
Thyroid Hormone Induces Ca2+-Mediated Mitochondrial Activation in Brown Adipocytes

 , ,
, ,
Abstract
1. Introduction
2. Results
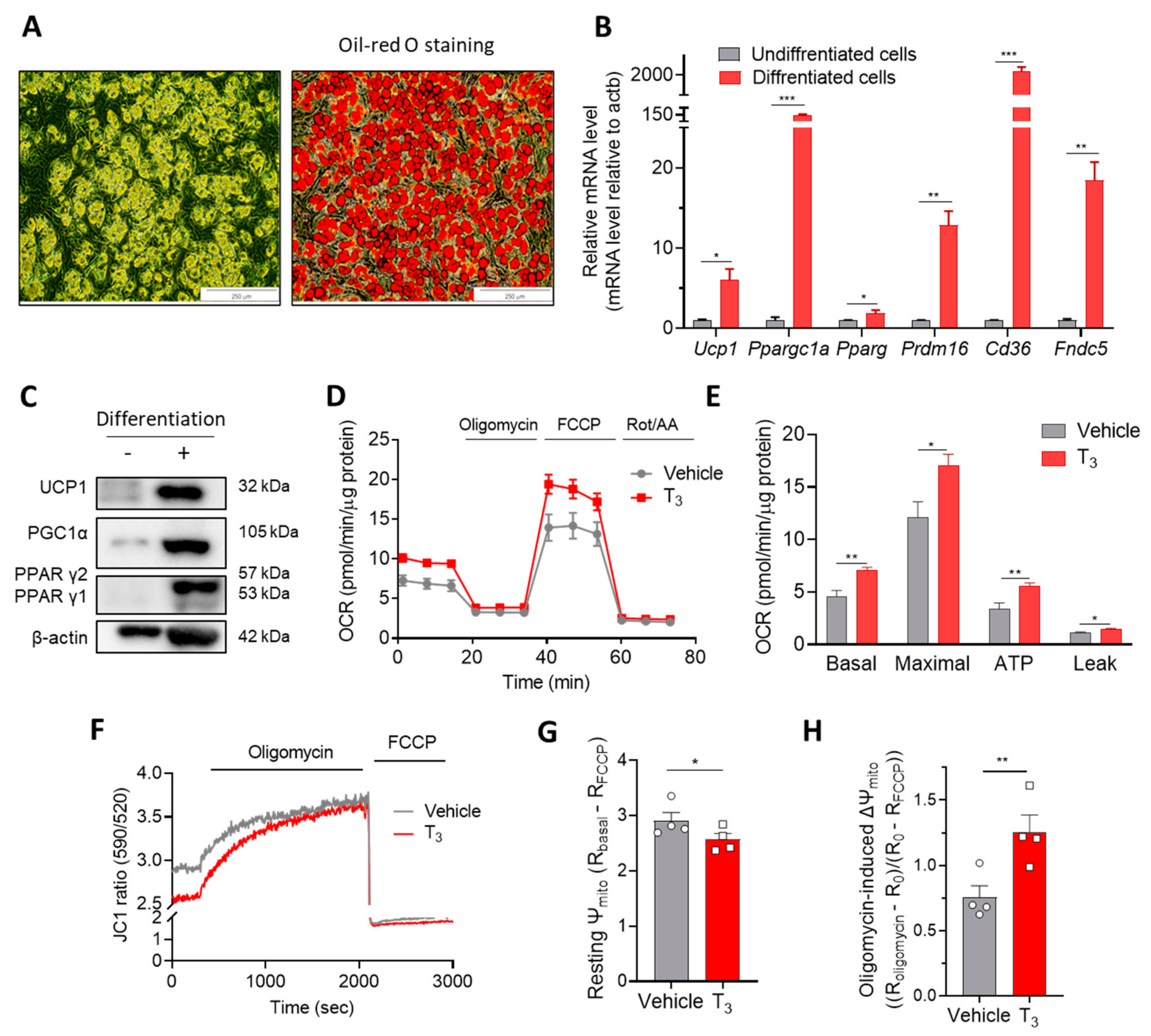
2.1. Acute T3 Exposure Induces Mitochondrial Activation in Brown Adipocytes
2.1.1. T3 Increases Mitochondrial Respiration
2.1.2. T3 Alters Mitochondrial Membrane Potential (Ψm)
2.2. Changes in Cellular Function by Acute T3 Treatment
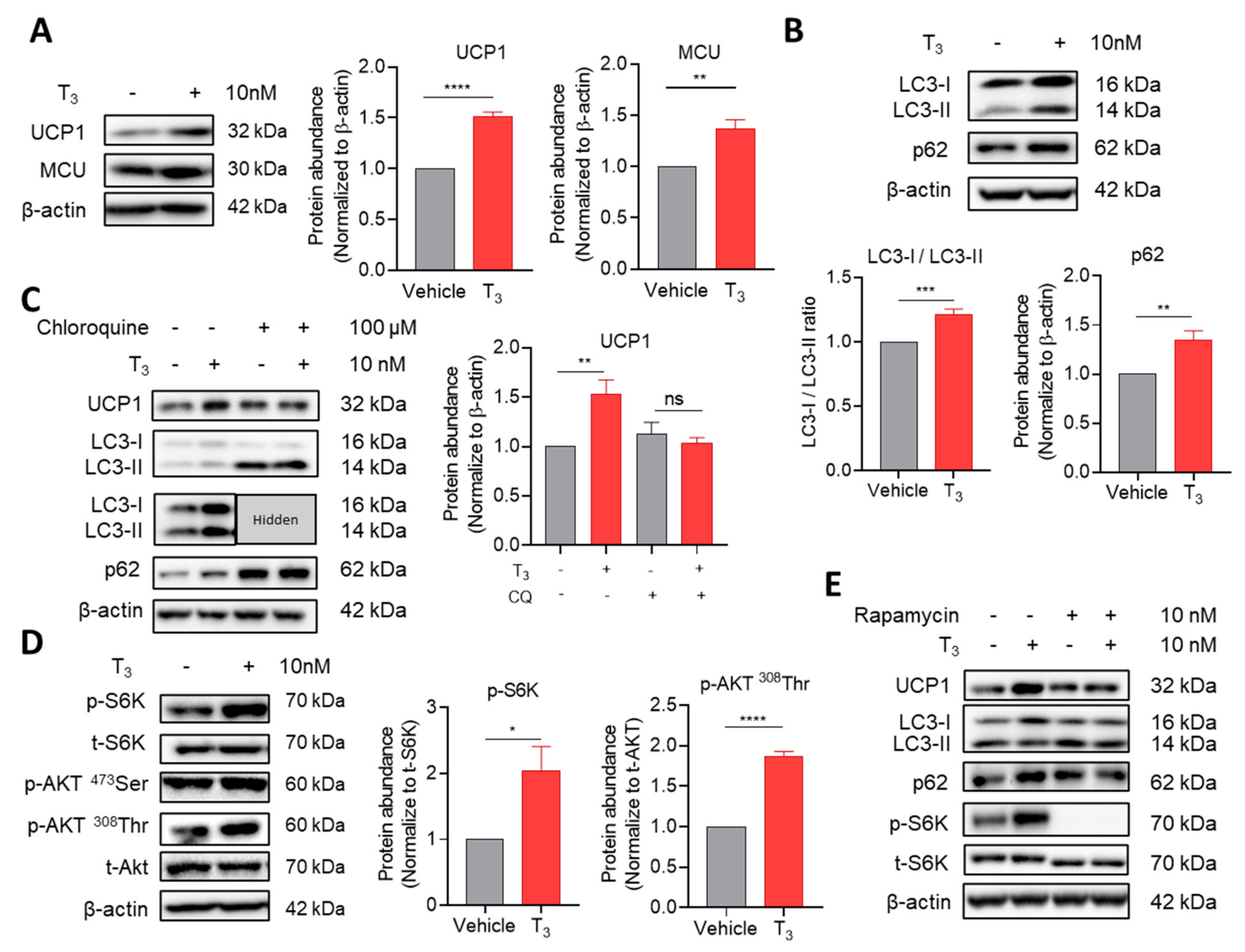
2.2.1. T3 Suppresses Autophagic Degradation
2.2.2. T3-Induced Mammalian Target of Rapamycin (mTOR) Activation Inhibits Autophagic Degradation
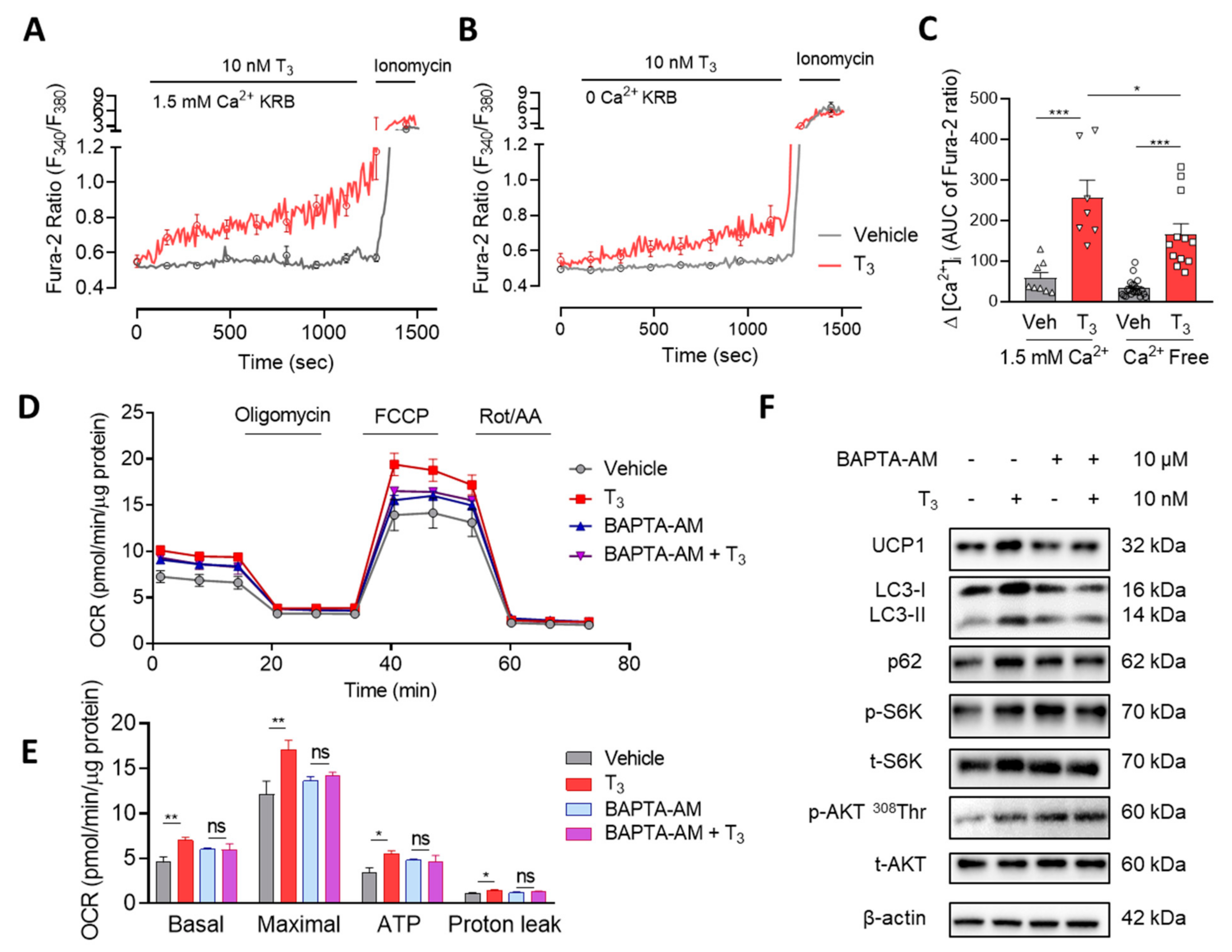
2.3. T3 Induces Cytosolic Ca2+ Increase and Mitochondrial Activation
2.3.1. T3 Induces a Rapid and Sustained [Ca2+]i Elevation
2.3.2. T3 Activates Mitochondrial Respiration via Ca2+ Signaling
2.3.3. T3-Induced mTOR Activation via Ca2+ Signaling
2.3.4. Cytosolic and Mitochondrial Ca2+ Mediate T3-Activated Mitochondrial Respiration
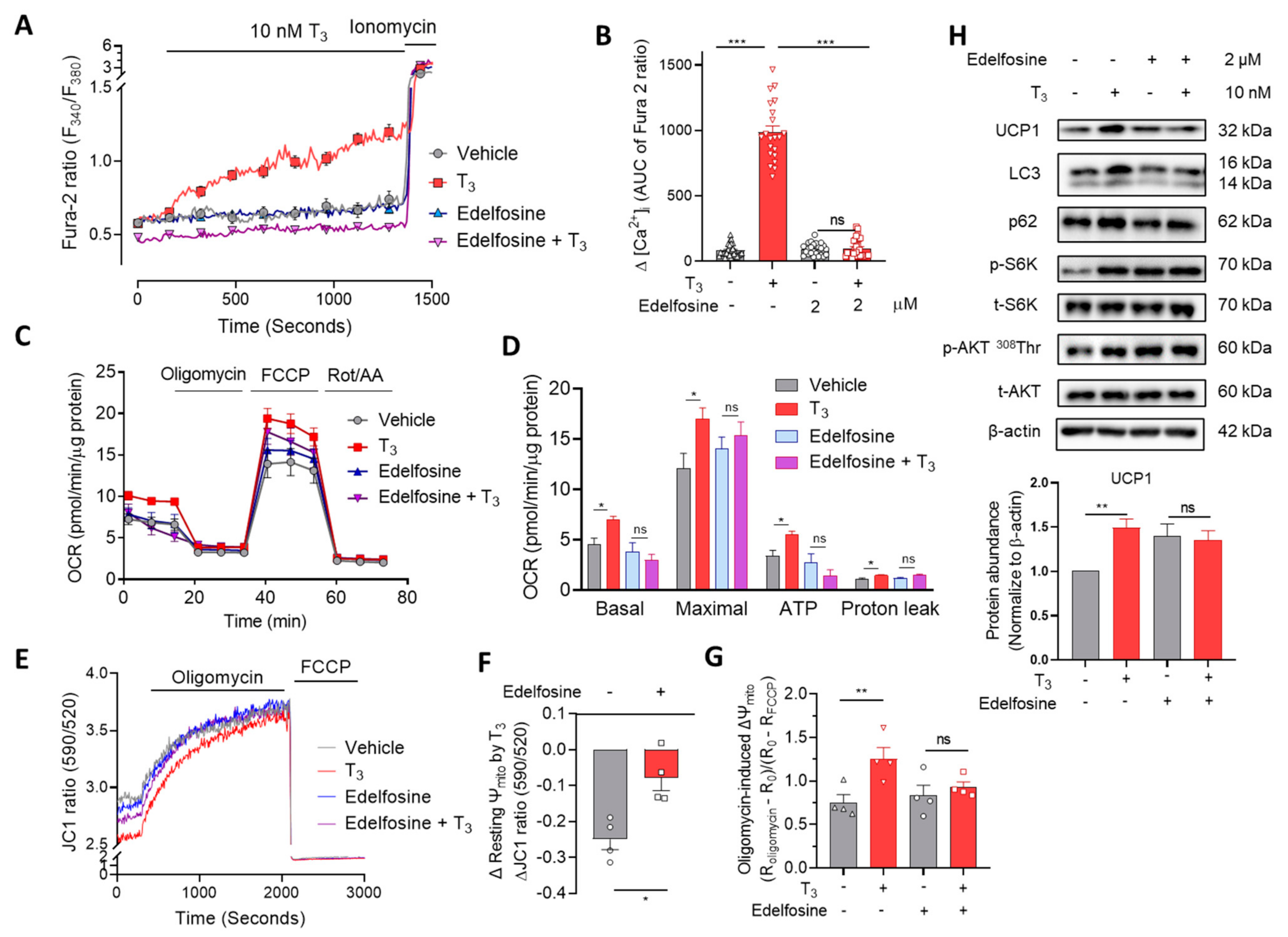
2.4. Phospholipase C (PLC) Is Involved in T3-Induced [Ca2+]i Increase and Mitochondrial Activation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Immortalized Cell Culture
4.3. Primary Cell Isolation
4.4. Oxygen Consumption Rate (OCR) Measurement
4.5. Calcium Measurement by Live-Cell Imaging
4.6. SDS-PAGE/Western Blotting
4.7. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
4.8. Mitochondrial Membrane Potential Measurement
