
Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis


Abstract
:1. Introduction
2. Fundamental Roles of ER Stress and Unfolded Protein Responses
3. Homeostatic Role of Proteolysis through Adaptive UPRs Activation in Disease Progression
4. ER Stress-Mediated Autophagy and Proteostasis
5. UPRs and Systemic Fibrosis Progression
6. UPRs in Renal Disease and Fibrosis
6.1. Disturbance of UPR Contributes to AKI-to-CKD Transition
6.2. Dysregulation of UPR Mediates Renal Fibrosis in Diabetic Nephropathy and Podocyte Defect Mice Model
7. Therapeutic Strategies: Targeting the IRE1-XBP1 and PERK-eIF2α Axis
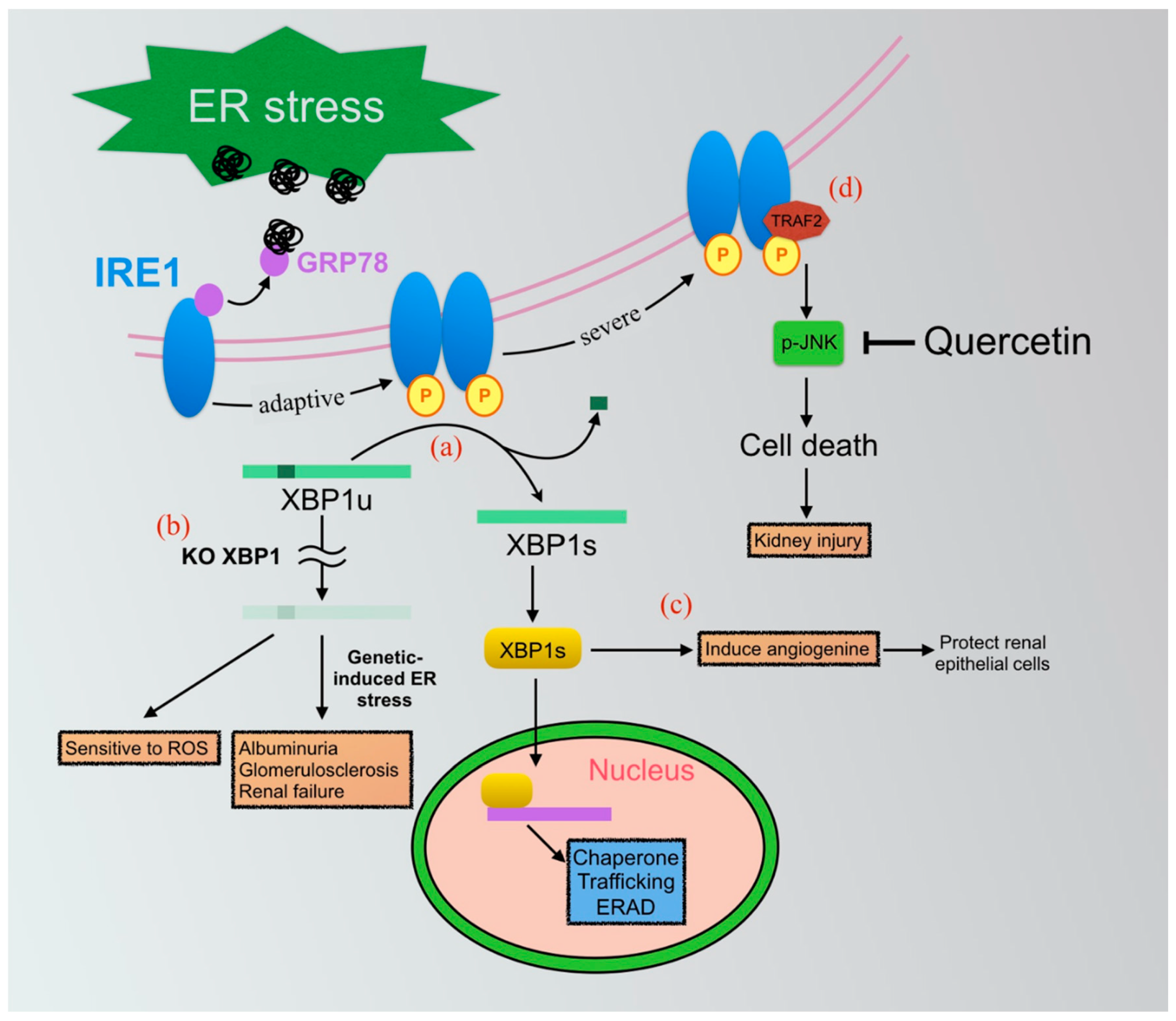
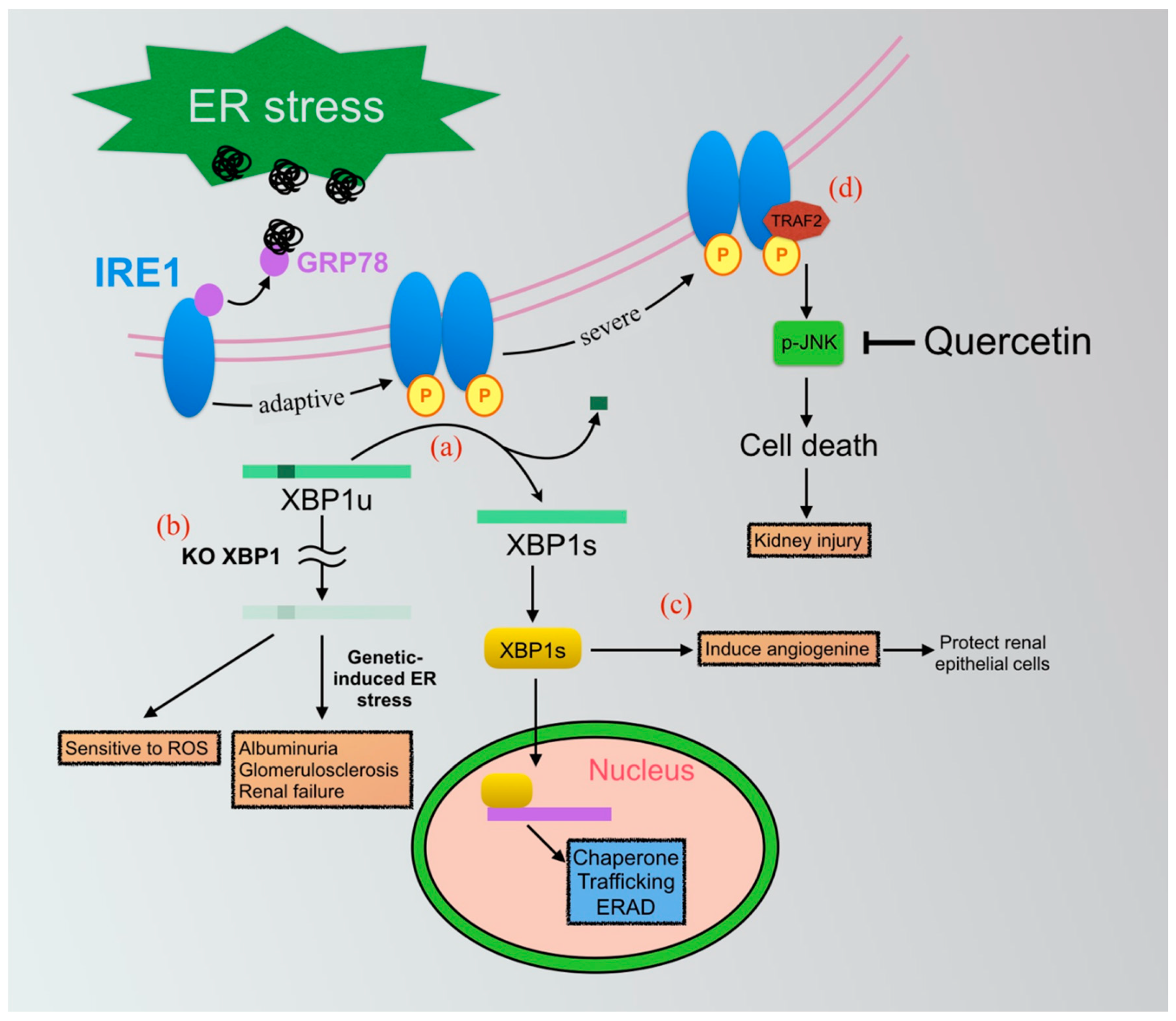
7.1. IRE1-XBP1 Axis of UPRs
7.1.1. IRE1-XBP1-Mediated Inflammasome in Renal Fibrosis
7.1.2. Application of Small Molecular Compounds Targeting the IRE1-XBP1 Pathway
Kinase-Inhibiting RNase Attenuators (KIRA)
Quercetin
XBP1 Agonists
7.2. PERK-eIF2α Axis
7.2.1. Application of Salubrinal
7.2.2. CHOP Is the Most Potential Therapeutic Target
8. Other Beneficial Effects in Moderating the ER Protein Homeostasis
8.1. Pre-Conditioning ER Stress
8.2. AMPK Activation
8.3. Roles of Chemical Chaperone 4-PBA and TUDCA

{kind=link}
| Chemical | Mechanism of Action | Animal Model | Therapeutic Effect | Ref. |
|---|---|---|---|---|
| 4-PBA | unfolded protein folding | UIRI, UUO, Dahl salt-sensitive rat, STZ-DN | Yes | [57,69,140,141] |
| GSK2606414 | PERK inhibitor | Prion infected mice | Not clear | [143] |
| Guanabenz | eIF2α phosphatase inhibitor | N/A | Not clear | [7,123] |
| KIRA6 | IRE1 RNase inhibitor | Akita Mouse | Yes | [103] |
| Salubrinal | eIF2α phosphatase inhibitor | Cisplatin, Cadmium, Arsenic, Paraquat, Cyclosporine | Not clear | [64,120,121] |
| TUDCA | unfolded protein folding | UIRI, STZ-induced DN | Yes | [87,140] |
| HLJ2 | XBP1s agonist | UC | Yes | [115,144] |
| (±)-8-ADC | XBP1s agonist | UC | Yes | [116] |
| Quercetin | IRE1 RNase activator | UUO, DN, Cadmium, ADAM | Yes | [104,105,106,107,108,109,110,111,112] |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenylbutyric acid |
| ACEI | Angiotensin-converting enzyme inhibitor |
| AKI | Acute kidney injury |
| AMPK | AMP-activated protein kinase |
| ANG | Angiogenin |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| BiP | Binding immunoglobulin protein |
| CHOP | CAAT/enhancer-binding protein (C/EBP) homologous protein |
| CKD | Chronic kidney disease |
| COX-2 | Cyclo-oxygenase-2 |
| CREB | cAMP response element-binding protein |
| DN | Diabetic nephropathy |
| EDEM | ER degradation-enhancing alpha-mannosidase-like protein |
| eIF2α | Eukaryotic translation initiation factor 2α |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| FIP | Familial interstitial pneumonia |
| GADD34 | Growth arrest and DNA-damage-inducible 34 |
| GRP78 | Glucose-regulated protein 78 |
| HDAC3 | Histone deacetylase 3 |
| IBD | Inflammatory bowel disease |
| IRE1α | Inositol-requiring enzyme 1 α |
| JNK | c-Jun amino-terminal kinases |
| KIRA | Kinase-Inhibiting RNase Attenuators |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| PDI | Protein disulfide isomerase |
| PP1 | Phosphatase-1 |
| RIDD | Regulated IRE1-dependent decay |
| ROS | Reactive oxygen species |
| SPC | Surfactant protein C |
| STZ | Streptozotocin |
| TNRF | Tumor necrosis factor receptor |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| TUDCA | Tauroursodeoxycholic acid |
| UC | Ulcerative colitis |
| UPR | Unfolded protein response |
| UUO | Unilateral ureteral obstruction |
| XBP1 | X-box binding protein 1 |
| XBP1s | Spliced isoform of XBP1 |
