
Mitochondrial Metabolism behind Region-Specific Resistance to Ischemia-Reperfusion Injury in Gerbil Hippocampus. Role of PKCβII and Phosphate-Activated Glutaminase

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PKCβII and GLS1 Glutaminase Isoform GAC but Not KGA Co-Localize in the Postischemic Mitochondria In Vivo
2.2. Glutaminase Activity Is Higher in CA2-4,DG than in CA1 and Is Regulated by PKCβII
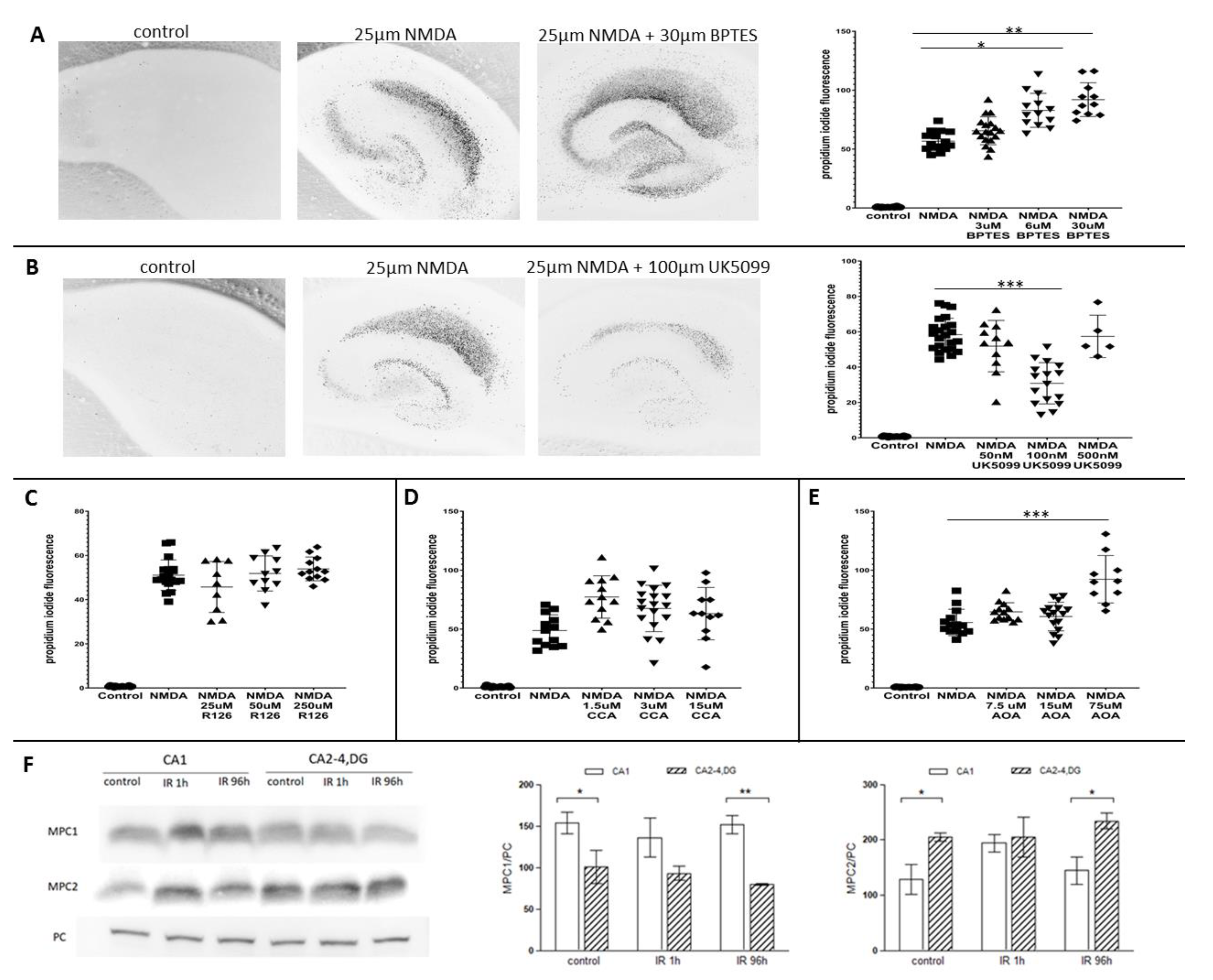
2.3. GAC, One of GLS1 Isoforms, Is Responsible for Higher GLS1 Activity in CA2-4,DG
2.4. Glutaminase and Mitochondrial Pyruvate Carrier Contribute to the Survival Rate of Hippocampal Slices
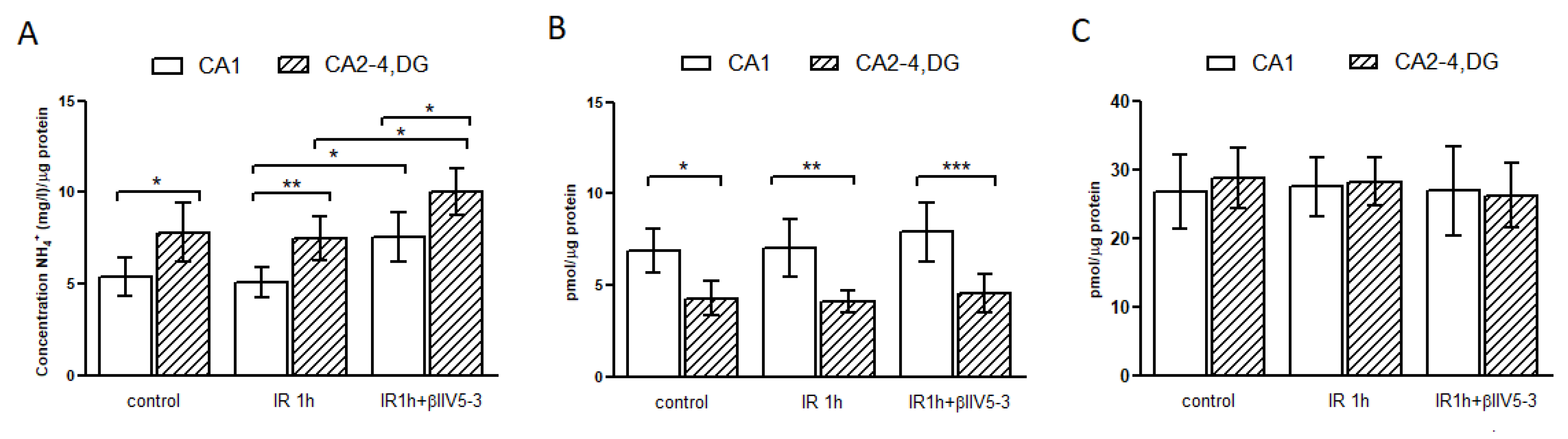
2.5. Metabolite Analysis
3. Discussion
4. Materials and Methods
4.1. Ethical Statement and Animals
4.2. Transient Brain Ischemia in Gerbils
4.3. Sample Preparation and Isolation of Pure Mitochondria
4.4. Co-Immunoprecipitations
4.5. GLS1 Phosphorylation by PKCβII in In Vitro Assay
4.6. Glutaminase Activity Measurement
4.7. Glutamine and Glutamate Concentration Measurement
4.8. Western Blot Analysis
4.9. Organotypic Hippocampal Slice Culture
4.10. Untargeted Metabolomics Analysis
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cronberg, T.; Jensen, K.; Rytter, A.; Wieloch, T. Selective sparing of hippocampal CA3 cells following in vitro ischemia is due to selective inhibition by acidosis. Eur. J. Neurosci. 2005, 22, 310–316. [Google Scholar] [CrossRef]
- Lifshitz, J.; Friberg, H.; Neumar, R.W.; Raghupathi, R.; Welsh, F.A.; Janmey, P.; Saatman, K.E.; Wieloch, T.; Grady, M.S.; McIntosh, T.K. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: Evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow Metab. 2003, 23, 219–231. [Google Scholar] [CrossRef]
- Hu, B.R.; Wieloch, T. Persistent translocation of Ca2+/calmodulin-dependent protein kinase II to synaptic junctions in the vulnerable hippocampal CA1 region following transient ischemia. J. Neurochem. 1995, 64, 277–284. [Google Scholar] [CrossRef]
