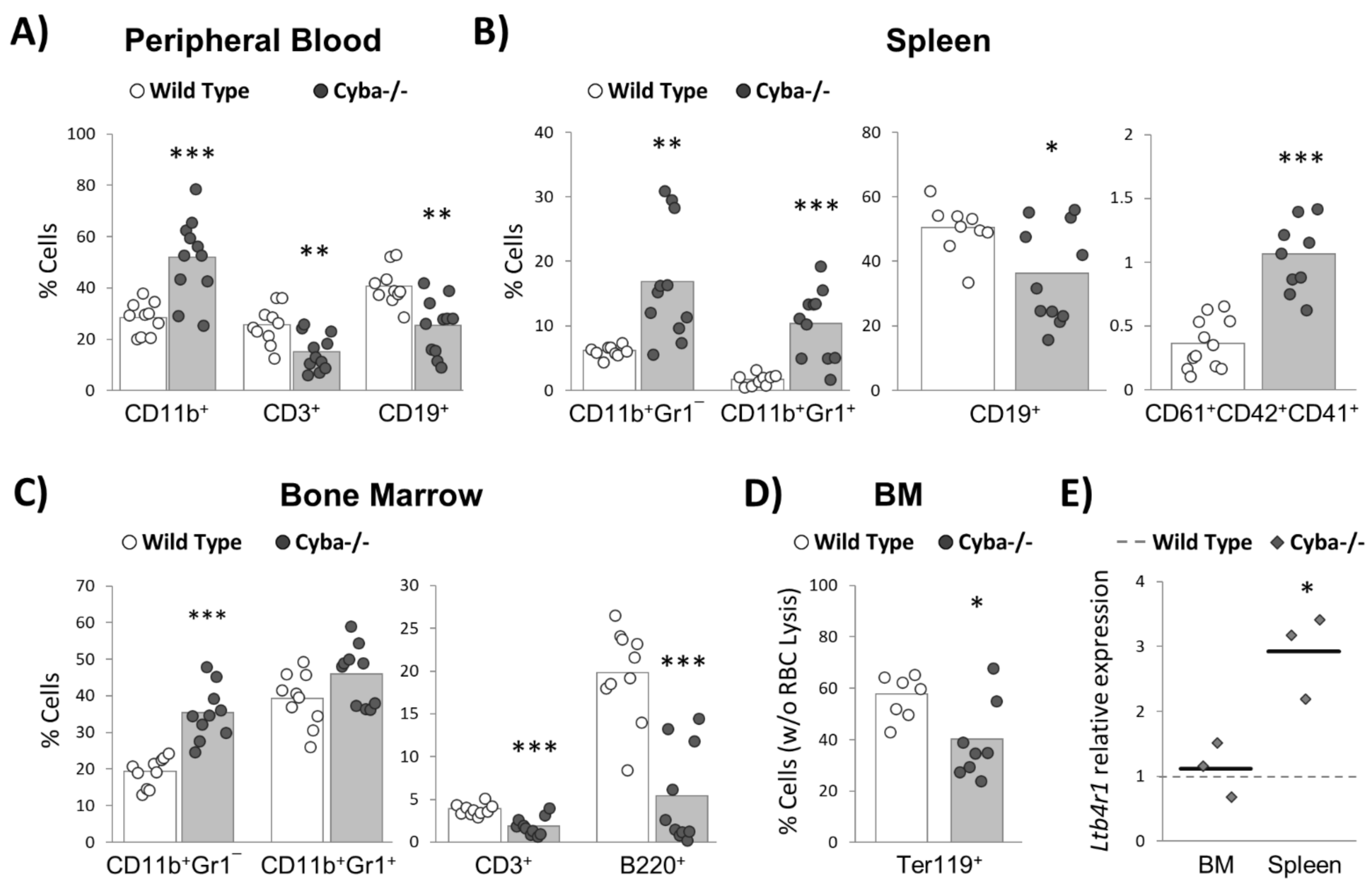
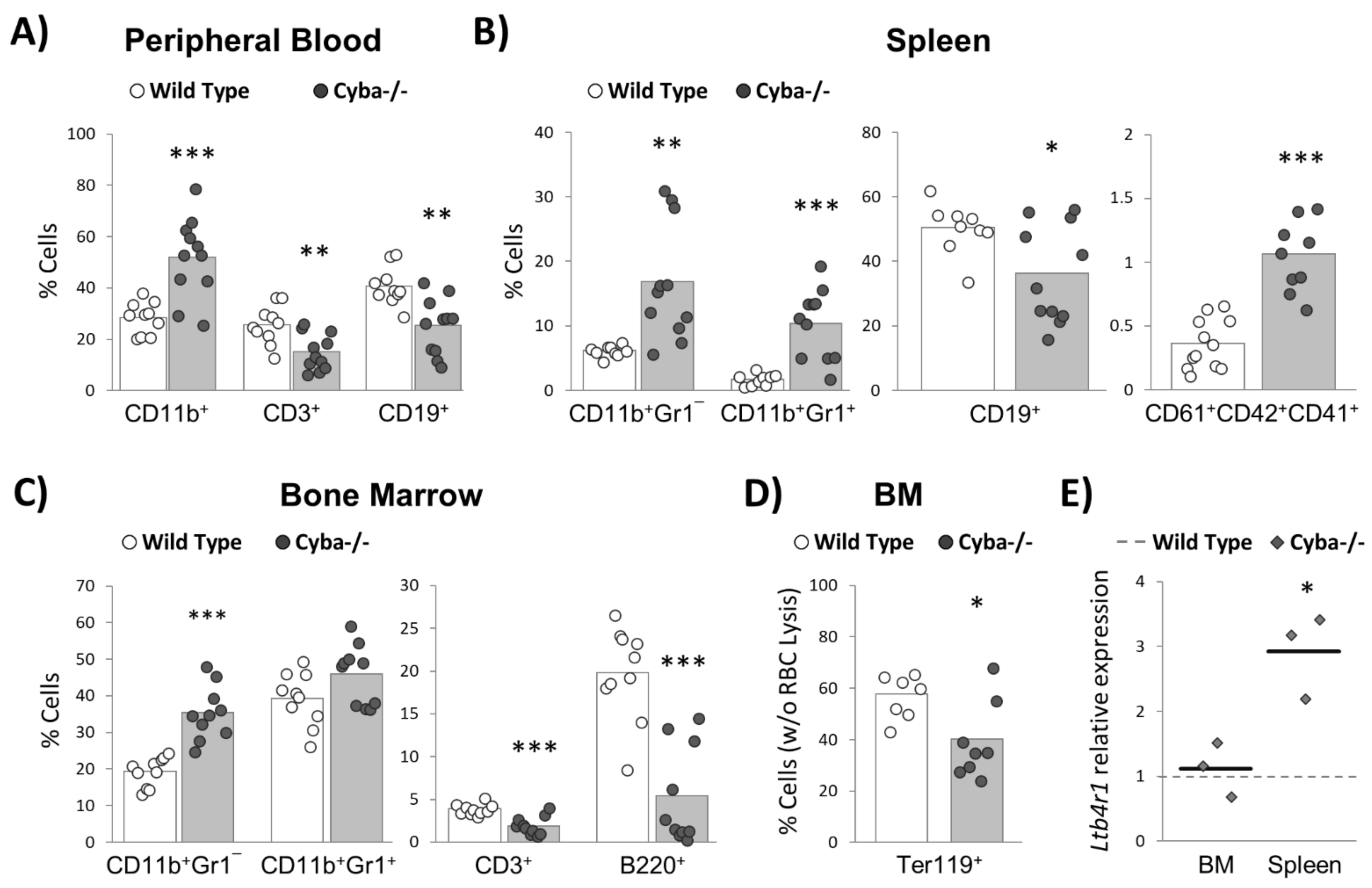
2.3. Cyba−/− Mice Bearing Abscesses Undergo Drastic Changes in Haematopoiesis Homeostasis
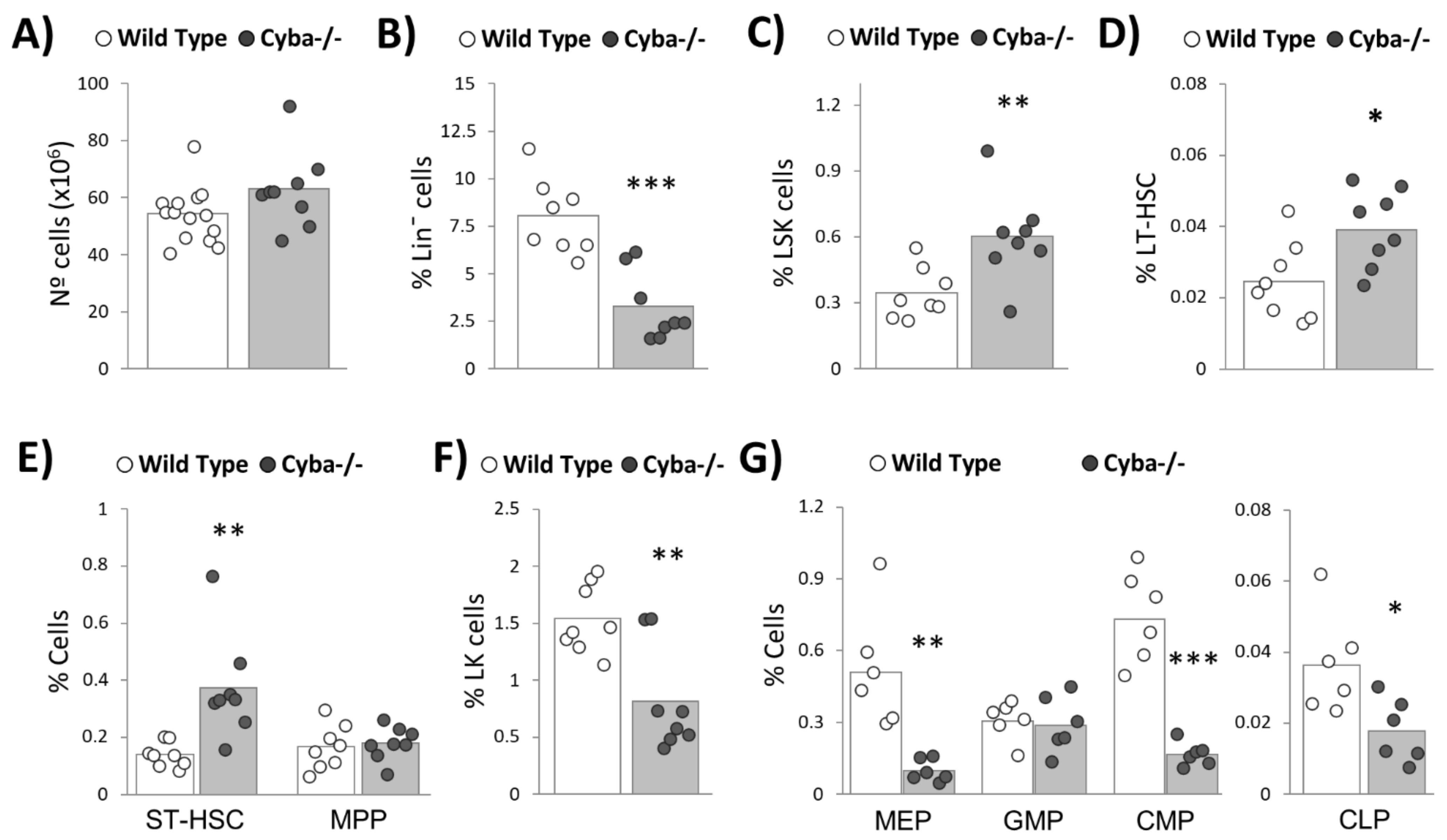
2.4. Granuloma Induces a Sharp Decrease in Haematopoietic Progenitor Cells
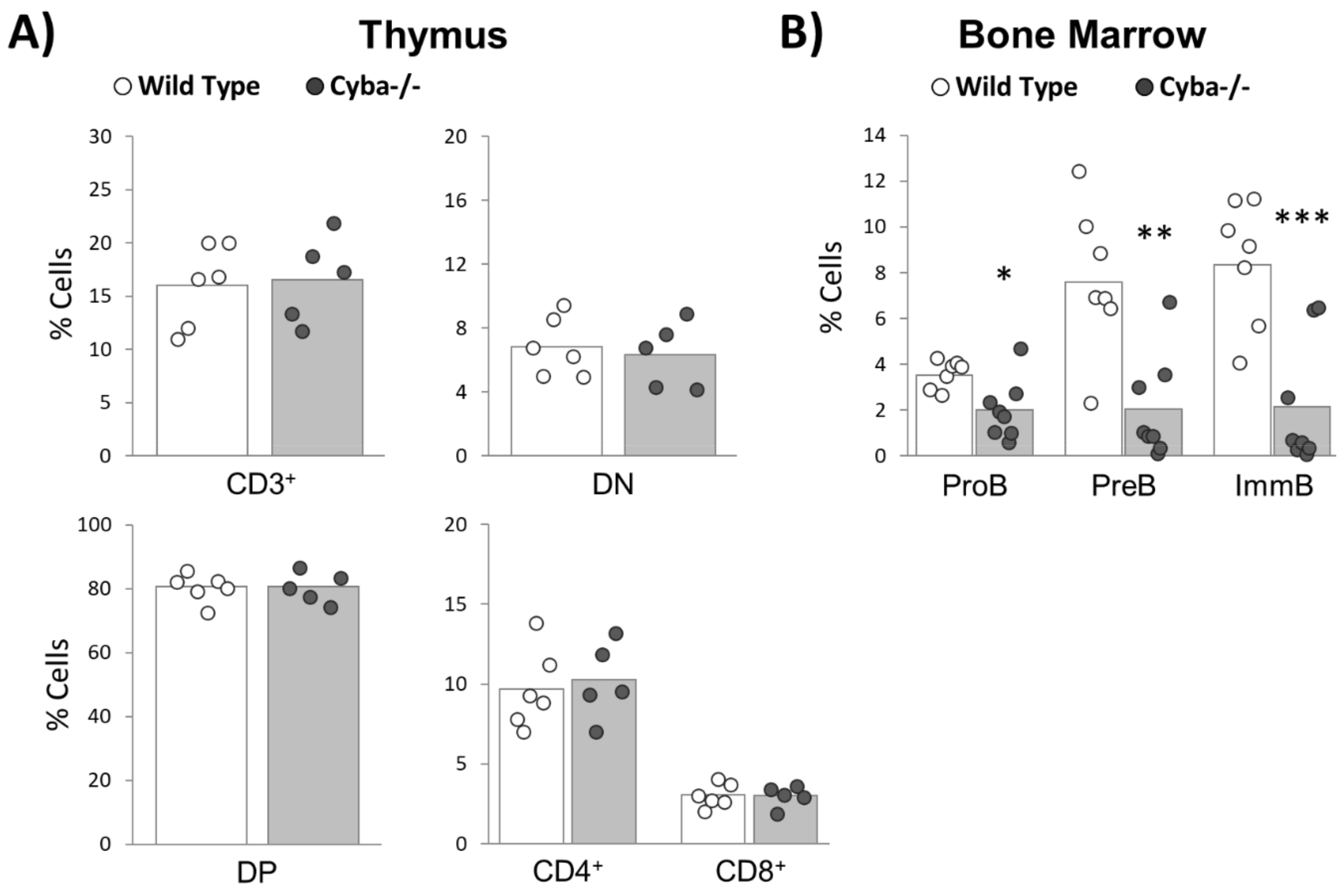
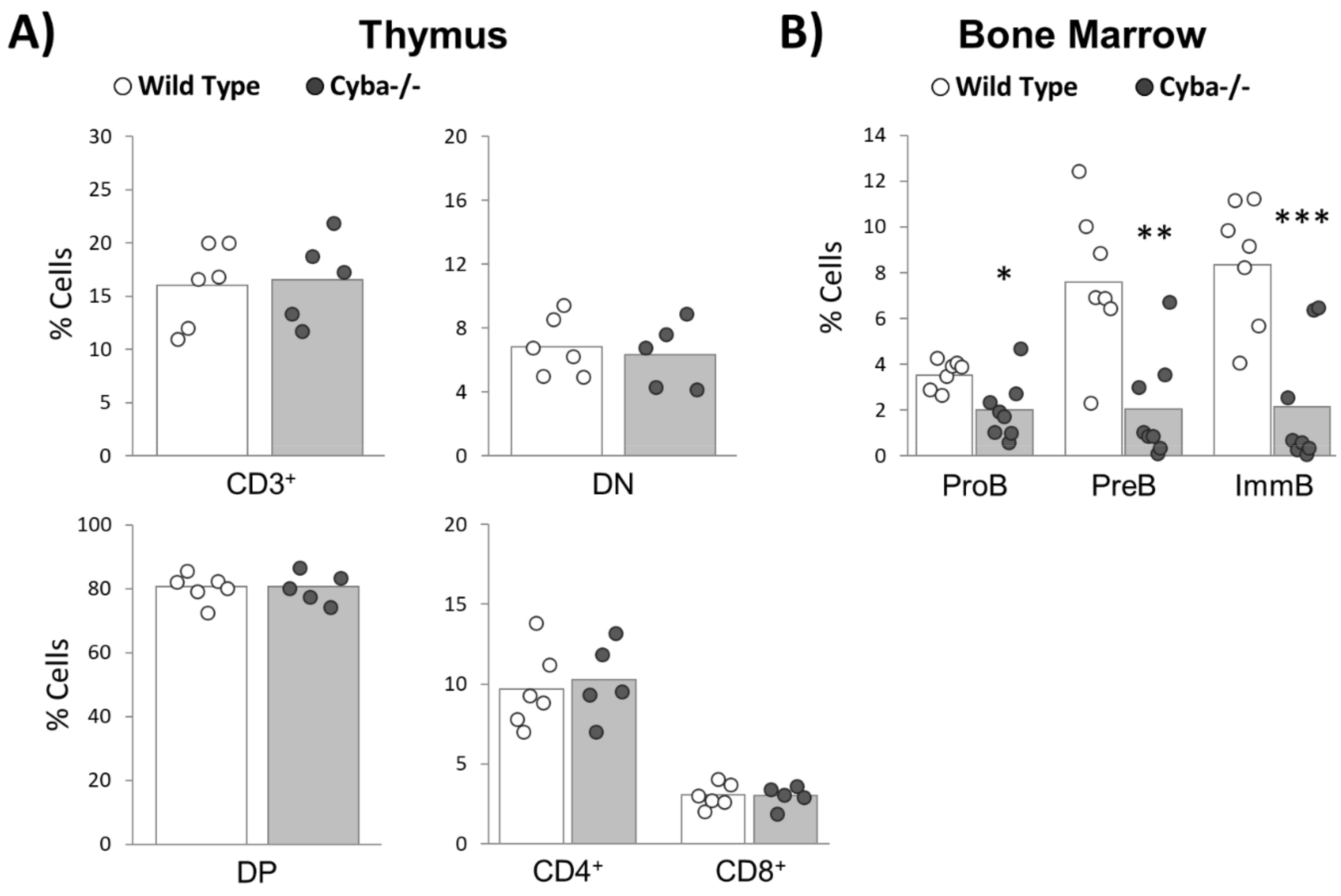
2.5. Granuloma Alters B-Cell Differentiation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Histological Analysis
4.4. Haematopoietic Lineages Analysis
4.5. Quantitative RT-PCR
4.6. Statistical Analyses and Data Report
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef] [Green Version]
- Roos, D. Chronic granulomatous disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, W.F.; Zhang, Y.D.; Chen, T.X. Clinical Features and Genetic Analysis of 48 Patients with Chronic Granulomatous Disease in a Single Center Study from Shanghai, China (2005-2015): New Studies and a Literature Review. J. Immunol. Res. 2017, 2017, 8745254. [Google Scholar] [CrossRef] [Green Version]
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.L.; Soule, B.P.; et al. Residual NADPH Oxidase and Survival in Chronic Granulomatous Disease. N. Engl. J. Med. 2010, 363, 2600–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouy, R.; Fischer, A.; Vilmer, E.; Seger, R.; Griscelli, C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J. Pediatr. 