
Protective Effects of Flavonoids Against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention

 ,
,  , , ,
, , ,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Mitochondrial Damage and Associated Impairments
2.1. Mitochondiopaties Are Involved in Cancer Development
2.2. Mitochondrial Dysfunction in Cardiovascular Diseases
2.3. Mitochondriopathies in the Neurodegeneration
3. Flavonoids Classification and Functions
4. Protective Effects of Flavonoids against Pathologies Associated with Mitochondriopathies
4.1. Preclinical Research
4.1.1. Cancer
4.1.2. Cardiovascular Diseases
4.1.3. Neurodegenerative Disorders
4.2. Clinical Data
4.2.1. Cancer
4.2.2. Cardiovascular Diseases
4.2.3. Neurodegenerative Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Natarajan, V.; Chawla, R.; Mah, T.; Vivekanandan, R.; Tan, S.Y.; Sato, P.Y.; Mallilankaraman, K. Mitochondrial Dysfunction in Age-Related Metabolic Disorders. Proteom. 2020, 20, e1800404. [Google Scholar] [CrossRef]
- Calvo, S.; Jain, M.; Xie, X.; A Sheth, S.; Chang, B.; A Goldberger, O.; Spinazzola, A.; Zeviani, M.; A Carr, S.; Mootha, V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006, 38, 576–582. [Google Scholar] [CrossRef]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Hum. Hypothal. Neuropsychiatr. Disord. 2018, 145, 147–155. [Google Scholar] [CrossRef]
- Golubnitschaja, O.; Topolcan, O.; Kucera, R.; Costigliola, V. Anniversary of the European Association for Predictive, Preventive and Personalised (3P) Medicine–EPMA World Congress Supplement 2020. EPMA J. 2020, 11, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Crigna, A.T.; Samec, M.; Koklesova, L.; Liskova, A.; Giordano, F.A.; Kubatka, P.; Golubnitschaja, O. Cell-free nucleic acid patterns in disease prediction and monitoring—hype or hope? EPMA J. 2020, 11, 603–627. [Google Scholar] [CrossRef]
- Stastny, I.; Zubor, P.; Kajo, K.; Kubatka, P.; Golubnitschaja, O.; Dankova, Z. Aberrantly Methylated cfDNA in Body Fluids as a Promising Diagnostic Tool for Early Detection of Breast Cancer. Clin. Breast Cancer 2020, 20, e711–e722. [Google Scholar] [CrossRef]
- Gerner, C.; Costigliola, V.; Golubnitschaja, O. Multiomic patterns in body fluids: Technological challenge with a great potential to implement the advanced paradigm of 3P medicine. Mass Spectrom. Rev. 2020, 39, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Sica, D.A. Drug Absorption in the Management of Congestive Heart Failure: Loop Diuretics. Congest. Hear. Fail. 2003, 9, 287–292. [Google Scholar] [CrossRef]
- Barrett, M.; Boyne, J.; Brandts, J.; Rocca, H.-P.B.-L.; De Maesschalck, L.; De Wit, K.; Dixon, L.; Eurlings, C.; Fitzsimons, D.; Golubnitschaja, O.; et al. Artificial intelligence supported patient self-care in chronic heart failure: a paradigm shift from reactive to predictive, preventive and personalised care. EPMA J. 2019, 10, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Golubnitschaja, O.; Costigliola, V. Common origin but individual outcomes: time for new guidelines in personalized healthcare. Pers. Med. 2010, 7, 561–568. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Koklesova, L.; Samec, M.; Liskova, A.; Zhai, K.; Büsselberg, D.; Giordano, F.A.; Kubatka, P.; Golunitschaja, O. Mitochondrial impairments in aetiopathology of multifactorial diseases: common origin but individual outcomes in context of 3P medicine. EPMA J. 2021, 12, 27–40. [Google Scholar] [CrossRef]
- Liu, W.; Li, W.; Liu, H.; Yu, X. Xanthohumol inhibits colorectal cancer cells via downregulation of Hexokinases II-mediated glycolysis. Int. J. Biol. Sci. 2019, 15, 2497–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; Mackenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-M.; Dong, X.; Xue, X.-D.; Zhang, J.; Li, Z.; Wu, H.-J.; Yang, Z.-L.; Yang, Y.; Wang, H.-S. Naringenin improves mitochondrial function and reduces cardiac damage following ischemia-reperfusion injury: the role of the AMPK-SIRT3 signaling pathway. Food Funct. 2019, 10, 2752–2765. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Song, H.; Fan, M.; You, F.; Zhang, L.; Luo, J.; Li, J.; Wang, L.; Li, C.; Yuan, M. Luteolin attenuates sepsis-induced myocardial injury by enhancing autophagy in mice. Int. J. Mol. Med. 2020, 45, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Karuppagounder, S.; Madathil, S.; Pandey, M.; Haobam, R.; Rajamma, U.; Mohanakumar, K. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience 2013, 236, 136–148. [Google Scholar] [CrossRef]
- Chen, L.; Feng, P.; Peng, A.; Qiu, X.; Lai, W.; Zhang, L. Protective effects of isoquercitrin on streptozotocin-induced neurotoxicity. J. Cell. Mol. Med. 2020, 24, 10458–10467. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Bakhoda, M.R.; Bahmanpour, Z.; Ilkhani, K.; Zarrabi, A.; Makvandi, P.; Khan, H.; Mazaheri, S.; Darvish, M.; Mirzaei, H. Apigenin as Tumor Suppressor in Cancers: Biotherapeutic Activity, Nanodelivery, and Mechanisms With Emphasis on Pancreatic Cancer. Front. Chem. 2020, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, J.C.S.; Srirattana, K.; Tsai, T.-S.; Sun, X. The mitochondrial genome: how it drives fertility. Reprod. Fertil. Dev. 2018, 30, 118–139. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Mandavilli, B.S.; Santos, J.H.; Van Houten, B. Mitochondrial DNA repair and aging. Mutat. Res. Mol. Mech. Mutagen. 2002, 509, 127–151. [Google Scholar] [CrossRef]
- Bajpai, R.; Sharma, A.; Achreja, A.; Edgar, C.L.; Wei, C.; Siddiqa, A.A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Czarny, P.; Wigner, P.; Galecki, P.; Sliwinski, T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatr. 2018, 80, 309–321. [Google Scholar] [CrossRef]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- A Butow, R.; Avadhani, N.G. Mitochondrial Signaling: The Retrograde Response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Picard, M.; Hirano, M. Disentangling (Epi)Genetic and Environmental Contributions to the Mitochondrial 3243A>G Mutation Phenotype. JAMA Neurol. 2016, 73, 923–925. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- Wei, Y.-H.; Wu, S.-B.; Ma, Y.-S.; Lee, H.-C. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang. Gung Med. J. 2009, 32, 113–132. [Google Scholar]