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brestoff, J.; Artis, D. Immune Regulation of Metabolic Homeostasis in Health and Disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Kozak, L.P.; Anunciado-Koza, R. UCP1: Its involvement and utility in obesity. Int. J. Obes. 2008, 32 (Suppl. S7), S32–S38. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Stock, M.J. Luxuskonsumption, diet-induced thermogenesis and brown fat: The case in favour. Clin. Sci. 1983, 64, 19–23. [Google Scholar] [CrossRef]
- Cypess, A.M.; Kahn, C.R. Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 143–149. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef]
- Mendel, C.M.; Weisiger, R.A.; Jones, A.L.; Cavalieri, R.R. Thyroid hormone-binding proteins in plasma facilitate uniform distribution of thyroxine within tissues: A perfused rat liver study. Endocrinology 1987, 120, 1742–1749. [Google Scholar] [CrossRef]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef]
- Lin, H.Y.; Cody, V.; Davis, F.B.; Hercbergs, A.A.; Luidens, M.K.; Mousa, S.A.; Davis, P.J. Identification and functions of the plasma membrane receptor for thyroid hormone analogues. Discov. Med. 2011, 11, 337–347. [Google Scholar]
- Lin, H.Y.; Landersdorfer, C.B.; London, D.; Meng, R.; Lim, C.U.; Lin, C.; Lin, S.; Tang, H.Y.; Brown, D.; Van Scoy, B.; et al. Pharmacodynamic modeling of anti-cancer activity of tetraiodothyroacetic acid in a perfused cell culture system. PLoS Comput. Biol. 2011, 7, e1001073. [Google Scholar] [CrossRef]