- Krupska, O.; Sarnowska, A.; Fedorczyk, B.; Gewartowska, M.; Misicka, A.; Zablocka, B.; Beresewicz, M. Ischemia/Reperfusion-Induced Translocation of PKCβII to Mitochondria as an Important Mediator of a Protective Signaling Mechanism in an Ischemia-Resistant Region of the Hippocampus. Neurochem. Res. 2017, 42, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Man, H.-Y. Synaptic Activity and Bioenergy Homeostasis: Implications in Brain Trauma and Neurodegenerative Diseases. Front. Neurol. 2013, 4, 199. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine Oxidation Maintains the TCA Cycle and Cell Survival during Impaired Mitochondrial Pyruvate Transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Ferrari, F.; Gorini, A.; Hoyer, S.; Villa, R.F. Glutamate metabolism in cerebral mitochondria after ischemia and post-ischemic recovery during aging: Relationships with brain energy metabolism. J. Neurochem. 2018, 146, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Daikhin, Y.; Yudkoff, M. Compartmentation of brain glutamate metabolism in neurons and glia. J. Nutr. 2000, 130 (Suppl. 4), 1026S–1031S. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, C.; Zhu, J.; Wang, Y.; Huang, Y.; Zhao, S.; Sheng, S.; Song, Y.; Ji, C.; Li, C.; et al. Glutaminase 1 Regulates Neuroinflammation After Cerebral Ischemia Through Enhancing Microglial Activation and Pro-Inflammatory Exosome Release. Front. Immunol. 2020, 11, 161. [Google Scholar] [CrossRef]
- Martín-Rufián, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gomez, F.J.M.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. 2013, 92, 277–290. [Google Scholar] [CrossRef]
- Rink, C.; Gnyawali, S.; Stewart, R.; Teplitsky, S.; Harris, H.; Roy, S.; Sen, C.K.; Khanna, S. Glutamate oxaloacetate transaminase enables anaplerotic refilling of TCA cycle intermediates in stroke-affected brain. FASEB J. 2016, 31, 1709–1718. [Google Scholar] [CrossRef]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.-L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.-C. Identification and Functional Expression of the Mitochondrial Pyruvate Carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Zangari, J.; Petrelli, F.; Maillot, B.; Martinou, J.-C. The Multifaceted Pyruvate Metabolism: Role of the Mitochondrial Pyruvate Carrier. Biomolecules 2020, 10, 1068. [Google Scholar] [CrossRef]