1989, 114, 555–560. [Google Scholar] [CrossRef]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.B.K.R.; McGrogan, P.; Flood, T.J.; Gennery, A.R.; Morton, L.; Thrasher, A.; Goldblatt, D.; Parker, L.; Cant, A.J. Special Article: Chronic granulomatous disease in the United Kingdom and Ireland: A comprehensive national patient-based registry. Clin. Exp. Immunol. 2008, 152, 211–218. [Google Scholar] [CrossRef]
- Blancas-Galicia, L.; Santos-Chávez, E.; Deswarte, C.; Mignac, Q.; Medina-Vera, I.; León-Lara, X.; Roynard, M.; Scheffler-Mendoza, S.C.; Rioja-Valencia, R.; Alvirde-Ayala, A.; et al. Genetic, Immunological, and Clinical Features of the First Mexican Cohort of Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2020, 40, 475–493. [Google Scholar] [CrossRef]
- Boettcher, S.; Manz, M.G. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol. 2017, 38, 345–357. [Google Scholar] [CrossRef]
- Takizawa, H.; Manz, M.G. Impact of inflammation on early hematopoiesis and the microenvironment. Int. J. Hematol. 2017, 106, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460. [Google Scholar] [CrossRef] [PubMed]
- Rider, N.L.; Jameson, M.B.; Creech, C.B. Chronic granulomatous disease: Epidemiology, pathophysiology, and genetic basis of disease. J. Pediatric Infect. Dis. Soc. 2018, 7, S2–S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijpers, T.; Lutter, R. Inflammation