- Chassande, O.; Fraichard, A.; Gauthier, K.; Flamant, F.; Legrand, C.; Savatier, P.; Laudet, V.; Samarut, J. Identification of transcripts initiated from an internal promoter in the c-erbA alpha locus that encode inhibitors of retinoic acid receptor-alpha and triiodothyronine receptor activities. Mol. Endocrinol. 1997, 11, 1278–1290. [Google Scholar] [CrossRef][Green Version]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef]
- Obregon, M.J. Thyroid hormone and adipocyte differentiation. Thyroid Off. J. Am. Thyroid Assoc. 2008, 18, 185–195. [Google Scholar] [CrossRef]
- Bianco, A.C.; Sheng, X.Y.; Silva, J.E. Triiodothyronine amplifies norepinephrine stimulation of uncoupling protein gene transcription by a mechanism not requiring protein synthesis. J. Biol. Chem. 1988, 263, 18168–18175. [Google Scholar] [CrossRef]
- Guerra, C.; Roncero, C.; Porras, A.; Fernández, M.; Benito, M. Triiodothyronine induces the transcription of the uncoupling protein gene and stabilizes its mRNA in fetal rat brown adipocyte primary cultures. J. Biol. Chem. 1996, 271, 2076–2081. [Google Scholar] [CrossRef]
- Yau, W.W.; Yen, P.M. Thermogenesis in Adipose Tissue Activated by Thyroid Hormone. Int. J. Mol. Sci. 2020, 21, 3020. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.-Y.; et al. Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef]
- Sterling, K.; Brenner, M.A.; Sakurada, T. Rapid effect of triiodothyronine on the mitochondrial pathway in rat liver in vivo. Science 1980, 210, 340–342. [Google Scholar] [CrossRef]