- Boggan, R.; Lim, A.; Taylor, R.W.; McFarland, R.; Pickett, S.J. Resolving complexity in mitochondrial disease: Towards precision medicine. Mol. Genet. Metab. 2019, 128, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W.G. Mitochondrial apoptosis: killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.M.; Kim, K. Protein kinase CK2 modulation of pyruvate kinase M isoforms augments the Warburg effect in cancer cells. J. Cell. Biochem. 2018, 119, 8501–8510. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosom. Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Horton, T.M.; Petros, J.A.; Heddi, A.; Shoffner, J.; Kaufman, A.E.; Graham, S.D.; Gramlich, T.; Wallace, D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosom. Cancer 1996, 15, 95–101. [Google Scholar] [CrossRef]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Pustylnikov, S.; Costabile, F.; Beghi, S.; Facciabene, A. Targeting mitochondria in cancer: current concepts and immunotherapy approaches. Transl. Res. 2018, 202, 35–51. [Google Scholar] [CrossRef]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K.-Y. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Farhood, B.; Ashrafizadeh, M.; Khodamoradi, E.; Hoseini-Ghahfarokhi, M.; Afrashi, S.; Musa, A.E.; Najafi, M. Targeting of cellular redox metabolism for mitigation of radiation injury. Life Sci. 2020, 250, 117570. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of Mitochondrial Genetics and Metabolism in Cancer Development. Cold Spring Harb. Perspect. Biol. 2013, 5, a011411. [Google Scholar] [CrossRef] [Green Version]
- Maaliki, D.; A Shaito, A.; Pintus, G.; El-Yazbi, A.; Eid, A.H. Flavonoids in hypertension: a brief review of the underlying mechanisms. Curr. Opin. Pharmacol. 2019, 45, 57–65. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pinto, A.; Popolo, A. Mitochondria and Cardiovascular Disease: A Brief Account. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Suomalainen-Wartiovaara, A. Mouse models of mitochondrial DNA defects and their relevance for human disease. EMBO Rep. 2009, 10, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbria, M.; Ramos, A.; Aluja, M.P.; Santos, C. The role of control region mitochondrial DNA mutations in cardiovascular disease: stroke and myocardial infarction. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-J.; Li, C.G. Tanshinone IIA and Cryptotanshinone Prevent Mitochondrial Dysfunction in Hypoxia-Induced H9c2 Cells: Association to Mitochondrial ROS, Intracellular Nitric Oxide, and Calcium Levels. Evid. Based Complement. Altern. Med. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R.; Barros, M.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.B.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.; Vercesi, A.E. Mitochondria as a Source of Reactive Oxygen and Nitrogen Species: From Molecular Mechanisms to Human Health. Antioxidants Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q.; et al. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free. Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Palaniyandi, S.S.; Qi, X.; Yogalingam, G.; Ferreira, J.C.B.; Mochly-Rosen, D. Regulation of mitochondrial processes: A target for heart failure. Drug Discov. Today: Dis. Mech. 2010, 7, e95–e102. [Google Scholar] [CrossRef] [Green Version]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1544–1554. [Google Scholar] [CrossRef]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [Green Version]
- Samarghandian, S.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Folgado, S.L.; Rajabpour-Sanati, A.; Khazdair, M.R. Green tea catechins inhibit microglial activation which prevents the development of neurological disorders. Neural Regen. Res. 2020, 15, 1792–1798. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial Diseases of the Brain. Free. Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Liskova, A.; Samec, M.; Koklesova, L.; Kudela, E.; Kubatka, P.; Golubnitschaja, O. Mitochondriopathies as a Clue to Systemic Disorders—Analytical Tools and Mitigating Measures in Context of Predictive, Preventive, and Personalized (3P) Medicine. Int. J. Mol. Sci. 2021, 22, 2007. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S. Molecular and Cellular Basis of Neurodegeneration in Alzheimer’s Disease. Mol. Cells 2017, 40, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Bai, F. The Association of Tau With Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [Green Version]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [Green Version]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Balestrino, R.; Schapira, A.H. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: an overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, M.; Koketsu, M. Minor Flavonoids (Chalcones, Flavanones, Dihydrochalcones, and Aurones). Nat. Prod. 2013, 1867–1900. [Google Scholar]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef] [PubMed]
- Ciumărnean, L.; Milaciu, M.V.; Runcan, O.; Vesa, S.C.; Răchișan, A.L.; Negrean, V.; Perné, M.-G.; Donca, V.I.; Alexescu, T.-G.; Para, I.; et al. The effects of favonoids in cardiovascular diseases. Molecules 2020, 25, 4320. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Líšková, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The Immunomodulatory and Anti-Inflammatory Role of Polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Guan, L.-P.; Liu, B.-Y. Antidepressant-like effects and mechanisms of flavonoids and related analogues. Eur. J. Med. Chem. 2016, 121, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Mirossay, L.; Varinská, L.; Mojžiš, J. Antiangiogenic Effect of Flavonoids and Chalcones: An Update. Int. J. Mol. Sci. 2017, 19, 27. [Google Scholar] [CrossRef] [Green Version]
- Manish, P.; Wei Ling, L.; Seong Lin, T.; Mohamad Fairuz, Y. Flavonoids and its Neuroprotective Effects on Brain Ischemia and Neurodegenerative Diseases. Curr. Drug Targets 2018, 19, 1710–1720. [Google Scholar] [CrossRef]
- Kozłowska, A.; Szostak-Wegierek, D. Flavonoids--food sources and health benefits. Rocz. Państw. Zakł. Hig. 2014, 65, 79–85. [Google Scholar]