| XBP1u | Unspliced isoform of XBP1 |
References
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G. Chronic Dialysis and Death Among Survivors of Acute Kidney Injury Requiring Dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Risdon, R.A.; Sloper, J.C.; De Wardener, H.E. Relationship between Renal Function and Histological Changes Found in Renal-Biopsy Specimens from Patients with Persistent Glomerular Nephritis. Lancet 1968, 2, 363–366. [Google Scholar] [CrossRef]
- Clements, M.E.; Chaber, C.J.; Ledbetter, S.R.; Zuk, A. Increased Cellular Senescence and Vascular Rarefaction Exacerbate the Progression of Kidney Fibrosis in Aged Mice Following Transient Ischemic Injury. PLoS ONE 2013, 8, e70464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum—New mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef]
- Tanjore, H.; Lawson, W.E.; Blackwell, T.S. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1832, 1832, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Chevet, E.; Harding, H. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-independent Dimerization Activates the Stress Response Kinases IRE1 and PERK in the Lumen of the Endoplasmic Reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Lerner, A.G.; Walle, L.V.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A Time-Dependent Phase Shift in the Mammalian Unfolded Protein Response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, S.; Yoshida, H. The essential biology of the endoplasmic reticulum stress response for structural and computational biologists. Comput. Struct. Biotechnol. J. 2013, 6, e201303010. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Tam, A.B.; Koong, A.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wek, R.C.; Cavener, D.R. Translational control and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2357–2372. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two Distinct Stress Signaling Pathways Converge Upon the CHOP Promoter During the Mammalian Unfolded Protein Response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, S.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Li, M.; Mao, C.; Lee, A.S. Endoplasmic reticulum stress triggers an acute proteasome-dependent degradation of ATF. J. Cell. Biochem. 2004, 92, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Prywes, R. Dependence of Site-2 Protease Cleavage of ATF6 on Prior Site-1 Protease Digestion Is Determined by the Size of the Luminal Domain of ATF. J. Biol. Chem. 2004, 279, 43046–43051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366, 585–594. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Galindo, I.; Hernaez, B.; Muñoz-Moreno, R.; Cuesta-Geijo, M.A.; Dalmau-Mena, I.; Alonso, C. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012, 3, e341. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.C.; Qi, J.Y.; Thamsen, M.; Ghosh, R.; Peng, J.; Gliedt, M.J.; Meza-Acevedo, R.; Warren, R.E.; Hiniker, A.; Kim, G.E.; et al. Parallel Signaling through IRE1α and PERK Regulates Pancreatic Neuroendocrine Tumor Growth and Survival. Cancer Res. 2019, 79, 6190–6203. [Google Scholar] [CrossRef] [Green Version]
- Duwaerts, C.C.; Siao, K.; Soon, R.K.; Her, C.; Iwawaki, T.; Kohno, K.; Mattis, A.N.; Maher, J.J. Hepatocyte-specific deletion of XBP1 sensitizes mice to liver injury through hyperactivation of IRE1α. Cell Death Differ. 2021, 28, 1455–1465. [Google Scholar] [CrossRef]