- Oeckl, J.; Bast-Habersbrunner, A.; Fromme, T.; Klingenspor, M.; Li, Y. Isolation, Culture, and Functional Analysis of Murine Thermogenic Adipocytes. STAR Protoc. 2020, 1, 100118. [Google Scholar] [CrossRef]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef]
- Chen, S.C.; Brooks, R.; Houskeeper, J.; Bremner, S.K.; Dunlop, J.; Viollet, B.; Logan, P.J.; Salt, I.P.; Ahmed, S.F.; Yarwood, S.J. Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. Mol. Cell. Endocrinol. 2017, 440, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Blommaart, E.F.; Luiken, J.J.; Blommaart, P.J.; van Woerkom, G.M.; Meijer, A.J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995, 270, 2320–2326. [Google Scholar] [CrossRef]
- Missiaen, L.; Callewaert, G.; De Smedt, H.; Parys, J.B. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium 2001, 29, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Collins, T.J.; Mackenzie, L.; Roderick, H.L.; Berridge, M.J.; Peppiatt, C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1145–1150. [Google Scholar] [CrossRef]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 1324–1333. [Google Scholar] [CrossRef]
- Essen, L.O.; Perisic, O.; Katan, M.; Wu, Y.; Roberts, M.F.; Williams, R.L. Structural mapping of the catalytic mechanism for a mammalian phosphoinositide-specific phospholipase C. Biochemistry 1997, 36, 1704–1718. [Google Scholar] [CrossRef]