- Prochazkova, D.; Boušová, I.; Wilhelmová, N. Antioxidant and prooxidant properties of flavonoids. In Fitoterapia; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Airoldi, C.; La Ferla, B.; D’Orazio, G.; Ciaramelli, C.; Palmioli, A. Flavonoids in the Treatment of Alzheimer’s and Other Neurodegenerative Diseases. Curr. Med. Chem. 2018, 25, 3228–3246. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a Flavonoid with Potential for Cancer Prevention and Therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Constantin, R.P.; Constantin, J.; Pagadigorria, C.L.S.; Ishii-Iwamoto, E.L.; Bracht, A.; De Castro, C.V.; Yamamoto, N.S. Prooxidant activity of fisetin: Effects on energy metabolism in the rat liver. J. Biochem. Mol. Toxicol. 2010, 25, 117–126. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. BioMed Res. Int. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Lee-Hilz, Y.Y.; Boerboom, A.-M.J.F.; Westphal, A.H.; Van Berkel, W.J.H.; Aarts, J.M.M.J.G.; Rietjens, I.M.C.M. Pro-Oxidant Activity of Flavonoids Induces EpRE-Mediated Gene Expression. Chem. Res. Toxicol. 2006, 19, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Eren-Guzelgun, B.; Ince, E.; Gurer-Orhan, H. In vitro antioxidant/prooxidant effects of combined use of flavonoids. Nat. Prod. Res. 2017, 32, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Minihane, A.M. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-García, C.; Sánchez-Quesada, C.; Gaforio, J.J. Dietary Flavonoids as Cancer Chemopreventive Agents: An Updated Review of Human Studies. Antioxidants 2019, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Koes, R.; Verweij, W.; Quattrocchio, F.M. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant. Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Rees, A.; Dodd, G.F.; Spencer, J.P.E. The Effects of Flavonoids on Cardiovascular Health: A Review of Human Intervention Trials and Implications for Cerebrovascular Function. Nutrients 2018, 10, 1852. [Google Scholar] [CrossRef] [Green Version]
- Shan, S.; Shi, J.; Yang, P.; Jia, B.; Wu, H.; Zhang, X.; Li, Z. Apigenin Restrains Colon Cancer Cell Proliferation via Targeted Blocking of Pyruvate Kinase M2-Dependent Glycolysis. J. Agric. Food Chem. 2017, 65, 8136–8144. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.-X.; Wang, J.-J.; Leung, T.H.Y.; Han, C.Y.; Tsang, B.K.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers 2019, 11, 813. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, P.L. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211–222. [Google Scholar] [CrossRef]
- Luo, Q.; Wu, X.; Zhao, P.; Nan, Y.; Chang, W.; Zhu, X.; Su, D.; Liu, Z. OTUD1 Activates Caspase-Independent and Caspase-Dependent Apoptosis by Promoting AIF Nuclear Translocation and MCL1 Degradation. Adv. Sci. 2021, 8, 2002874. [Google Scholar] [CrossRef]
- Hou, S.; Song, Y.; Sun, D.; Zhu, S.; Wang, Z. Xanthohumol-Induced Rat Glioma C6 Cells Death by Triggering Mitochondrial Stress. Int. J. Mol. Sci. 2021, 22, 4506. [Google Scholar] [CrossRef]
- Deng, X.; Wang, Q.; Cheng, M.; Chen, Y.; Yan, X.; Guo, R.; Sun, L.; Li, Y.; Liu, Y. Pyruvate dehydrogenase kinase 1 interferes with glucose metabolism reprogramming and mitochondrial quality control to aggravate stress damage in cancer. J. Cancer 2020, 11, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K.; Ji, J.; Haam, K.; Han, T.-H.; Lim, S.; Kang, M.-J.; Lim, S.S.; Ban, H.S. Licochalcone A inhibits hypoxia-inducible factor-1α accumulation by suppressing mitochondrial respiration in hypoxic cancer cells. Biomed. Pharmacother. 2021, 133, 111082. [Google Scholar] [CrossRef]
- Phan, T.N.; Kim, O.; Ha, M.T.; Hwangbo, C.; Min, B.-S.; Lee, J.-H. Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth. Int. J. Mol. Sci. 2020, 21, 9502. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhang, P.; Sun, Z.; Liu, X.; Zhang, X.; Liu, X.; Wang, D.; Meng, Z. Lysionotin induces apoptosis of hepatocellular carcinoma cells via caspase-3 mediated mitochondrial pathway. Chem. Interact. 2021, 344, 109500. [Google Scholar] [CrossRef]
- Maués, L.; Alves, G.; Couto, N.; Da Silva, B.; Arruda, M.; Macchi, B.; Sena, C.; Prado, A.; Crespo-Lopez, M.; Silva, E.; et al. Flavonoids from the Amazon plant Brosimum acutifolium induce C6 glioma cell line apoptosis by disrupting mitochondrial membrane potential and reducing AKT phosphorylation. Biomed. Pharmacother. 2019, 113, 108728. [Google Scholar] [CrossRef]
- Won, Y.-S.; Kim, J.-H.; Lizardo, R.C.M.; Min, H.-J.; Cho, H.-D.; Hong, S.-M.; Seo, K.-I. The Flavonol Isoquercitrin Promotes Mitochondrial-Dependent Apoptosis in SK-Mel-2 Melanoma Cell via the PI3K/AKT/mTOR Pathway. Nutrients 2020, 12, 3683. [Google Scholar] [CrossRef]
- Rajendran, P.; Maheshwari, U.; Muthukrishnan, A.; Muthuswamy, R.; Anand, K.; Ravindran, B.; Dhanaraj, P.; Balamuralikrishnan, B.; Chang, S.W.; Chung, W.J. Myricetin: versatile plant based flavonoid for cancer treatment by inducing cell cycle arrest and ROS–reliant mitochondria-facilitated apoptosis in A549 lung cancer cells and in silico prediction. Mol. Cell. Biochem. 2021, 476, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Iyangar, R.M.; Devaraj, E. Silibinin Triggers the Mitochondrial Pathway of Apoptosis in Human Oral Squamous Carcinoma Cells. Asian Pac. J. Cancer Prev. 2020, 21, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, I.G.; Perkins, N.D. Hypoxia induces rapid, STAT3 and ROS dependent, mitochondrial translocation of RelA(p65) and IκBα. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Zapolska-Downar, D.; Bryk, D.; Małecki, M.; Hajdukiewicz, K.; Sitkiewicz, D. Aronia melanocarpa fruit extract exhibits anti-inflammatory activity in human aortic endothelial cells. Eur. J. Nutr. 2012, 51, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free. Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Sozański, T.; Kucharska, A.; Szumny, A.; Magdalan, J.; Bielska, K.; Merwid-Ląd, A.; Woźniak-Biel, A.; Dzimira, S.; Piórecki, N.; Trocha, M. The protective effect of the Cornus mas fruits (cornelian cherry) on hypertriglyceridemia and atherosclerosis through PPARα activation in hypercholesterolemic rabbits. Phytomedicine 2014, 21, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ku, H.J.; Kim, J.K.; Park, J.-W.; Lee, J.H. Amelioration of High Fructose-Induced Cardiac Hypertrophy by Naringin. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.-P.; Shao, Q.; Li, P.-F.; Sun, Y.; Luo, L.-Z.; Yan, X.-Q.; Fan, Z.-Y.; Hu, J.; Zhao, J.; et al. Brain-derived neurotrophic factor mimetic, 7,8-dihydroxyflavone, protects against myocardial ischemia by rebalancing optic atrophy 1 processing. Free. Radic. Biol. Med. 2019, 145, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Du, J.; Pan, Y.; Chen, T.; Zhao, L.; Zhu, Y.; Chen, Y.; Zheng, Y.; Liu, Y.; Sun, L.; et al. Activation of cardiac TrkB receptor by its small molecular agonist 7,8-dihydroxyflavone inhibits doxorubicin-induced cardiotoxicity via enhancing mitochondrial oxidative phosphorylation. Free. Radic. Biol. Med. 2019, 130, 557–567. [Google Scholar] [CrossRef]