- Meusser, B.; Hirsch, C.R.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Vanslyke, J.K.; Musil, L.S. Regulation of Ubiquitin-Proteasome System–mediated Degradation by Cytosolic Stress. Mol. Biol. Cell 2007, 18, 4279–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Lourie, R.; Cohen, S.B.; Ji, Y.; Goodrich, J.K.; Poole, A.C.; Ley, R.E.; Denkers, E.Y.; McGuckin, M.A.; Long, Q.; et al. Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol. Biol. Cell 2016, 27, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Gao, Y.; Zhang, M.; Deng, K.-Y.; Singh, R.; Tian, Q.; Gong, Y.; Pan, Z.; Liu, Q.; Boisclair, Y.R.; et al. Endoplasmic Reticulum–Associated Degradation (ERAD) Has a Critical Role in Supporting Glucose-Stimulated Insulin Secretion in Pancreatic β-Cells. Diabetes 2019, 68, 733–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Yagishita, N.; Aratani, S.; Saito-Fujita, T.; Morota, S.; Yamano, Y.; Hansson, M.; Inazu, M.; Kokuba, H.; Sudo, K.; et al. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC -1β. EMBO J. 2015, 34, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; Von Soly, S.K.; et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Deng, M.; Zhang, Y.; Yin, F.; Liu, J. Geniposide Increases Unfolded Protein Response-Mediating HRD1 Expression to Accelerate APP Degradation in Primary Cortical Neurons. Neurochem. Res. 2018, 43, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-Mediated Protein Degradation Causes Amyloid Precursor Protein Accumulation and Amyloid- Generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef]
- Yan, M.-M.; Ni, J.-D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Hoyerhansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA Splicing Triggers an Autophagic Response in Endothelial Cells through BECLIN-1 Transcriptional Activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Hino, S.-I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic ReticulumStress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Zhou, Y.; Cao, H.; Wen, P.; Jiang, L.; He, W.; Dai, C.; Yang, J. Autophagy Attenuates Diabetic Glomerular Damage through Protection of Hyperglycemia-Induced Podocyte Injury. PLoS ONE 2013, 8, e60546. [Google Scholar] [CrossRef]
- Qi, W.; Mu, J.; Luo, Z.-F.; Zeng, W.; Guo, Y.-H.; Pang, Q.; Ye, Z.-L.; Liu, L.; Yuan, F.-H.; Feng, B. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism 2011, 60, 594–603. [Google Scholar] [CrossRef]
- Lee, T.-H.; Yeh, C.-F.; Lee, Y.-T.; Shih, Y.-C.; Chen, Y.-T.; Hung, C.-T.; You, M.-Y.; Wu, P.-C.; Shentu, T.-P.; Huang, R.-T.; et al. Fibroblast-enriched endoplasmic reticulum protein TXNDC5 promotes pulmonary fibrosis by augmenting TGFβ signaling through TGFBR1 stabilization. Nat. Commun. 2020, 11, 4254. [Google Scholar] [CrossRef] [PubMed]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A Mutation in the Surfactant Protein C Gene Associated with Familial Interstitial Lung Disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A Surfactant Protein C Precursor Protein BRICHOS Domain Mutation Causes Endoplasmic Reticulum Stress, Proteasome Dysfunction, and Caspase 3 Activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, P.; Montenegro, J.; Vivar, R.; Letelier, A.; Urroz, P.A.; Copaja, M.; Pivet, D.; Humeres, C.; Troncoso, R.; Vicencio, J.M.; et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol. 2012, 92, 97–104. [Google Scholar] [CrossRef]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic Reticulum Stress Is Involved in Cardiac Damage and Vascular Endothelial Dysfunction in Hypertensive Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [Green Version]