- D’Arezzo, S.; Incerpi, S.; Davis, F.B.; Acconcia, F.; Marino, M.; Farias, R.N.; Davis, P.J. Rapid Nongenomic Effects of 3,5,3′-Triiodo-l-Thyronine on the Intracellular pH of L-6 Myoblasts Are Mediated by Intracellular Calcium Mobilization and Kinase Pathways. Endocrinology 2004, 145, 5694–5703. [Google Scholar] [CrossRef] [PubMed]
- Saelim, N.; John, L.M.; Wu, J.; Park, J.S.; Bai, Y.; Camacho, P.; Lechleiter, J.D. Nontranscriptional modulation of intracellular Ca2+ signaling by ligand stimulated thyroid hormone receptor. J. Cell Biol. 2004, 167, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Kambe, F.; Cao, X.; Lu, X.; Kozaki, Y.; Oiso, Y.; Seo, H. Thyroid hormone activates adenosine 5’-monophosphate-activated protein kinase via intracellular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase kinase-beta. Mol. Endocrinol. 2008, 22, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.M.; Baker, L.; Quinlan, P.R.; Robertson, K.E.; Bray, S.E.; Thomson, G.; Kellock, D.; Jordan, L.B.; Purdie, C.A.; Hardie, D.G.; et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer 2009, 9, 307. [Google Scholar] [CrossRef]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. CMLS 2005, 62, 1784–1803. [Google Scholar] [CrossRef]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008, 7, 456–465. [Google Scholar] [CrossRef]
- Li, R.-J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5, e19360. [Google Scholar] [CrossRef]