- Wu, B.; Lin, J.; Luo, J.; Han, D.; Fan, M.; Guo, T.; Tao, L.; Yuan, M.; Yi, F. Dihydromyricetin Protects against Diabetic Cardiomyopathy in Streptozotocin-Induced Diabetic Mice. BioMed Res. Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Alexandre, J.V.D.L.; Viana, Y.I.P.; David, C.E.B.; Cunha, P.L.O.; Albuquerque, A.C.; Varela, A.L.N.; Kowaltowski, A.J.; Facundo, H.T. Quercetin treatment increases H2O2 removal by restoration of endogenous antioxidant activity and blocks isoproterenol-induced cardiac hypertrophy. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 217–226. [Google Scholar] [CrossRef]
- Song, Y.-H.; Cai, H.; Zhao, Z.-M.; Chang, W.-J.; Gu, N.; Cao, S.-P.; Wu, M.-L. Icariin attenuated oxidative stress induced-cardiac apoptosis by mitochondria protection and ERK activation. Biomed. Pharmacother. 2016, 83, 1089–1094. [Google Scholar] [CrossRef]
- Li, F.; Lang, F.; Wang, Y.; Zhai, C.; Zhang, C.; Zhang, L.; Hao, E. Cyanidin ameliorates endotoxin-induced myocardial toxicity by modulating inflammation and oxidative stress through mitochondria and other factors. Food Chem. Toxicol. 2018, 120, 104–111. [Google Scholar] [CrossRef]
- Jiang, H.; Xing, J.; Fang, J.; Wang, L.; Wang, Y.; Zeng, L.; Li, Z.; Liu, R. Tilianin Protects against Ischemia/Reperfusion-Induced Myocardial Injury through the Inhibition of the Ca2+/Calmodulin-Dependent Protein Kinase II-Dependent Apoptotic and Inflammatory Signaling Pathways. BioMed Res. Int. 2020, 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, K.; Ravindran, S.; Kurian, G.A.; Rajesh, M. Fisetin Confers Cardioprotection against Myocardial Ischemia Reperfusion Injury by Suppressing Mitochondrial Oxidative Stress and Mitochondrial Dysfunction and Inhibiting Glycogen Synthase Kinase 3β Activity. Oxidative Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, K.; Boovarahan, S.R.; Prem, P.; Sivakumar, B.; A Kurian, G. Fisetin Attenuates Myocardial Ischemia-Reperfusion Injury by Activating the Reperfusion Injury Salvage Kinase (RISK) Signaling Pathway. Front. Pharmacol. 2021, 12, 566470. [Google Scholar] [CrossRef]
- Wu, J.; Chen, H.; Qin, J.; Chen, N.; Lu, S.; Jin, J.; Li, Y. Baicalin Improves Cardiac Outcome and Survival by Suppressing Drp1-Mediated Mitochondrial Fission after Cardiac Arrest-Induced Myocardial Damage. Oxidative Med. Cell. Longev. 2021, 2021, 1–14. [Google Scholar] [CrossRef]
- Bondy, S.C. The neurotoxicity of environmental aluminum is still an issue. NeuroToxicology 2010, 31, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Shur, B.; Kumar, A. Naringin protects memory impairment and mitochondrial oxidative damage against aluminum-induced neurotoxicity in rats. Int. J. Neurosci. 2013, 123, 636–645. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guáqueta, A.M.; Manco, J.I.M.; Ramírez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gómez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacol. 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Godoy, J.; Lindsay, C.B.; Quintanilla, R.A.; Carvajal, F.J.; Cerpa, W.; Inestrosa, N.C. Quercetin Exerts Differential Neuroprotective Effects Against H2O2 and Aβ Aggregates in Hippocampal Neurons: the Role of Mitochondria. Mol. Neurobiol. 2017, 54, 7116–7128. [Google Scholar] [CrossRef]
- Markham, A.; Bains, R.; Franklin, P.; Spedding, M. Changes in mitochondrial function are pivotal in neurodegenerative and psychiatric disorders: How important is BDNF? Br. J. Pharmacol. 2014, 171, 2206–2229. [Google Scholar] [CrossRef] [Green Version]
- Arnould, T.; Vankoningsloo, S.; Renard, P.; Houbion, A.; Ninane, N.; Demazy, C.; Remacle, J.; Raes, M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 2002, 21, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Molecular mechanisms underlying protective effects of quercetin against mitochondrial dysfunction and progressive dopaminergic neurodegeneration in cell culture and MitoPark transgenic mouse models of Parkinson’s Disease. J. Neurochem. 2017, 141, 766–782. [Google Scholar] [CrossRef]
- Kim, E.; Park, M.; Jeong, J.; Kim, H.; Lee, S.K.; Lee, E.; Oh, B.H.; Namkoong, K. Cholinesterase Inhibitor Donepezil Increases Mitochondrial Biogenesis through AMP-Activated Protein Kinase in the Hippocampus. Neuropsychobiology 2016, 73, 81–91. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Wiehager, B.; Sakai, J.; Frykman, S.; Behbahani, H.; Winblad, B.; Ankarcrona, M. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 2011, 25, 78–88. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, Q.; Chen, J.-Y.; OuYang, D.; Lu, J.-H. The pharmacological activity of epigallocatechin-3-gallate (EGCG) on Alzheimer’s disease animal model: A systematic review. Phytomedicine 2020, 79, 153316. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Gomez, M.V.S.; Ruiz, A.; Cavaliere, F.; Ortiz-Sanz, C.; Quintela, T.; Capetillo-Zarate, E.; Solé-Domènech, S.; Matute, C. Mangiferin and Morin Attenuate Oxidative Stress, Mitochondrial Dysfunction, and Neurocytotoxicity, Induced by Amyloid Beta Oligomers. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Wang, W.-W.; Han, R.; He, H.-J.; Li, J.; Chen, S.-Y.; Gu, Y.; Xie, C. Administration of quercetin improves mitochondria quality control and protects the neurons in 6-OHDA-lesioned Parkinson’s disease models. Aging 2021, 13, 11738–11751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Xuan, Z.-H.; Tian, S.; He, G.-R.; Du, G.-H. Myricitrin attenuates 6-hydroxydopamine-induced mitochondrial damage and apoptosis in PC12 cells via inhibition of mitochondrial oxidation. J. Funct. Foods 2013, 5, 337–345. [Google Scholar] [CrossRef]
- Cai, Z.; Zeng, W.; Tao, K.; Lu, F.; Gao, G.; Yang, Q. Myricitrin alleviates MPP+-induced mitochondrial dysfunction in a DJ-1-dependent manner in SN4741 cells. Biochem. Biophys. Res. Commun. 2015, 458, 227–233. [Google Scholar] [CrossRef]