- Nangaku, M. Mechanisms of Tubulointerstitial Injury in the Kidney: Final Common Pathways to End-stage Renal Failure. Intern. Med. 2004, 43, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.; Forni, L.G. Acute kidney injury: Short-term and long-term effects. Crit. Care 2016, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Coca, S.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term Risk of Mortality and Other Adverse Outcomes After Acute Kidney Injury: A Systematic Review and Meta-analysis. Am. J. kidney Dis. 2009, 53, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Inagi, R.; Kumagai, T.; Nishi, H.; Kawakami, T.; Miyata, T.; Fujita, T.; Nangaku, M. Preconditioning with Endoplasmic Reticulum Stress Ameliorates Mesangioproliferative Glomerulonephritis. J. Am. Soc. Nephrol. 2008, 19, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.T.; Weng, T.I.; Chen, L.P.; Chiang, C.K.; Liu, S.H. Involvement of caspase-12-dependent apoptotic pathway in ionic radiocontrast urografin-induced renal tubular cell injury. Toxicol. Appl. Pharmacol. 2013, 266, 167–175. [Google Scholar] [CrossRef]
- Wu, C.T.; Sheu, M.-L.; Tsai, K.-S.; Chiang, C.K.; Liu, S.H. Salubrinal, an eIF2α dephosphorylation inhibitor, enhances cisplatin-induced oxidative stress and nephrotoxicity in a mouse model. Free. Radic. Biol. Med. 2011, 51, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-K.; Hsu, S.-P.; Wu, C.-T.; Huang, J.-W.; Cheng, H.-T.; Chang, Y.-W.; Hung, K.-Y.; Wu, K.-D.; Liu, S.-H. Endoplasmic Reticulum Stress Implicated in the Development of Renal Fibrosis. Mol. Med. 2011, 17, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.L.; Sheu, M.-L.; Tsai, K.-S.; Lan, K.C.; Guan, S.S.; Wu, C.T.; Chen, L.P.; Hung, K.-Y.; Huang, J.-W.; Chiang, C.-K.; et al. CCAAT-Enhancer-Binding Protein Homologous Protein Deficiency Attenuates Oxidative Stress and Renal Ischemia-Reperfusion Injury. Antioxid. Redox Signal. 2015, 23, 1233–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Maekawa, H.; Inagi, R. Organelle crosstalk in the kidney. Kidney Int. 2019, 95, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-H.; Zhang, Z.-Y.; Sun, S.; Wu, X.-D. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress. Shock 2008, 30, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tian, J.; Qiao, X.; Su, X.; Mi, Y.; Zhang, R.; Li, R. Intermedin protects against renal ischemia-reperfusion injury by inhibiting endoplasmic reticulum stress. BMC Nephrol. 2015, 16, 169. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Xiao, W.; Lee, K.; Salem, F.; Wen, J.; He, L.; Zhang, J.; Fei, Y.; Cheng, D.; Bao, H.; et al. Inhibition of Reticulon-1A–Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 2007–2021. [Google Scholar] [CrossRef] [Green Version]
- Jao, T.-M.; Nangaku, M.; Wu, C.-H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α downregulation of PPARα promotes lipotoxicity-induced tubulointerstitial fibrosis. Kidney Int. 2019, 95, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular Interactions Between Reactive Oxygen Species and Autophagy in Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, M. Antioxidants for chronic kidney disease. Nephrology 2013, 18, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, M.; Levitin, F.; Beucken, J.V.D.; Rumantir, R.A.; Harding, N.J.; Chu, K.C.; Boutros, P.; Braakman, I.; Wouters, B. Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 2013, 203, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, Z.; Cai, L. Diabetic Cardiomyopathy and Its Prevention by Nrf2: Current Status. Diabetes Metab. J. 2014, 38, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Okamura, M.; Du, S.; Kasai, A.; Takano, Y.; Shitamura, A.; Shimada, T.; Yao, J.; et al. Involvement of Selective Reactive Oxygen Species Upstream of Proapoptotic Branches of Unfolded Protein Response. J. Biol. Chem. 2008, 283, 4252–4260. [Google Scholar] [CrossRef] [Green Version]