and repeated infections in CGD: Two sides of a coin. Cell. Mol. Life Sci. 2012, 69, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Romani, L.; Fallarino, F.; De Luca, A.; Montagnoli, C.; D’Angelo, C.; Zelante, T.; Vacca, C.; Bistoni, F.; Fioretti, M.C.; Grohmann, U.; et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 2008, 451, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Chiriaco, M.; Salfa, I.; Di Matteo, G.; Rossi, P.; Finocchi, A. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr. Allergy Immunol. 2016, 27, 242–253. [Google Scholar] [CrossRef]
- Pollock, J.D.; Williams, D.A.; Gifford, M.A.; Lin, L.L.; Du, X.; Fisherman, J.; Orkin, S.H.; Doerschuk, C.M.; Dinauer, M.C. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet. 1995, 9, 202–209. [Google Scholar] [CrossRef]
- Jackson, S.H.; Gallin, J.I.; Holland, S.M. The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 1995, 182, 751–758. [Google Scholar] [CrossRef]
- Kume, A.; Dinauer, M.C. Gene therapy for chronic granulomatous disease. J. Lab. Clin. Med. 2000, 135, 122–128. [Google Scholar] [CrossRef]
- Nakano, Y.; Longo-Guess, C.M.; Bergstrom, D.E.; Nauseef, W.M.; Jones, S.M.; Banfi, B. Mutation of the Cyba gene encoding p22phox causes vestibular and immune defects in mice. J. Clin. Investig. 2008, 118, 1176–1185. [Google Scholar] [CrossRef]