- Tamilselvam, K.; Braidy, N.; Manivasagam, T.; Essa, M.M.; Prasad, R.; Karthikeyan, S.; Thenmozhi, A.J.; Selvaraju, S.; Guillemin, G.J. Neuroprotective Effects of Hesperidin, a Plant Flavanone, on Rotenone-Induced Oxidative Stress and Apoptosis in a Cellular Model for Parkinson’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fang, J.; Xing, J.; Wang, L.; Wang, Q.; Wang, Y.; Li, Z.; Liu, R. Tilianin mediates neuroprotection against ischemic injury by attenuating CaMKII-dependent mitochondrion-mediated apoptosis and MAPK/NF-κB signaling. Life Sci. 2018, 216, 233–245. [Google Scholar] [CrossRef]
- Chen, S.; Sun, M.; Zhao, X.; Yang, Z.; Liu, W.; Cao, J.; Qiao, Y.; Luo, X.; Wen, A. Neuroprotection of hydroxysafflor yellow A in experimental cerebral ischemia/reperfusion injury via metabolic inhibition of phenylalanine and mitochondrial biogenesis. Mol. Med. Rep. 2019, 19, 3009–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Wu, S.-P.; Wang, N.; Seto, S.; Chang, D. Hydroxysafflor yellow A alleviates cerebral ischemia reperfusion injury by suppressing apoptosis via mitochondrial permeability transition pore. Phytomedicine 2021, 85, 153532. [Google Scholar] [CrossRef] [PubMed]
- Amarsanaa, K.; Kim, H.-J.; Ko, E.-A.; Jo, J.; Jung, J.J.A.S.-C. Nobiletin Exhibits Neuroprotective Effects against Mitochondrial Complex I Inhibition via Regulating Apoptotic Signaling. Exp. Neurobiol. 2021, 30, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Teleman, A.A. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg phenotype—concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J. 2020, 11, 377–398. [Google Scholar] [CrossRef]
- Hurley, D.M.; Williams, E.R.; Cross, J.M.; Riedinger, B.R.; Meyer, R.A.; Abela, G.S.; Slade, J.M. Aerobic Exercise Improves Microvascular Function in Older Adults. Med. Sci. Sports Exerc. 2019, 51, 773–781. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Qiao, Y.; Xiang, S.; Li, W.; Gan, Y.; Chen, Y. Work stress and the risk of cancer: A meta-analysis of observational studies. Int. J. Cancer 2019, 144, 2390–2400. [Google Scholar] [CrossRef]
- Kunin, A.; Polivka, J.; Moiseeva, N.; Golubnitschaja, O. “Dry mouth” and “Flammer” syndromes—neglected risks in adolescents and new concepts by predictive, preventive and personalised approach. EPMA J. 2018, 9, 307–317. [Google Scholar] [CrossRef]
- Tidwell, T.R.; Søreide, K.; Hagland, H.R. Aging, Metabolism, and Cancer Development: from Peto’s Paradox to the Warburg Effect. Aging Dis. 2017, 8, 662–676. [Google Scholar] [CrossRef] [Green Version]
- Murata, M. Inflammation and cancer. Environ. Heal. Prev. Med. 2018, 23, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrino, M.; Cooke-Barber, J.; Dasgupta, R.; Geller, J.I. Genetic predisposition to cancer: Surveillance and intervention. Semin. Pediatr. Surg. 2019, 28, 150858. [Google Scholar] [CrossRef]
- Winnard, P.T.; Pathak, A.P.; Dhara, S.; Cho, S.Y.; Raman, V.; Pomper, M.G. Molecular Imaging of Metastatic Potential. J. Nucl. Med. 2008, 49, 96S–112S. [Google Scholar] [CrossRef] [Green Version]
- Golubnitschaja, O.; Flammer, J. Individualised patient profile: clinical utility of Flammer syndrome phenotype and general lessons for predictive, preventive and personalised medicine. EPMA J. 2018, 9, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grech, G.; Zhan, X.; Yoo, B.C.; Bubnov, R.; Hagan, S.; Danesi, R.; Vittadini, G.; Desiderio, D.M. EPMA position paper in cancer: current overview and future perspectives. EPMA J. 2015, 6, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhan, X. Signaling pathway network alterations in human ovarian cancers identified with quantitative mitochondrial proteomics. EPMA J. 2019, 10, 153–172. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, H.; Wang, Y.; Cao, L.; Zhan, X. Quantitative proteomics revealed energy metabolism pathway alterations in human epithelial ovarian carcinoma and their regulation by the antiparasite drug ivermectin: data interpretation in the context of 3P medicine. EPMA J. 2020, 11, 661–694. [Google Scholar] [CrossRef]
- Chan, Y.-H.; Lau, K.-K.; Yiu, K.-H.; Li, S.-W.; Chan, H.-T.; Fong, D.; Tam, S.; Lau, C.-P.; Tse, H.-F. Reduction of C-reactive protein with isoflavone supplement reverses endothelial dysfunction in patients with ischaemic stroke. Eur. Hear. J. 2008, 29, 2800–2807. [Google Scholar] [CrossRef] [Green Version]
- Grosso, G.; Micek, A.; Godos, J.; Pajak, A.; Sciacca, S.; Galvano, F.; Giovannucci, E.L. Dietary Flavonoid and Lignan Intake and Mortality in Prospective Cohort Studies: Systematic Review and Dose-Response Meta-Analysis. Am. J. Epidemiol. 2017, 185, 1304–1316. [Google Scholar] [CrossRef]
- Peters, U.; Poole, C.; Arab, L. Does Tea Affect Cardiovascular Disease? A Meta-Analysis. Am. J. Epidemiol. 2001, 154, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkenheim, A.; Szabó, B.; Áron, J.; Sztaniszláv. Hereditary transthyretin amyloidosis caused by the rare Phe33Leu mutation. BMJ Case Rep. 2020, 13, e232756. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Stone, N.; Robinson, J.G.; Lichtenstein, A.H.; Merz, C.N.B.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.; et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.J.; Carvalho, R.A.; Portincasa, P.; Bonfrate, L.; Sardao, V.A. Fatty Acid Oxidation and Cardiovascular Risk during Menopause: A Mitochondrial Connection? J. Lipids 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sathyapalan, T.; Aye, M.; Rigby, A.; Thatcher, N.; Dargham, S.; Kilpatrick, E.; Atkin, S. Soy isoflavones improve cardiovascular disease risk markers in women during the early menopause. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 691–697. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Vigo, D.E. Melatonin, mitochondria, and the metabolic syndrome. Cell. Mol. Life Sci. 2017, 74, 3941–3954. [Google Scholar] [CrossRef]