- Molitch, M.E.; DeFronzo, R.A.; Franz, M.J.; Keane, W.F. American Diabetes Association Nephropathy in Diabetes. Diabetes Care 2004, 27, S79–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Zhang, R.; Torreggiani, M.; Ting, A.; Xiong, H.; Striker, G.E.; Vlassara, H.; Zheng, F. Induction of Diabetes in Aged C57B6 Mice Results in Severe Nephropathy: An Association with Oxidative Stress, Endoplasmic Reticulum Stress, and Inflammation. Am. J. Pathol. 2010, 176, 2163–2176. [Google Scholar] [CrossRef]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and Renal Fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Sun, L.; Li, Y.; Shao, M.; Cheng, X.; Ge, N.; Lu, J.; Li, S. ACE-inhibitor Suppresses the Apoptosis Induced by Endoplasmic Reticulum Stress in Renal Tubular in Experimental Diabetic Rats. Exp. Clin. Endocrinol. Diabetes 2009, 117, 336–344. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Rastaldi, M.P.; Ikehata, M.; Neusser, M.A.; Kretzler, M.; Cohen, C.D.; Schlöndorff, D. Proteinuria and Hyperglycemia Induce Endoplasmic Reticulum Stress. J. Am. Soc. Nephrol. 2008, 19, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Madhusudhan, T.; Wang, H.; Dong, W.; Ghosh, S.; Bock, F.; Thangapandi, V.R.; Ranjan, S.; Wolter, J.; Kohli, S.; Shahzad, K.; et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat. Commun. 2015, 6, 6496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Betancourt, J.R.; Papillon, J.; Guillemette, J.; Iwawaki, T.; Chung, C.-F.; Cybulsky, A.V. Role of IRE1α in podocyte proteostasis and mitochondrial health. Cell Death Discov. 2020, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.; Tian, X.; Inoue, K.; Chai, N.; Liu, C.; Soda, K.; Moeckel, G.; Tufro, A.; Lee, A.-H.; Somlo, S.; et al. Essential Role of X-Box Binding Protein-1 during Endoplasmic Reticulum Stress in Podocytes. J. Am. Soc. Nephrol. 2015, 27, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Fedeles, S.; Marlier, A.; Zhang, C.; Gallagher, A.-R.; Lee, A.-H.; Somlo, S. Spliced XBP1 Rescues Renal Interstitial Inflammation Due to Loss of Sec63 in Collecting Ducts. J. Am. Soc. Nephrol. 2019, 30, 443–459. [Google Scholar] [CrossRef] [Green Version]
- Ozbek, E. Induction of Oxidative Stress in Kidney. Int. J. Nephrol. 2012, 2012, 465897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.; Luo, D.; Rando, T.; Imai, K.; Shinomura, Y. Preventing oxidative stress: A new role for XBP. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef]
- Martin, D.; Li, Y.; Yang, J.; Wang, G.; Margariti, A.; Jiang, Z.; Yu, H.; Zampetaki, A.; Hu, Y.; Xu, Q.; et al. Unspliced X-box-binding Protein 1 (XBP1) Protects Endothelial Cells from Oxidative Stress through Interaction with Histone Deacetylase. J. Biol. Chem. 2014, 289, 30625–30634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Feng, J.; Liu, Q.; Sun, F.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2008, 583, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mami, I.; Bouvier, N.; El Karoui, K.; Gallazzini, M.; Rabant, M.; Laurent-Puig, P.; Li, S.; Tharaux, P.-L.; Beaune, P.; Thervet, E.; et al. Angiogenin Mediates Cell-Autonomous Translational Control under Endoplasmic Reticulum Stress and Attenuates Kidney Injury. J. Am. Soc. Nephrol. 2015, 27, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurts, C.; Panzer, U.; Anders, H.-J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Tampe, D.; Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Vesey, D.; Cheung, C.W.; Cuttle, L.; Endre, Z.; Gobé, G.; Johnson, D. Interleukin-1β induces human proximal tubule cell injury, α-smooth muscle actin expression and fibronectin production. Kidney Int. 2002, 62, 31–40. [Google Scholar] [CrossRef]