- Aviello, G.; Singh, A.K.; O’Neill, S.; Conroy, E.; Gallagher, W.; D’Agostino, G.; Walker, A.W.; Bourke, B.; Scholz, D.; Knaus, U.G. Colitis susceptibility in mice with reactive oxygen species deficiency is mediated by mucus barrier and immune defense defects. Mucosal Immunol. 2019, 12, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; García-Tuñón, I.; Sánchez-Martín, M.; Hernández-Hernández, Á. Cyba-deficient mice display an increase in hematopoietic stem cells and an overproduction of immunoglobulins. Haematologica 2021, 106, 142–153. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, L.; Speak, A.O.; Swiatkowska, A.; Clare, S.; Schejtman, A.; Santilli, G.; Arends, M.J.; Adams, D.J. Pulmonary metastatic colonisation and granulomas in NOX2-deficient mice. J. Pathol. 2018, 246, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Huang, G.; Paracatu, L.C.; Grimes, D.; Gu, J.; Luke, C.J.; Clemens, R.A.; Dinauer, M.C. NADPH oxidase controls pulmonary neutrophil infiltration in the response to fungal cell walls by limiting LTB4. Blood 2020, 135, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Claesson, H.E.; Dahlberg, N.; Gahrton, G. Stimulation of human myelopoiesis by leukotriene B4. Biochem. Biophys. Res. Commun. 1985, 131, 579–585. [Google Scholar] [CrossRef]
- Rosenzweig, S.D. Inflammatory manifestations in chronic granulomatous disease (CGD). J. Clin. Immunol. 2008, 28, 67–72. [Google Scholar] [CrossRef]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef] [Green Version]