- Vacanti, N.M.; Divakaruni, A.S.; Green, C.; Parker, S.; Henry, R.R.; Ciaraldi, T.P.; Murphy, A.N.; Metallo, C.M. Regulation of Substrate Utilization by the Mitochondrial Pyruvate Carrier. Mol. Cell 2014, 56, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Targeting the Mitochondrial Pyruvate Carrier for Neuroprotection. Brain Sci. 2019, 9, 238. [Google Scholar] [CrossRef] [PubMed]
- Ascenção, C.F.R.; Nagampalli, R.S.K.; Islam, Z.; Pinheiro, M.P.; dos Reis, L.M.; Pauletti, B.A.; Cassago, C.A.D.G.; Granato, D.C.; Leme, A.P.; Dias, S.M.G. N-terminal phosphorylation of glutaminase C decreases its enzymatic activity and cancer cell migration. Biochimie 2018, 154, 69–76. [Google Scholar] [CrossRef]
- Han, T.; Zhan, W.; Gan, M.; Liu, F.; Yu, B.; Chin, Y.E.; Wang, J.-B. Phosphorylation of glutaminase by PKCε is essential for its enzymatic activity and critically contributes to tumorigenesis. Cell Res. 2018, 28, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Khoury, N.; Jackson, C.W.; Escobar, I.; Stegelmann, S.D.; Dave, K.R.; Perez-Pinzon, M.A. Ischemic Neuroprotectant PKCε Restores Mitochondrial Glutamate Oxaloacetate Transaminase in the Neuronal NADH Shuttle after Ischemic Injury. Transl. Stroke Res. 2019, 11, 418–432. [Google Scholar] [CrossRef]
- Dave, K.R.; Defazio, R.A.; Raval, A.P.; Torraco, A.; Saul, I.; Barrientos, A.; Perez-Pinzon, M.A. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J. Neurosci. 2008, 28, 4172–4182. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef]
- Morris-Blanco, K.C.; Cohan, C.H.; Neumann, J.T.; Sick, T.J.; Perez-Pinzon, M.A. Protein Kinase C Epsilon Regulates Mitochondrial Pools of Nampt and NAD Following Resveratrol and Ischemic Preconditioning in the Rat Cortex. Br. J. Pharmacol. 2014, 34, 1024–1032. [Google Scholar] [CrossRef]
- Wang, P.; Xu, T.-Y.; Guan, Y.-F.; Tian, W.-W.; Viollet, B.; Rui, Y.-C.; Zhai, Q.-W.; Su, D.-F.; Miao, C.-Y. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef]
- Shiino, A.; Matsuda, M.; Handa, J.; Chance, B. Poor Recovery of Mitochondrial Redox State in CA1 after Transient Forebrain Ischemia in Gerbils. Stroke 1998, 29, 2421–2425. [Google Scholar] [CrossRef][Green Version]
- Halestrap, A.; Denton, R.M. The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by α-Cyano-4-hydroxycinnamate and related compounds. Biochem. J. 1975, 148, 97–106. [Google Scholar] [CrossRef]