- Turner, C.M.; Arulkumaran, N.; Singer, M.; Unwin, R.J.; Tam, F.W. Is the inflammasome a potential therapeutic target in renal disease? BMC Nephrol. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [Green Version]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 Inflammasome Promotes Renal Inflammation and Contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric Inhibition of the IRE1α RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [Green Version]
- De Vries, J.H.; Hollman, P.C.; Meyboom, S.; Buysman, M.N.; Zock, P.L.; van Staveren, W.A.; Katan, M.B. Plasma concentra-tions and urinary excretion of the antioxidant flavonols quercetin and kaempferol as biomarkers for dietary intake. Am. J. Clin. Nutr. 1998, 68, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, L.; Zhang, Y.; Lee, K.P.; Harding, H.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol Activation Defines an Unanticipated Ligand-Binding Site in the Kinase-RNase Domain of IRE. Mol. Cell 2010, 38, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-M.; Zheng, G.H.; Ming, Q.L.; Sun, J.M.; Cheng, C. Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free. Radic. Res. 2013, 47, 192–201. [Google Scholar] [CrossRef]
- Anjaneyulu, M.; Chopra, K. Quercetin, an anti-oxidant bioflavonoid, attenuates diabetic nephropathy in rats. Clin. Exp. Pharmacol. Physiol. 2004, 31, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Vicente-Sánchez, C.; Sandoval, J.S.; Egido, J.; Mayoral, P.; Arévalo, M.; Fernández-Tagarro, M.; Lopez-Novoa, J.M.; Pérez-Barriocanal, F. Protective effect of quercetin on experimental chronic cadmium nephrotoxicity in rats is based on its antioxidant properties. Food Chem. Toxicol. 2006, 44, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Shahed, A.; Shoskes, D.A. Modulation of apoptotic and inflammatory genes by bioflavonoids and angiotensin II inhibition in ureteral obstruction. Urololgy 2000, 56, 346–351. [Google Scholar] [CrossRef]
- Hu, Q.; Noor, M.; Wong, Y.F.; Hylands, P.J.; Simmonds, M.S.J.; Xu, Q.; Jiang, D.; Hendry, B.M.; Xu, Q. In vitro anti-fibrotic activities of herbal compounds and herbs. Nephrol. Dial. Transplant. 2009, 24, 3033–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Feng, Y.; Li, W.; Che, J.-P.; Wang, G.-C.; Liu, M.; Zheng, J.-H. Protective effects of quercetin and hyperoside on renal fibrosis in rats with unilateral ureteral obstruction. Exp. Ther. Med. 2014, 8, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Ding, J.; Zhang, A.; Dai, W.; Liu, S.; Diao, Z.; Wang, L.; Han, X.; Liu, W. The Inhibitory Effect of Quercetin on Asymmetric Dimethylarginine-Induced Apoptosis Is Mediated by the Endoplasmic Reticulum Stress Pathway in Glomerular Endothelial Cells. Int. J. Mol. Sci. 2014, 15, 484–503. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, Q.W.; Ye, W.C.; Wang, Y.T. Isoquinoline alkaloids from the rhizoma of Coptis chinensis. J. Nat. Med. 2007, 5, 348. [Google Scholar]
- Zhang, H.; Zhang, Z.; Song, G.; Tang, X.; Song, H.; Deng, A.; Wang, W.; Wu, L.; Qin, H. Development of an XBP1 agonist, HLJ2, as a potential therapeutic agent for ulcerative colitis. Eur. J. Pharm. Sci. 2017, 109, 56–64. [Google Scholar] [CrossRef]