- Weisser, M.; Demel, U.M.; Stein, S.; Chen-Wichmann, L.; Touzot, F.; Santilli, G.; Sujer, S.; Brendel, C.; Siler, U.; Cavazzana, M.; et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J. Allergy Clin. Immunol. 2016, 138, 219–228.e9. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Goldblatt, D.; Buddle, J.; Morton, L.; Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukoc. Biol. 2003, 73, 591–599. [Google Scholar] [CrossRef]
- Meissner, F.; Seger, R.A.; Moshous, D.; Fischer, A.; Reichenbach, J.; Zychlinsky, A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 2010, 116, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Segal, B.H.; Han, W.; Bushey, J.J.; Joo, M.; Bhatti, Z.; Feminella, J.; Dennis, C.G.; Vethanayagam, R.R.; Yull, F.E.; Capitano, M.; et al. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS ONE 2010, 5, e9631. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Kondo, M.; Kelsoe, G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 2005, 201, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Yang, K.; Foster, S.J.; Kondo, M.; Kelsoe, G. Inflammation Controls B Lymphopoiesis by Regulating Chemokine CXCL12 Expression. J. Exp. Med. 2004, 199, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Dong, C.; Cooper, M.D. Impairment of T and B cell development by treatment with a type I interferon. J. Exp. Med. 1998, 187, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Cain, D.; Kondo, M.; Chen, H.; Kelsoe, G. Effects of acute and chronic inflammation on B-cell development and differentiation. J. Investig. Dermatol. 2009, 129, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Couldwell, G.; Machlus, K.R. Modulation of megakaryopoiesis and platelet production during inflammation. Thromb. Res. 2019, 179, 114–120. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Jones, T.B.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.C.; Ward, J.M.; Anver, M.R.; Berman, J.J.; Brayton, C.; Cardiff, R.D.; Carter, J.S.; De Coronado, S.; Downing, J.R.; Fredrickson, T.N.; et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood 2002, 100, 238–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.N.; Bejar, R. MDS overlap disorders and diagnostic boundaries. Blood 2019, 133, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Björkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; García-Macías, M.C.; Sánchez-Bernal, C.; García-Tuñón, I.; Sánchez-Yagüe, J.; Sánchez-Martín, M.; Hernández-Hernández, Á. Granuloma Formation in a Cyba-Deficient Model of Chronic Granulomatous Disease Is Associated with Myeloid Hyperplasia and the Exhaustion of B-Cell Lineage. Int. J. Mol. Sci. 2021, 22, 8701. https://doi.org/10.3390/ijms22168701