- Basu, A.; Du, M.; Leyva, M.J.; Sanchez, K.; Betts, N.M.; Wu, M.; Aston, C.E.; Lyons, T.J. Blueberries Decrease Cardiovascular Risk Factors in Obese Men and Women with Metabolic Syndrome. J. Nutr. 2010, 140, 1582–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Betts, N.M.; Ortiz, J.; Simmons, B.; Wu, M.; Lyons, T.J. Low-energy cranberry juice decreases lipid oxidation and increases plasma antioxidant capacity in women with metabolic syndrome. Nutr. Res. 2011, 31, 190–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, A.; Bertoia, M.; Chiuve, S.; Flint, A.; Forman, J.; Rimm, E.B. Habitual intake of anthocyanins and flavanones and risk of cardiovascular disease in men. Am. J. Clin. Nutr. 2016, 104, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Naruszewicz, M.; Łaniewska, I.; Millo, B.; Dłużniewski, M. Combination therapy of statin with flavonoids rich extract from chokeberry fruits enhanced reduction in cardiovascular risk markers in patients after myocardial infraction (MI). Atherosclerosis 2007, 194, e179–e184. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-J.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhouafli, Z.; Cuanalo-Contreras, K.; Hayouni, E.A.; Mays, C.E.; Soto, C.; Moreno-Gonzalez, I. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; The PROMESA study group; Maaß, S.; Schuberth, M.; Giese, A.; Oertel, W.H.; Poewe, W.; Trenkwalder, C.; Wenning, G.K.; Mansmann, U.; et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2019, 18, 724–735. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Morillas-Ruiz, J.; Rubio-Perez, J.M.; Albaladejo, M.; Zafrilla, P.; Parra, S.; Vidal-Guevara, M. Effect of an antioxidant drink on homocysteine levels in Alzheimer’s patients. J. Neurol. Sci. 2010, 299, 175–178. [Google Scholar] [CrossRef]
- Le Bars, P.L.; Kieser, M.; Itil, K.Z. A 26-Week Analysis of a Double-Blind, Placebo-Controlled Trial of the Ginkgo biloba Extract EGb 761® in Dementia. Dement. Geriatr. Cogn. Disord. 2000, 11, 230–237. [Google Scholar] [CrossRef]
- Levin, J.; Maaß, S.; Schuberth, M.; Respondek, G.; Paul, F.; Mansmann, U.; Oertel, W.H.; Lorenzl, S.; Krismer, F.; et al.; The PROMESA study group The PROMESA-protocol: progression rate of multiple system atrophy under EGCG supplementation as anti-aggregation-approach. J. Neural Transm. 2016, 123, 439–445. [Google Scholar] [CrossRef]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef]
- Francula-Zaninovic, S.; Nola, S.F.Z.A.I.A. Management of Measurable Variable Cardiovascular Disease’ Risk Factors. Curr. Cardiol. Rev. 2018, 14, 153–163. [Google Scholar] [CrossRef]
- Laconi, E.; Marongiu, F.; DeGregori, J. Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br. J. Cancer 2020, 122, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 419–438. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Fraga, C.G.; Croft, K.D.; Kennedy, D.; Tomás-Barberán, F.A. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusi, F.; Trezza, A.; Tramaglino, M.; Sgaragli, G.; Saponara, S.; Spiga, O. The beneficial health effects of flavonoids on the cardiovascular system: Focus on K+ channels. Pharmacol. Res. 2020, 152, 104625. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [Green Version]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Afshar, E.G. Tangeretin: a mechanistic review of its pharmacological and therapeutic effects. J. Basic Clin. Physiol. Pharmacol. 2020, 31. [Google Scholar] [CrossRef]
- Koklesova, L.; Liskova, A.; Samec, M.; Qaradakhi, T.; Zulli, A.; Smejkal, K.; Kajo, K.; Jakubikova, J.; Behzadi, P.; Pec, M.; et al. Genoprotective activities of plant natural substances in cancer and chemopreventive strategies in the context of 3P medicine. EPMA J. 2020, 11, 261–287. [Google Scholar] [CrossRef] [PubMed]
- Sabel, B.A.; Wang, J.; Fähse, S.; Cárdenas-Morales, L.; Antal, A. Personality and stress influence vision restoration and recovery in glaucoma and optic neuropathy following alternating current stimulation: implications for personalized neuromodulation and rehabilitation. EPMA J. 2020, 11, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J.; Pesta, M.; Rohan, V.; Celedova, L.; Mahajani, S.; Topolcan, O.; Golubnitschaja, O. Risks associated with the stroke predisposition at young age: facts and hypotheses in light of individualized predictive and preventive approach. EPMA J. 2019, 10, 81–99. [Google Scholar] [CrossRef]
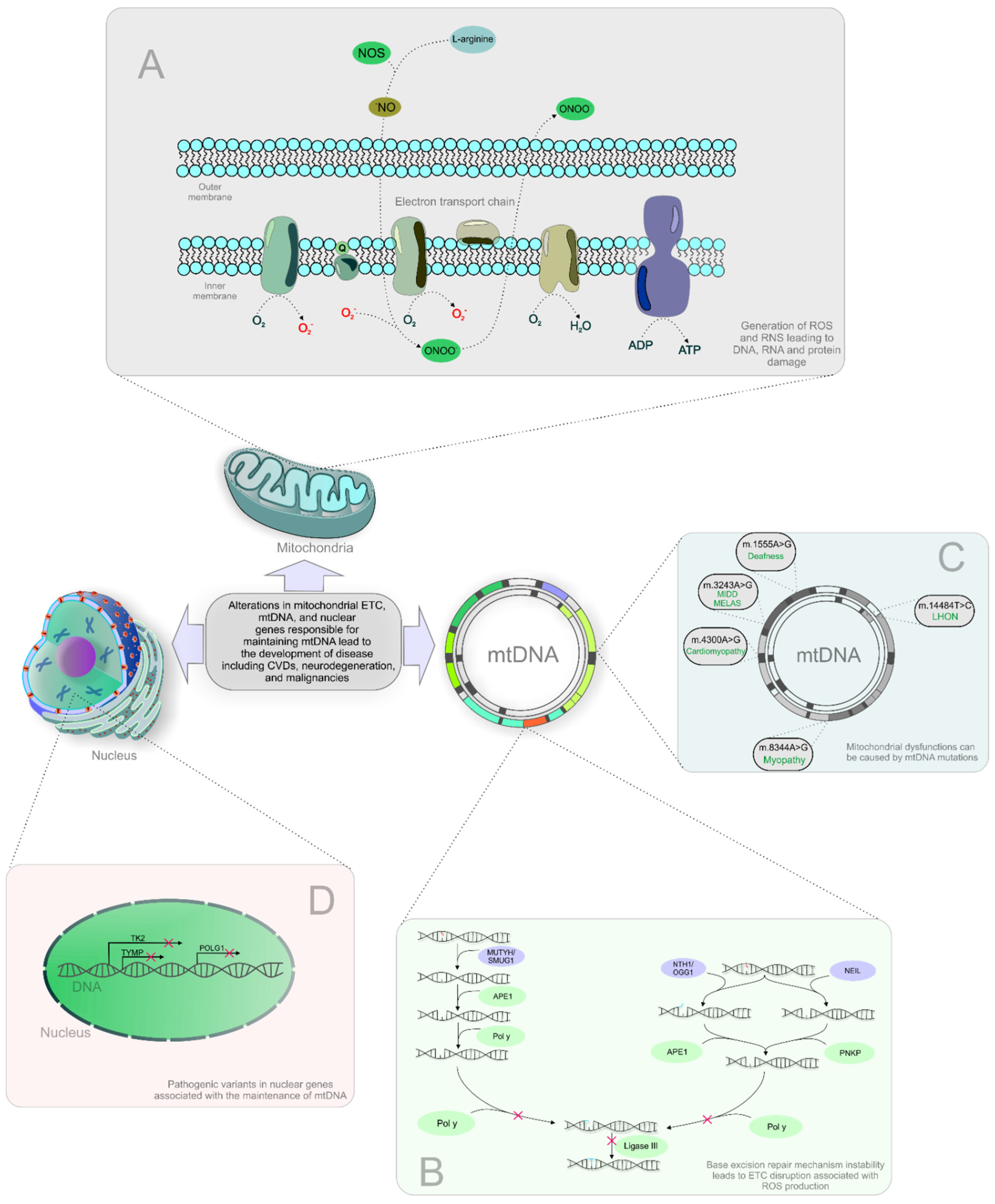


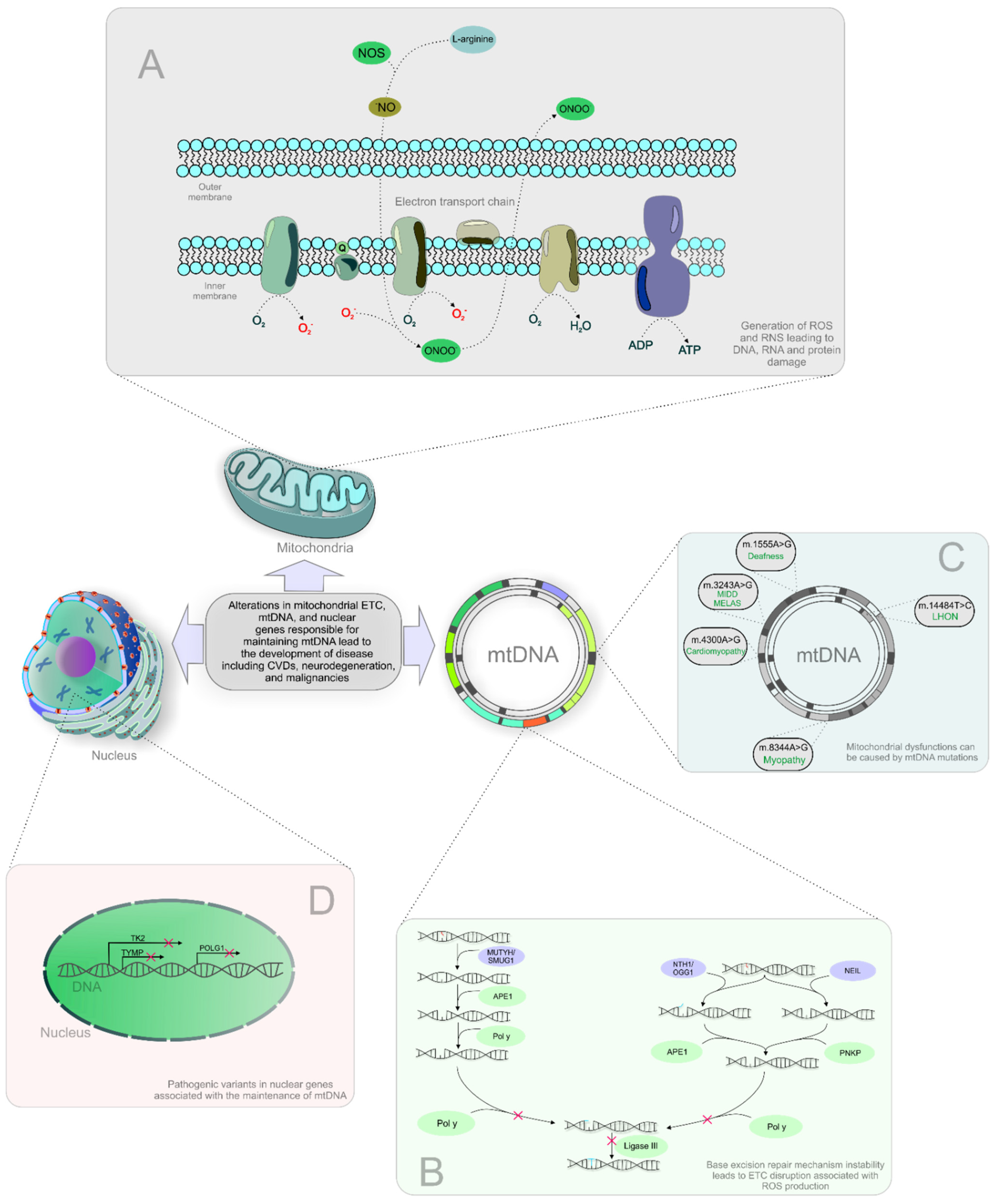{kind=link}
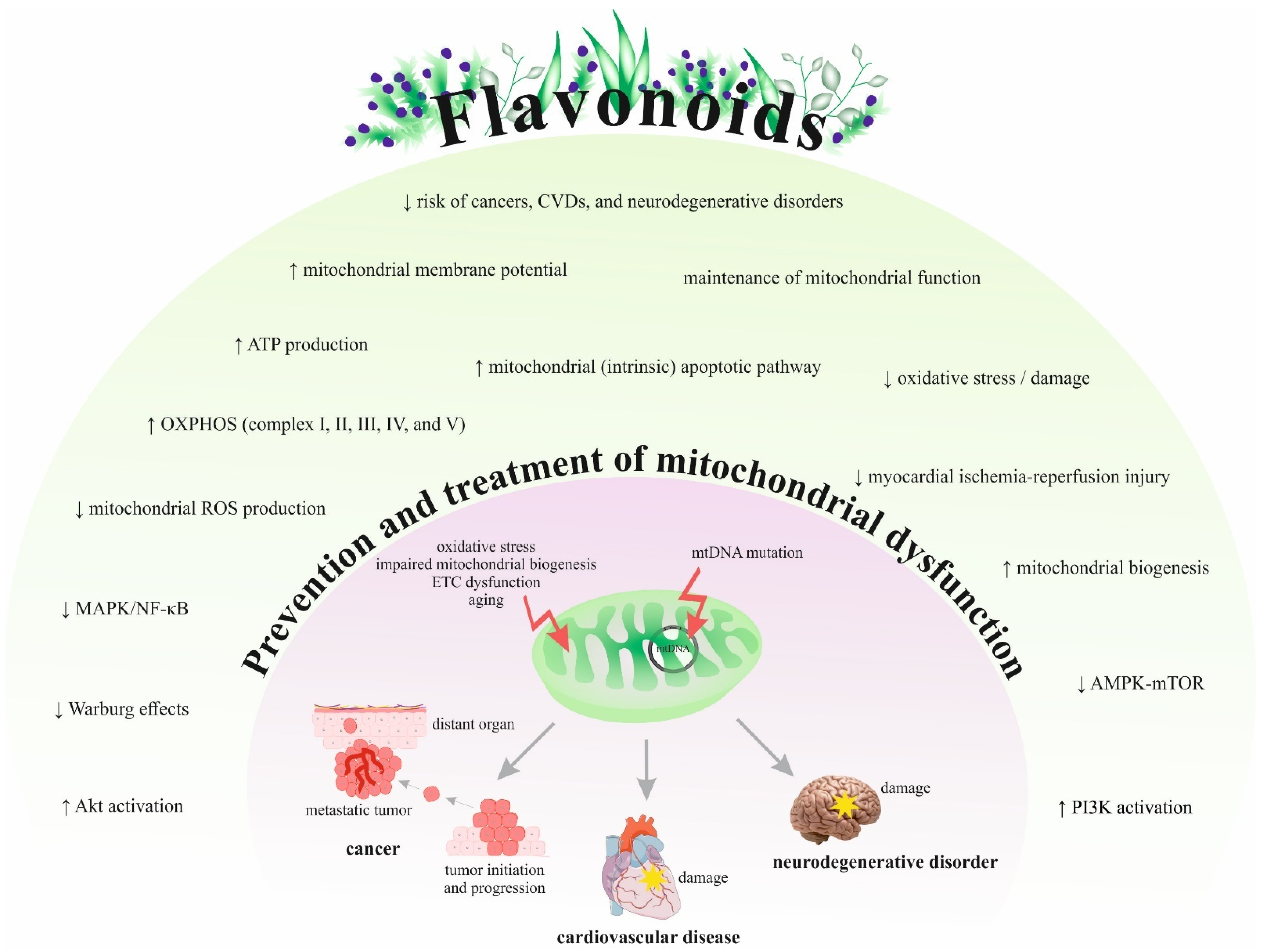
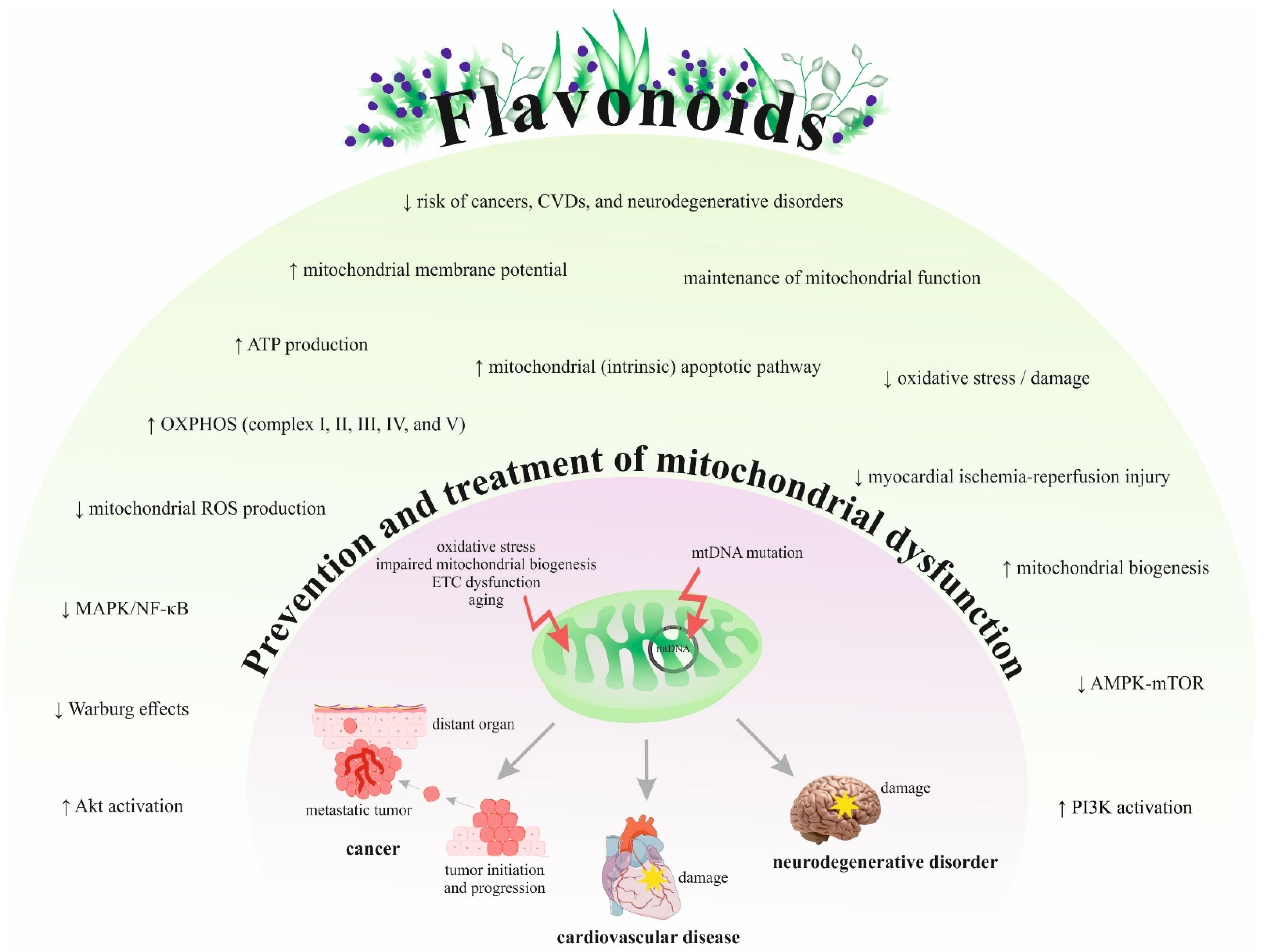{kind=link}
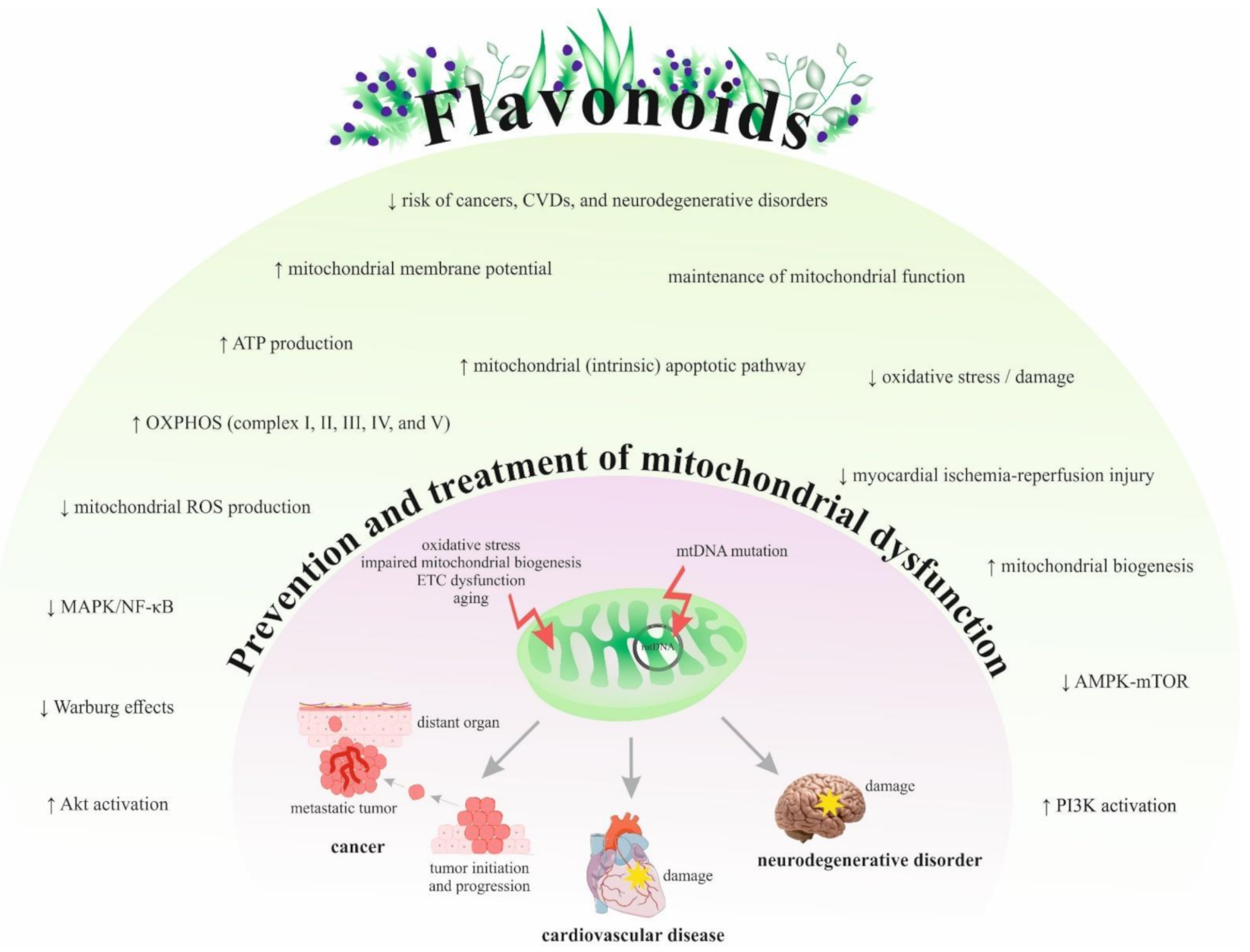{kind=link}
| Flavonoid | Mitochondrial Disorder | Study Design | Effects | Ref |
|---|---|---|---|---|
| Cancers | ||||
| Apigenin | colon cancer | HCT116, HT29, and DLD1 colon cancer cells | ↓ PKM2 activity and expression, ↓ PKM2/PKM1 ratio, blocked β-catenin/c-Myc/PTBP1 | [105] |
| Quercetin | breast cancer | MCF-7 and MDA-MB-231 human breast cancer cell lines; murine MCF-7 xenografts | ↓ PKM2, ↓ GLUT1, ↓ LDHA, ↓ p-Akt/Akt | [106] |
| Shikonin | lung carcinoma and melanoma | Lewis lung carcinoma and B16 melanoma cells | ↓ glucose uptake, ↓ lactate production, ↓ tumor cell ATP production, ↓ PKM2 | [107] |
| Xanthohumol | colorectal cancer | HT29, SW480, LOVO, HCT116, and SW620 colorectal cancer cell lines | ↓ HK2, ↓ glycolysis, ↑ cytochrome c, ↑ intrinsic (mitochondrial) apoptotic pathway | [13] |
| Glioma | rat glioma C6 cells | ↓ proliferation, ↑ apoptosis, ↑ AIF | [111] | |
| Licochalcone | colorectal cancer, non-small cell lung carcinoma, and primary bronchioalveolar carcinoma | HCT116 colorectal cancer, H1299 non-small cell lung carcinoma, and H322 primary bronchioalveolar carcinoma cells. | ↓ HIF-1α, ↓ GLUT1, ↓ PDK1, ↑ intracellular oxygen content, ↓ mitochondrial respiration | [113] |
| EGCG | breast cancer | 4T1 mouse breast cancer cells | ↓ glucose, ↓ lactate, ↓ ATP levels, ↓ HIF1α, ↓ GLUT1, ↑ mitochondrial depolarization, ↓ HK, ↓ phosphofructokinase, ↓ LDH, ↓ PK | [14] |
| Albanol B | lung cancer | A549, BZR, H1975, and H226 human lung cancer cell lines | ↑ mtROS production, ↑ phosphorylation of Akt, ↑ phosphorylation of ERK1/2, ↑ apoptosis, ↑ cell cycle arrest at G2/M phase | [114] |
| Lysionotin | liver cancer | HepG2 and SMMC-7721 cells and HepG2 and SMMC-7721-xenoghraft tumor mouse model | ↑ mitochondrial apoptosis pathway, ↑ caspase 3, control oxidative stress | [115] |
| BAS-4 | Glioma | C6 glioma cells | ↑ apoptosis, loss of mitochondrial membrane potential, Akt pathway disruption | [116] |
| Isoquercetin | Melanoma | SK-Mel-2 human melanoma cells | ↑ mitochondrial apoptosis, ↓ procaspase-8 and -9, ↓ Bcl-2 protein, ↑ cleaved caspase, ↑ Bax, ↑ AIF, ↑ Endo G, ↓ PI3K/Akt/mTOR signaling | [117] |
| Myricetin | lung cancer | in vitro (A549 lung cancer cells) and in silico study | ↑ cell cycle arrest, ↑ ROS-reliant mitochondrial apoptosis | [118] |