- Bricker, D.K.; Taylor, E.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.-C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Safi, R.; Liu, X.; Baldi, R.; Liu, W.; Liu, J.; Locasale, J.W.; Chang, C.-Y.; McDonnell, D.P. Inhibition of ERRα Prevents Mitochondrial Pyruvate Uptake Exposing NADPH-Generating Pathways as Targetable Vulnerabilities in Breast Cancer. Cell Rep. 2019, 27, 3587–3601.e4. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wang, J.; Wu, G.; Lu, Z.-H.; Chakraborty, G.; Meyenhofer, M.; Tyring, S.K.; Matalon, R. Physiological Role of N-Acetylaspartate. N-Acetylaspartate 2006, 576, 131–143. [Google Scholar] [CrossRef]
- Dluzniewska, J.; Sarnowska, A.; Beresewicz, M.; Johnson, I.P.; Srai, S.K.S.; Ramesh, B.; Goldspink, G.; Gorecki, D.C.; Zablocka, B. A strong neuroprotective effect of the autonomous C-terminal peptide of IGF-1 Ec (MGF) in brain ischemia. FASEB J. 2005, 19, 1896–1898. [Google Scholar] [CrossRef]
- Dluzniewska, J.; Beresewicz, M.; Wojewodzka, U.; Gajkowska, B.; Zablocka, B. Transient cerebral ischemia induces delayed proapoptotic bad translocation to mitochondria in CA1 sector of hippocampus. Mol. Brain Res. 2005, 133, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, E.G.; Mochly-Rosen, D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J. Biol. Chem. 2001, 276, 29644–29650. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Romero-Gó, M.; Mez, M.; Jover, D.D.A.; Gó, A.; Mez, L.C.d.T.; Rodrigo, R.; Camacho, M.E.S.; Felipo, V. Phosphate-activated glutaminase activity is enhanced in brain, intestine and kidneys of rats following portacaval anastomosis. World J. Gastroenterol. 2006, 12, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Zieminska, E.; Toczylowska, B.; Diamandakis, D.; Hilgier, W.; Filipkowski, R.K.; Polowy, R.; Orzel, J.; Gorka, M.; Lazarewicz, J. Glutamate, Glutamine and GABA Levels in Rat Brain Measured Using MRS, HPLC and NMR Methods in Study of Two Models of Autism. Front. Mol. Neurosci. 2018, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Garcia, A.; Barbas, C. Gas Chromatography-Mass Spectrometry (GC-MS)-Based Metabolomics. Metab. Profiling 2010, 708, 191–204. [Google Scholar] [CrossRef]