- Zhang, H.; Song, G.; Zhang, Z.; Song, H.; Tang, X.; Deng, A.; Wang, W.; Wu, L.; Qin, H. Colitis Is Effectively Ameliorated by (±)-8-Acetonyl-dihydrocoptisine via the XBP1-NF-κB Pathway. Front. Pharmacol. 2017, 8, 619. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Boyce, M.; Lin, H.; Yuan, J.; Ma, D. Structure–activity relationship studies of salubrinal lead to its active biotinylated derivative. Bioorganic Med. Chem. Lett. 2005, 15, 3849–3852. [Google Scholar] [CrossRef] [PubMed]
- Jacquillet, G.; Barbier, O.; Cougnon, M.; Tauc, M.; Namorado, M.C.; Martin, D.; Reyes, J.L.; Poujeol, P. Zinc protects renal function during cadmium intoxication in the rat. Am. J. Physiol. Ren. Physiol. 2006, 290, F127–F137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, S.; Cuypers, A.; Maringwa, J.; Smeets, K.; Horemans, N.; Lambrichts, I.; Van Kerkhove, E. Low cadmium exposure triggers a biphasic oxidative stress response in mice kidneys. Toxicology 2007, 236, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Komoike, Y.; Inamura, H.; Matsuoka, M. Effects of salubrinal on cadmium-induced apoptosis in HK-2 human renal proximal tubular cells. Arch. Toxicol. 2011, 86, 37–44. [Google Scholar] [CrossRef]
- Matsuoka, M.; Komoike, Y. Experimental Evidence Shows Salubrinal, an eIF2α Dephosphorylation Inhibitor, Reduces Xenotoxicant-Induced Cellular Damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef]
- Jeon, B.J.; Yang, H.M.; Lyu, Y.S.; Pae, H.O.; Ju, S.M.; Jeon, B.H. Apigenin inhibits indoxyl sulfate-induced endoplasmic retic-ulum stress and anti-proliferative pathways, CHOP and IL-6/p21, in human renal proximal tubular cells. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2303–2310. [Google Scholar]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Guo, Y.; Fu, H.; Hu, S.; Pan, J.; Wang, Y.; Cheng, J.; Song, J.; Yu, Q.; Zhang, S.; et al. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFκB/IL-1β signaling. Cell Death Dis. 2015, 6, e1847. [Google Scholar] [CrossRef] [Green Version]
- DeZwaan-McCabe, D.; Riordan, J.; Arensdorf, A.M.; Icardi, M.; Dupuy, A.; Rutkowski, D.T. The Stress-Regulated Transcription Factor CHOP Promotes Hepatic Inflammatory Gene Expression, Fibrosis, and Oncogenesis. PLoS Genet. 2013, 9, e1003937. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.-L.; Lebeau, J.; Guillaumot, P.; Petrilli, V.; Malek, M.; Chilloux, J.; Fauvet, F.; Payen, L.; Kfoury, A.; Renno, T.; et al. p58IPK-Mediated Attenuation of the Proapoptotic PERK-CHOP Pathway Allows Malignant Progression upon Low Glucose. Mol. Cell 2013, 49, 1049–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, V.; Grosjean, F.; Tan, J.; Huang, L.; Zhu, L.; Chen, J.; Xiong, H.; Striker, G.E.; Zheng, F. CHOP deficiency results in elevated lipopolysaccharide-induced inflammation and kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 304, F440–F450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.T.; George, S.K.; Zhang, P.L.; Nigam, S.K. Pretreatment with inducers of ER molecular chaperones protects epithelial cells subjected to ATP depletion. Am. J. Physiol. Content 1999, 277, F211–F218. [Google Scholar] [CrossRef]
- Prachasilchai, W.; Sonoda, H.; Yokota-Ikeda, N.; Oshikawa, S.; Aikawa, C.; Uchida, K.; Ito, K.; Kudo, T.; Imaizumi, K.; Ikeda, M. A protective role of unfolded protein response in mouse ischemic acute kidney injury. Eur. J. Pharmacol. 2008, 592, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.L.; Lun, M.; Teng, J.; Huang, J.; Blasick, T.M.; Yin, L.; Herrera, G.A.; Cheung, J.Y. Preinduced molecular chaperones in the endoplasmic reticulum protect cardiomyocytes from lethal injury. Ann. Clin. Lab. Sci. 2004, 34, 449–457. [Google Scholar] [PubMed]