Prieto-Bermejo R, Romo-González M, Pérez-Fernández A, García-Macías MC, Sánchez-Bernal C, García-Tuñón I, Sánchez-Yagüe J, Sánchez-Martín M, Hernández-Hernández Á. Granuloma Formation in a Cyba-Deficient Model of Chronic Granulomatous Disease Is Associated with Myeloid Hyperplasia and the Exhaustion of B-Cell Lineage. International Journal of Molecular Sciences. 2021; 22(16):8701. https://doi.org/10.3390/ijms22168701
Chicago/Turabian StylePrieto-Bermejo, Rodrigo, Marta Romo-González, Alejandro Pérez-Fernández, María Carmen García-Macías, Carmen Sánchez-Bernal, Ignacio García-Tuñón, Jesús Sánchez-Yagüe, Manuel Sánchez-Martín, and Ángel Hernández-Hernández. 2021. "Granuloma Formation in a Cyba-Deficient Model of Chronic Granulomatous Disease Is Associated with Myeloid Hyperplasia and the Exhaustion of B-Cell Lineage" International Journal of Molecular Sciences 22, no. 16: 8701. https://doi.org/10.3390/ijms22168701
APA StylePrieto-Bermejo, R., Romo-González, M., Pérez-Fernández, A., García-Macías, M. C., Sánchez-Bernal, C., García-Tuñón, I., Sánchez-Yagüe, J., Sánchez-Martín, M., & Hernández-Hernández, Á. (2021). Granuloma Formation in a Cyba-Deficient Model of Chronic Granulomatous Disease Is Associated with Myeloid Hyperplasia and the Exhaustion of B-Cell Lineage. International Journal of Molecular Sciences, 22(16), 8701. https://doi.org/10.3390/ijms22168701