- Gocher, A.M.; Azabdaftari, G.; Euscher, L.M.; Dai, S.; Karacosta, L.G.; Franke, T.F.; Edelman, A.M. Akt activation by Ca(2+)/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.-H.; Zhu, X.; Dong, X.-P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Lin, J.; Lim, W.; Jin, W.; Lee, H.J. Role of brown adipose tissue in metabolic syndrome, aging, and cancer cachexia. Front. Med. 2018, 12, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Heeren, J. The holy grail of metabolic disease: Brown adipose tissue. Curr. Opin. Lipidol. 2012, 23, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Aune, U.L.; Ruiz, L.; Kajimura, S. Isolation and differentiation of stromal vascular cells to beige/brite cells. J. Vis. Exp. JoVE 2013. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence |
|---|---|
| Ucp1 | F-5′ TGGAAAGGGACGACCCCTAA 3′ R-3′CAAAACCCGGCAACAAGAGC 5′ |
| Ppargc1a | F-5′ TGGAGTGACATAGAGTGTGCT 3′ R-3′ TTCCGATTGGTCGCTACACC 5′ |
| Pparg | F-5′ ACCATGGTAATTTCAGTAAAGG 3′ R-3′ GTCTCGGTTGAGGGGACG 5′ |
| Prdm16 | F-5′ CGACTTTGGATGGGAGCAGAT 3′ R-3′ ACGGATGTACTTGAGCCAGC 5′ |
| Fndc5 | F-5′ CTCTTCATGTGGGCAGCTGTTA 3′ R-3′ GCGCTCTTGGTTTTCTCCTTG 5′ |
| Cd36 | F-5′ TTGAAAGCAGTGGTCCTTC 3′ R-3′ GCTGTTATTGGTGCAGTCCT 5′ |
| mTOR | F-5′ CCGCCTTCACAGATACCCAG 3′ R-3′ TTAAACTCCGACCTCACGGC 5′ |
| Acaca | F-5′ CCTGACAAACGAGTCTGGCT 3′ R-3′ CATTCCATGCAGTGGTCCCT 5′ |
| Acacb | F-5′ GCCTGACCTTTTCCTGGCTA 3′ R-3′ TGAGGCTGAAAGGGACTCCT 5′ |
| Rps6 | F-5′GGAAGCCCTTAAACAAAGAAGGTAA 3′ R-3′ AATACGTCGGCGTTTGTGTT 5′ |
| Akt | F-5′ GACTTCCGATCAGGCTCACC3′ R-3′ACTCGTTCATGGTCACACGG 5′ |