- González-Peña, D.; Dudzik, D.; García, A.; De Ancos, B.; Barbas, C.; Sánchez-Moreno, C. Metabolomic Fingerprinting in the Comprehensive Study of Liver Changes Associated with Onion Supplementation in Hypercholesterolemic Wistar Rats. Int. J. Mol. Sci. 2017, 18, 267. [Google Scholar] [CrossRef] [PubMed]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.; Kuligowski, J.; Wilson, I.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kuligowski, J.; Sánchez-Illana, Á.; Sanjuán-Herráez, D.; Vento, M.; Quintás, G. Intra-batch effect correction in liquid chromatography-mass spectrometry using quality control samples and support vector regression (QC-SVRC). Analyst 2015, 140, 7810–7817. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beręsewicz-Haller, M.; Krupska, O.; Bochomulski, P.; Dudzik, D.; Chęcińska, A.; Hilgier, W.; Barbas, C.; Zablocki, K.; Zablocka, B. Mitochondrial Metabolism behind Region-Specific Resistance to Ischemia-Reperfusion Injury in Gerbil Hippocampus. Role of PKCβII and Phosphate-Activated Glutaminase. Int. J. Mol. Sci. 2021, 22, 8504. https://doi.org/10.3390/ijms22168504
Beręsewicz-Haller M, Krupska O, Bochomulski P, Dudzik D, Chęcińska A, Hilgier W, Barbas C, Zablocki K, Zablocka B. Mitochondrial Metabolism behind Region-Specific Resistance to Ischemia-Reperfusion Injury in Gerbil Hippocampus. Role of PKCβII and Phosphate-Activated Glutaminase. International Journal of Molecular Sciences. 2021; 22(16):8504. https://doi.org/10.3390/ijms22168504
Chicago/Turabian StyleBeręsewicz-Haller, Małgorzata, Olga Krupska, Paweł Bochomulski, Danuta Dudzik, Anita Chęcińska, Wojciech Hilgier, Coral Barbas, Krzysztof Zablocki, and Barbara Zablocka. 2021. "Mitochondrial Metabolism behind Region-Specific Resistance to Ischemia-Reperfusion Injury in Gerbil Hippocampus. Role of PKCβII and Phosphate-Activated Glutaminase" International Journal of Molecular Sciences 22, no. 16: 8504. https://doi.org/10.3390/ijms22168504
APA StyleBeręsewicz-Haller, M., Krupska, O., Bochomulski, P., Dudzik, D., Chęcińska, A., Hilgier, W., Barbas, C., Zablocki, K., & Zablocka, B. (2021). Mitochondrial Metabolism behind Region-Specific Resistance to Ischemia-Reperfusion Injury in Gerbil Hippocampus. Role of PKCβII and Phosphate-Activated Glutaminase. International Journal of Molecular Sciences, 22(16), 8504. https://doi.org/10.3390/ijms22168504