| Silibinin | oral squamous carcinoma | SCC-25 human oral squamous carcinoma cells | ↑ apoptosis, ↑ cytochrome c, ↑ caspase-3 and -9 | [119] |
| Cardiovascular Diseases | ||||
| Extract of Aronia melanocarpa | cardiovascular disease | 50 μg/mL of Aronia Melanocarpa fruit extract in human aortic endothelial cells | ↑ NF-κB, ↑ ROS production | [121] |
| Cornelian cherry fruits | hypertriglicerydemia and atherosclerosis | diet-induced hypertriglicerydemia and atherosclerosis in a New Zealand rabbit model | ↓ serum triglyceride levels, ↑ PPARα protein expression | [123] |
| Naringenin | myocardial ischemia-reperfusion injury | Sprague-Dawley rats and H9c2 cardiomyoblasts | ↓ mitochondrial oxidative stress damage, ↓ mitochondrial cytochrome c release, ↓ oxidative markers, ↑ mitochondrial biogenesis, ↑ NRF1, ↑ TFAM, ↑ OXPHOS II, III and IV subunits, ↓ myocardial ischemia-reperfusion injury | [15] |
| Naringin | cardiomyocyte hypertrophy | H9c2 rat myoblasts after fructose exposure, high fructose-induced cardiac hypertrophy | ↓ mtROS production, ↓ cardiomyocyte hypertrophy, ↓ AMPK-mTOR | [124] |
| 7,8-Dihydroxyflavone | myocardial ischemia | adult Kunming mice, H2O2-treated H9c2 Rattus norvegicus myoblasts | ↓ mitochondrial fission, ↓ Fis-1, ↑ mitochondrial membrane potential, ↓ mitochondrial superoxide, ↓ OPA1 | [126] |
| heart disease | doxorubicin—induced cardiotoxicity in Kunming mice; H9c2 cells | ↑ cardiac function, ↓ cardiac injury, ↑ OXPHOS, ↑ mitochondrial membrane potential, ↑ Akt activation, ↑ OPA1 | [127] | |
| Dihydromyricetin | diabetic cardiomyopathy | streptozotocin—induced diabetic C57BL/6J mice; dihydromyricetin treatment at 100 mg/kg/day | ↑ ATP content, ↑ citrate synthase activity, ↑ complex Ι, ΙΙ, ΙΙΙ, ΙV, and V activities | [128] |
| Quercetin | cardiac hypertrophy | isoproterenol—induced cardiac hypertrophy in male Swiss mice | ↑ protein sulfhydryls, ↑ superoxide dismutase activity, ↓ opening of mitochondrial permeability transition pore | [129] |
| Luteolin | sepsis-induced cardiomyopathy | intraperitoneal injection of luteolin in male C57BL/6 mice; lipopolysaccharide—induced myocardial injury | ↓ mitochondrial injury, ↓ oxidative stress, ↓ phosphorylation of AMPK in septic heart tissue, ↓ destabilized mitochondrial membrane potential | [16] |
| Icariin | cardiac oxidative stress injury | H9C2 cardiac myocytes; H2O2—induced oxidative stress injury | ↑ ROS scavenging, phosphorylation of ERK pathway, maintenance of Ca2+ homeostasis, prevention of mitochondrial membrane potential dissipation | [130] |
| Cyanidin | sepsis and myocardial oxidative or inflammation-induced injury | lipopolysaccharide—induced myocardial injury in male/female C57BL/6 mice | ↑ mitochondrial function, ↓ oxidative damage, ↓ Opa1, ↓ Trx1 | [131] |
| Tilialin | myocardial ischemia/reperfusion injury | oxygen-glucose deprivation/reperfusion-injured H9c2 cardiomyocytes; ischemia/reperfusion- (I/R-) injured isolated rat hearts | ↑ CaMKII-mediated mitochondrial apoptosis, ↑ JNK/NF-κB inflammation | [132] |
| Fisetin | acute myocardial infarction | in vitro (cardiomyocytes), in vivo (rat heart model), and in silico experiment | ↓ mitochondrial oxidative stress, ↓ mitochondrial dysfunction, ↓ GSK3β activity | [133] |
| ischemia/reperfusion injury | Male Wistar rat model | ↓ myocardial infarct size, apoptosis, lactate dehydrogenase, and creatine kinase in serum/perfusate, ↑ PI3K activation | [134] | |
| Baicalin | myocardial injury after cardiac arrest | Cardiac arrest-induced injury in male Sprague-Dawley rats | ↓ mitochondrial dysfunction, ↑ cardioprotective effect, ↓ phosphorylation at serine 616 and translocation of Drp1, and excessive fission of mitochondria | [135] |
| Neurodegenerative Disorders | ||||
| Naringin | neurodegenerative disorders and neurotoxicity | Six-week administration of aluminum (100 mg/kg) and naringin (40 and 80 mg/kg) to male Wistar rats | ↑ cognitive performance, ↓ mitochondrial oxidative damage, ↓ NADH dehydrogenase, ↓ succinate dehydrogenase, ↓ cytochrome oxidase | [137] |
| Quercetin | AD | Quercetin injection every 48 h for 3 months (25 mg/kg) in a murine triple transgenic AD model (3xTg-AD) | ↓ Aβ, ↓ BACE1-mediated cleavage of APP | [139] |
| H2O2—induced neuronal toxicity and Aβ—induced neurodegeneration of hippocampal neurons | ↓ ROS, ↑ recovery of normal mitochondrial morphology | [140] | ||
| EGCG | meta-analysis of 17 preclinical studies; animal models | ↓ Aβ42 level, ↓ acetylcholinesterase activity, ↓ tau phosphorylation, anti-oxidation, anti-inflammation, anti-apoptosis, ↑ α-, β-, and γ-secretase activity | [146] | |
| Isoquercitrin | streptozotocin—induced mitochondrial dysfunction and oxidative stress in murine Neuro-2a neuroblastoma cells | ↑ mitochondrial function, ↑ mitochondrial membrane potential, ↓ VDAC, ↓ mtROS | [18] | |
| Mangiferin and morin | Aβ—induced mitochondrial impairments in cortical neurons from E18 Sprague-Dawley rat embryos | ↓ mitochondrial impairments, ↑ respiratory capacity, ↓ mitochondrial membrane depolarization, ↓ cytochrome c release | [147] | |
| Quercetin | PD | Murine MN9D dopaminergic cells; 6-hydroxydopamine—induced neurotoxicity | ↑ phosphorylation of PKD1, Akt, CREB, BDNF; ↑ mitochondrial bioenergetics capacity | [143] |
| 6-hydroxydopamine—treated PC12 rat pheochromocytoma cells; 6-hydroxydopamine—lesioned rat model of PD | ↑ mitochondrial quality control, ↓ oxidative stress, ↑ Parkin, ↑ PINK1, ↓ α-syn | [148] | ||
| rotenone—induced rat model (inbred adult Sprague–Dawley rats) | ↑ mitochondrial complex-I activity, ↓ ROS | [17] | ||
| Myricitrin | PC12 rat adrenal gland cells, 6-OHDA—induced mitochondrial damage and neurotoxicity | ↓ mitochondrial damage, ↓ mitochondrial oxidation, ↓ ROS, ↓ lipid peroxidation | [149] | |
| 1-methyl-4-phenylpyridinium—induced mitochondrial dysfunction in SN4741 substantia nigra dopaminergic cells | ↑ maintenance of mitochondrial function, ↑ DJ-1 | [150] | ||
| Hesperidin | rotenone—induced apoptosis in SK-N-SH neuroblastoma cells | ↑ maintenance of mitochondrial function | [151] | |
| Tilialin | cerebral ischemia | oxygen-glucose deprivation protocol; in silico docking mode and SH-SY5Y human-derived thrice cloned cell line | ↑ mitochondrial function, ↓ inflammation, ↓ CaMKII-dependent mitochondrion-mediated apoptosis, ↓ MAPK/NF-κB inflammatory activation | [152] |