| Map1lca | F-5′ CCAAGATCCCAGTGATTATAGAGC 3′ R-3′ TGCAAGCGCCGTCTGATTAT 5′ |
| Map1lcb | F-5′ CCAAGATCCCAGTGATTATAGAGC 3′ R-3′ TGCAAGCGCCGTCTGATTAT 5′ |
| Actb | F-5′ CGCTACAAGGGTGAGAAGCA3′ R-3′ GCGGCGCCGGATGAT 5′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.-H.T.; Ly, D.D.; Nguyen, N.T.; Qi, X.-F.; Yi, H.-S.; Shong, M.; Cha, S.-K.; Park, S.; Park, K.-S. Thyroid Hormone Induces Ca2+-Mediated Mitochondrial Activation in Brown Adipocytes. Int. J. Mol. Sci. 2021, 22, 8640. https://doi.org/10.3390/ijms22168640
Nguyen M-HT, Ly DD, Nguyen NT, Qi X-F, Yi H-S, Shong M, Cha S-K, Park S, Park K-S. Thyroid Hormone Induces Ca2+-Mediated Mitochondrial Activation in Brown Adipocytes. International Journal of Molecular Sciences. 2021; 22(16):8640. https://doi.org/10.3390/ijms22168640
Chicago/Turabian StyleNguyen, Minh-Hanh Thi, Dat Da Ly, Nhung Thi Nguyen, Xu-Feng Qi, Hyon-Seung Yi, Minho Shong, Seung-Kuy Cha, Sangkyu Park, and Kyu-Sang Park. 2021. "Thyroid Hormone Induces Ca2+-Mediated Mitochondrial Activation in Brown Adipocytes" International Journal of Molecular Sciences 22, no. 16: 8640. https://doi.org/10.3390/ijms22168640
APA StyleNguyen, M.-H. T., Ly, D. D., Nguyen, N. T., Qi, X.-F., Yi, H.-S., Shong, M., Cha, S.-K., Park, S., & Park, K.-S. (2021). Thyroid Hormone Induces Ca2+-Mediated Mitochondrial Activation in Brown Adipocytes. International Journal of Molecular Sciences, 22(16), 8640. https://doi.org/10.3390/ijms22168640