- Hung, C.-C.; Ichimura, T.; Stevens, J.L.; Bonventre, J.V. Protection of Renal Epithelial Cells against Oxidative Injury by Endoplasmic Reticulum Stress Preconditioning Is Mediated by ERK1/2 Activation. J. Biol. Chem. 2003, 278, 29317–29326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Kemp, B. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Jeong, J.U.; Chang, J.W.; Yang, W.S.; Kim, S.B.; Kil Park, S.; Park, J.S.; Lee, S.K. Activation of AMP-Activated Protein Kinase Inhibits Albumin-Induced Endoplasmic Reticulum Stress and Apoptosis through Inhibition of Reactive Oxygen Species. Nephron Exp. Nephrol. 2012, 121, e38–e48. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, S.Y.; Kim, J.-S.; Baek, C.H.; Kim, M.; Min, J.Y.; Lee, S.K. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F226–F236. [Google Scholar] [CrossRef]
- Mimori, S.; Okuma, Y.; Kaneko, M.; Kawada, K.; Hosoi, T.; Ozawa, K.; Nomura, Y.; Hamana, H. Protective Effects of 4-Phenylbutyrate Derivatives on the Neuronal Cell Death and Endoplasmic Reticulum Stress. Biol. Pharm. Bull. 2012, 35, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carducci, M.A.; Nelson, J.B.; Chan-Tack, K.M.; Ayyagari, S.R.; Sweatt, W.H.; Campbell, P.; Nelson, W.G.; Simons, J.W. Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin. Cancer Res. 1996, 2, 379–387. [Google Scholar]
- Dyer, E.S.; Paulsen, M.T.; Markwart, S.M.; Goh, M.; Livant, D.L.; Ljungman, M. Phenylbutyrate inhibits the invasive properties of prostate and breast cancer cell lines in the sea urchin embryo basement membrane invasion assay. Int. J. Cancer 2002, 101, 496–499. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Mohamed, E.; Rodriguez, P.C. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J. Immunother. Cancer 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Özcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-H.; Yang, C.-C.; Chan, D.-C.; Wu, C.-T.; Chen, L.-P.; Huang, J.-W.; Hung, K.-Y.; Chiang, C.-K. Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wen, Y.; Lv, L.-L.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Liu, B.-C. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015, 36, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral Treatment Targeting the Unfolded Protein Response Prevents Neurodegeneration and Clinical Disease in Prion-Infected Mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Tang, X.; Li, X.; Wang, Y.; Deng, A.; Wang, W.; Zhang, H.; Qin, H.; Wu, L. HLJ2 Effectively Ameliorates Colitis-Associated Cancer via Inhibition of NF-κB and Epithelial–Mesenchymal Transition. Drug Des. Dev. Ther. 2020, 14, 4291–4302. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Maly, D.J.; Papa, F.R. Druggable sensors of the unfolded protein response. Nat. Chem. Biol. 2014, 10, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-H.; Wu, C.-H.; Chiang, C.-K. Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis. Int. J. Mol. Sci. 2021, 22, 8674. https://doi.org/10.3390/ijms22168674
Chen J-H, Wu C-H, Chiang C-K. Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis. International Journal of Molecular Sciences. 2021; 22(16):8674. https://doi.org/10.3390/ijms22168674
Chicago/Turabian StyleChen, Jia-Huang, Chia-Hsien Wu, and Chih-Kang Chiang. 2021. "Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis" International Journal of Molecular Sciences 22, no. 16: 8674. https://doi.org/10.3390/ijms22168674
APA StyleChen, J.-H., Wu, C.-H., & Chiang, C.-K. (2021). Therapeutic Approaches Targeting Proteostasis in Kidney Disease and Fibrosis. International Journal of Molecular Sciences, 22(16), 8674. https://doi.org/10.3390/ijms22168674