| Hydroxysafflor yellow A | ischemia/reperfusion injury | PC12 rat adrenal gland cells | ↓ ROS, ↓ suppress cellular apoptosis, ↓ phenylalanine levels, ↑ mitochondrial function, ↑ upregulation of mitochondrial fission protein Drp1 | [153] |
| cerebral ischemia/reperfusion injury | male Sprague Dawley rats | ↑ brain microvascular endothelial cells viability, ↓ ROS, ↓ opening of mitochondrial permeability transition pore, ↓ translocation of cytochrome c, ↑ phosphorylation of ERK, ↓ cyclophilin D | [154] | |
| Nobiletin | mitochondrial dysfunction in AD and PD | mitochondrial dysfunction mediated by the ETC system downregulation by hindering complex I and III in cortical neurons of rats | ↓ mtROS, ↓ apoptosis, ↑ ATP production, ↑ neuronal viability, ↓ translocation of AIF, ↑ complex I, ↑ Nrf2, ↑ HO-1 | [155] |
| Flavonoid | Supposed Mitochondriopathy | Clinical Trial | Study Design | Effect | Ref |
|---|---|---|---|---|---|
| Cardiovascular Diseases | |||||
| Isoflavone | ischemic stroke | randomized, double-blinded, placebo-controlled trial | 12-week treatment; 80 mg/day of isoflavone (n = 50) or placebo (n = 52) | ↓ serum hsCRP | [170] |
| Flavonols, flavones, flavanones, anthocyanidins, and proanthocyanidins | CVDs | meta-analysis (22 prospective studies) | 100 mg/day of flavonoids | ↓ risk of all-cause mortality, ↓ risk of CVD mortality | [171] |
| Tea consumption | CVDs, stroke, myocardial infarction | meta-analysis (10 cohort studies and 7 case-control studies) | 3 cups per day | in 4 studies: ↑ risk of coronary heart disease, ↑ myocardial infarction In the remaining studies: ↓ myocardial infarction, ↓ coronary heart disease | [172] |
| EGCG | amyloidotic transthyretin cardiomyopathy | randomized placebo-controlled clinical trial | 12-month treatment, patients with amyloidotic transthyretin cardiomyopathy (n = 19); 547 ± 49 mg/day of EGCG | no changes in cardiac wall thickness and mass | [174] |
| Soy protein with isoflavones | CVDs | randomized controlled trial | 6-month treatment with 15 g of soy protein and 66 mg of isoflavones/day during early menopause in Caucasian women from Hull and East Riding in Yorkshire (n = 200) | ↓ fasting glucose, ↓ fasting insulin, ↓ insulin resistance, ↓ systolic blood pressure | [177] |
| Blueberries | CVD risk | randomized controlled trial | 8-week treatment with 50 g of freeze-dried blueberries/daily in participants with metabolic syndrome (n = 48) | ↓ plasma oxidized LDL, ↓ serum malondialdehyde, ↓ hydroxynonenal | [179] |
| Cranberries | CVD risk | randomized, double-blind, placebo-controlled trial | 8-week treatment in women with metabolic syndrome (n = 15–16/group); 1st group: cranberry juice (480 mL/day); 2nd group: placebo (480 mL/day) | ↑ plasma antioxidant capacity ↓ lipid oxidation, ↓ oxidized LDL, ↓ malondialdehyde | [180] |
| Fruit-based flavonoids (flavanones and anthocyanins) | CVDs, myocardial infarction, and ischemic stroke | Health Professionals Follow-Up Study | men in analysis (n = 43,880) | ↓ risk of nonfatal myocardial infarction and ischemic stroke | [181] |
| Chokeberry flavonoid extract (Aronia melanocarpa E) and statin therapy | ischemic heart disease | double-blind, placebo-controlled, parallel trial | 6-week combination therapy with statin (simvastatin) and flavonoids from chokeberry extract (3 × 85 mg/day) or placebo in patients (n = 44) who survived myocardial infarction | ↓ serum 8-isoprostanes, ↓ oxidized LDL, ↓ hsCRP, ↓ MCP-1, ↑ adiponectin | [182] |
| Neurodegenerative Disorders | |||||
| EGCG | multiple system atrophy | randomized, double-blind, placebo-controlled clinical trial (NCT02008721) | 48-week treatment; 92 participants-EGCG treatment (n = 47) and placebo (n = 45) | no modification in disease progression | [185] |
| Antioxidant drink rich in polyphenols (apple and lemon juice concentrate, apple and green tea extracts, and vitamins B and C) | AD | multicenter, randomized, double-blind controlled clinical trial | 8-month treatment; 100 participants: AD patients (n = 24 in initial phase, n = 24 in moderate phase) and healthy controls (n = 52) | ↓ total plasma homocysteine levels in AD patients | [187] |
| EGb 761® | uncomplicated AD and multi-infarct dementia | double-blind, placebo-controlled trial | 26-week treatment with a 120-mg dose (40 mg t.i.d.) of EGb 761; placebo group (n = 161) and EGb treated group (n = 166) | ↑ cognitive assessment, ↑ daily living, ↑ social behavior | [188] |
| EGCG | multiple system atrophy | prospective, randomized, double-blind, placebo-controlled parallel-group phase III, multicenter study | 48-week treatment in patients (n = 86); PROMESA protocol | ↓ progression of multiple system atrophy-associated disabilities | [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koklesova, L.; Liskova, A.; Samec, M.; Zhai, K.; AL-Ishaq, R.K.; Bugos, O.; Šudomová, M.; Biringer, K.; Pec, M.; Adamkov, M.; et al. Protective Effects of Flavonoids Against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention. Int. J. Mol. Sci. 2021, 22, 8649. https://doi.org/10.3390/ijms22168649
Koklesova L, Liskova A, Samec M, Zhai K, AL-Ishaq RK, Bugos O, Šudomová M, Biringer K, Pec M, Adamkov M, et al. Protective Effects of Flavonoids Against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention. International Journal of Molecular Sciences. 2021; 22(16):8649. https://doi.org/10.3390/ijms22168649
Chicago/Turabian StyleKoklesova, Lenka, Alena Liskova, Marek Samec, Kevin Zhai, Raghad Khalid AL-Ishaq, Ondrej Bugos, Miroslava Šudomová, Kamil Biringer, Martin Pec, Marian Adamkov, and et al. 2021. "Protective Effects of Flavonoids Against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention" International Journal of Molecular Sciences 22, no. 16: 8649. https://doi.org/10.3390/ijms22168649
APA StyleKoklesova, L., Liskova, A., Samec, M., Zhai, K., AL-Ishaq, R. K., Bugos, O., Šudomová, M., Biringer, K., Pec, M., Adamkov, M., Hassan, S. T. S., Saso, L., Giordano, F. A., Büsselberg, D., Kubatka, P., & Golubnitschaja, O. (2021). Protective Effects of Flavonoids Against Mitochondriopathies and Associated Pathologies: Focus on the Predictive Approach and Personalized Prevention. International Journal of Molecular Sciences, 22(16), 8649. https://doi.org/10.3390/ijms22168649