
Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome

 , , ,
, , ,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
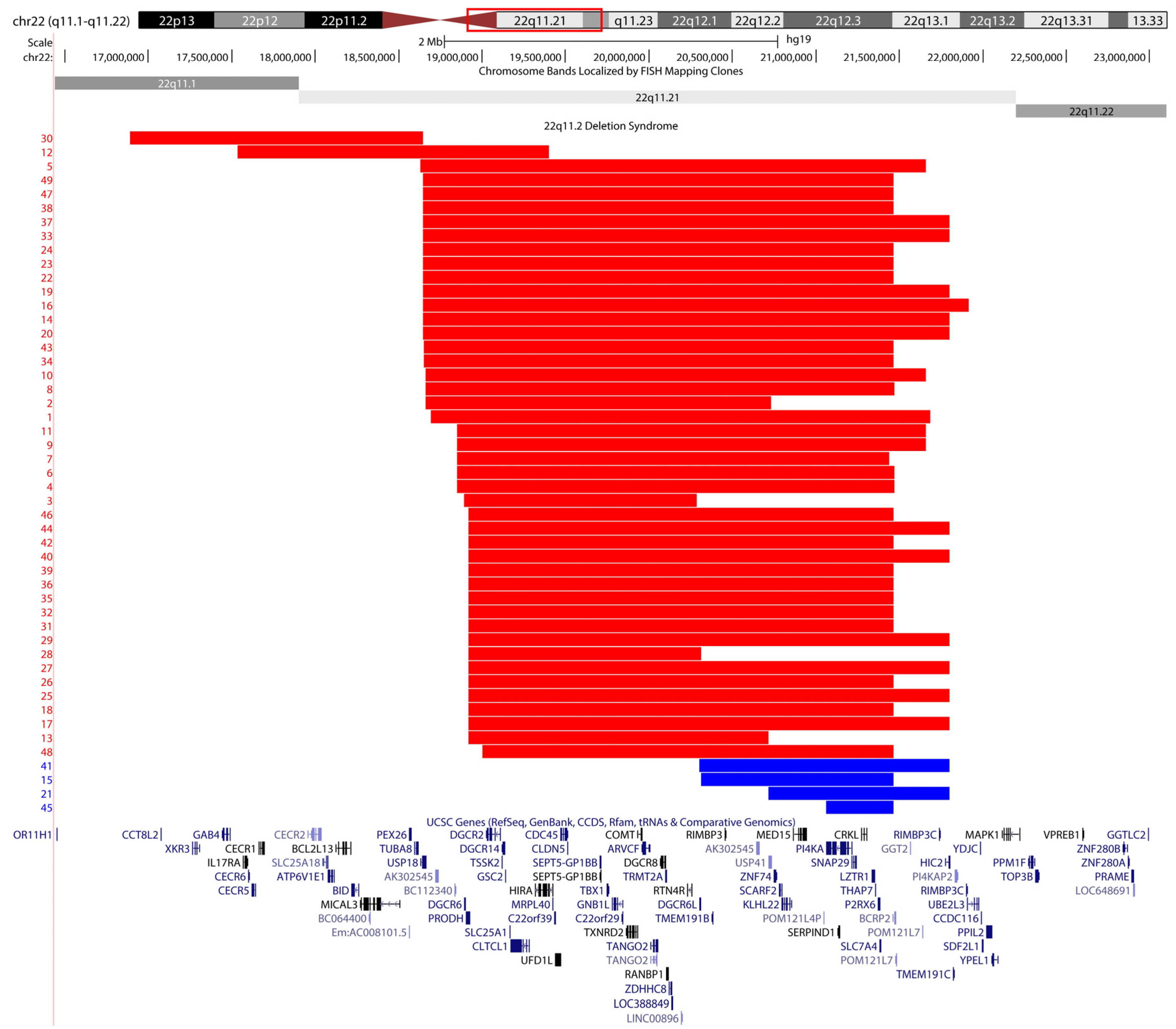
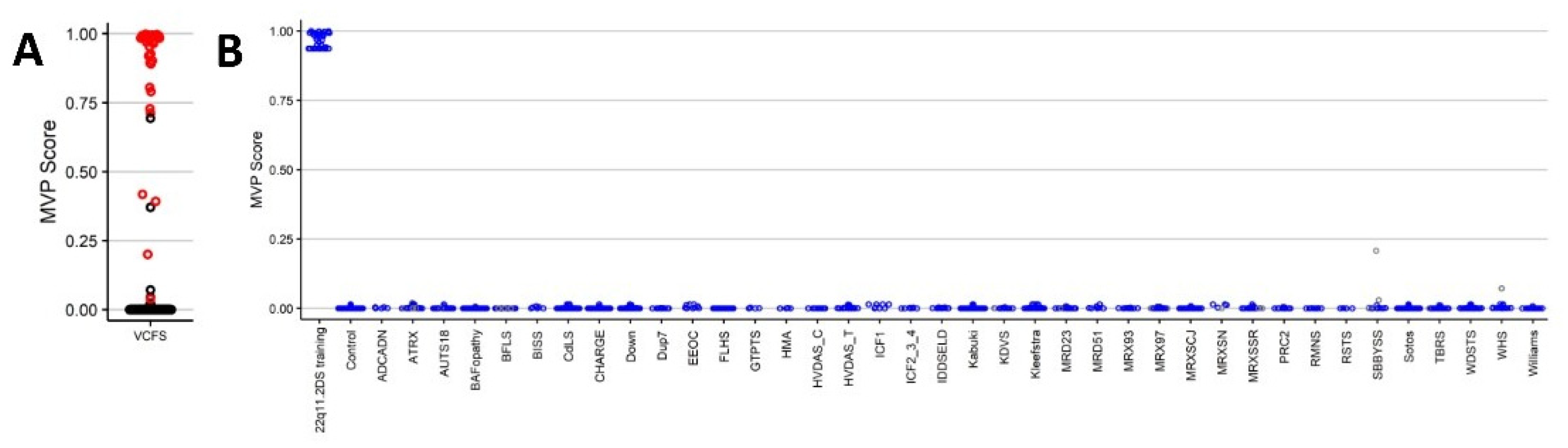
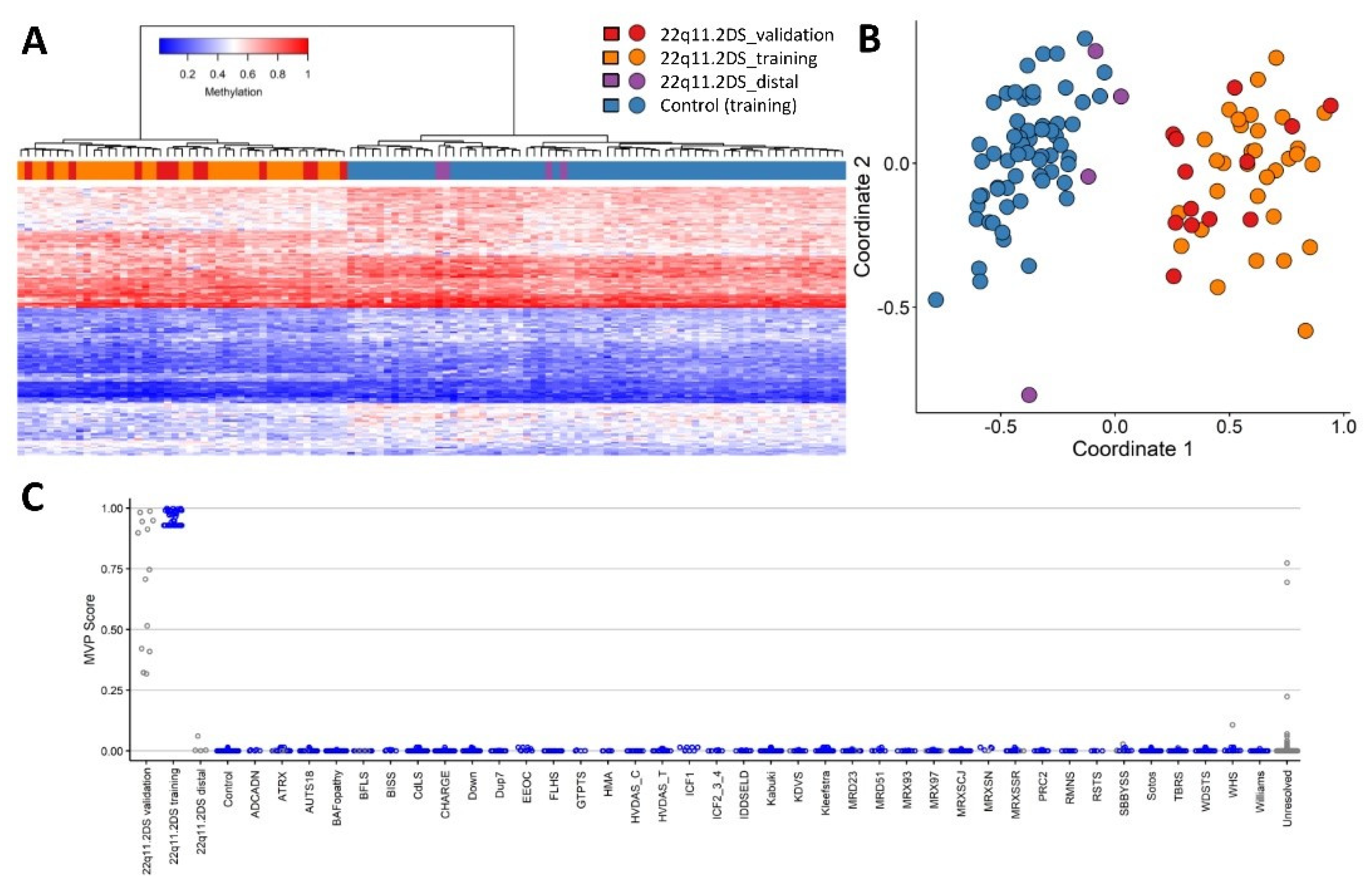
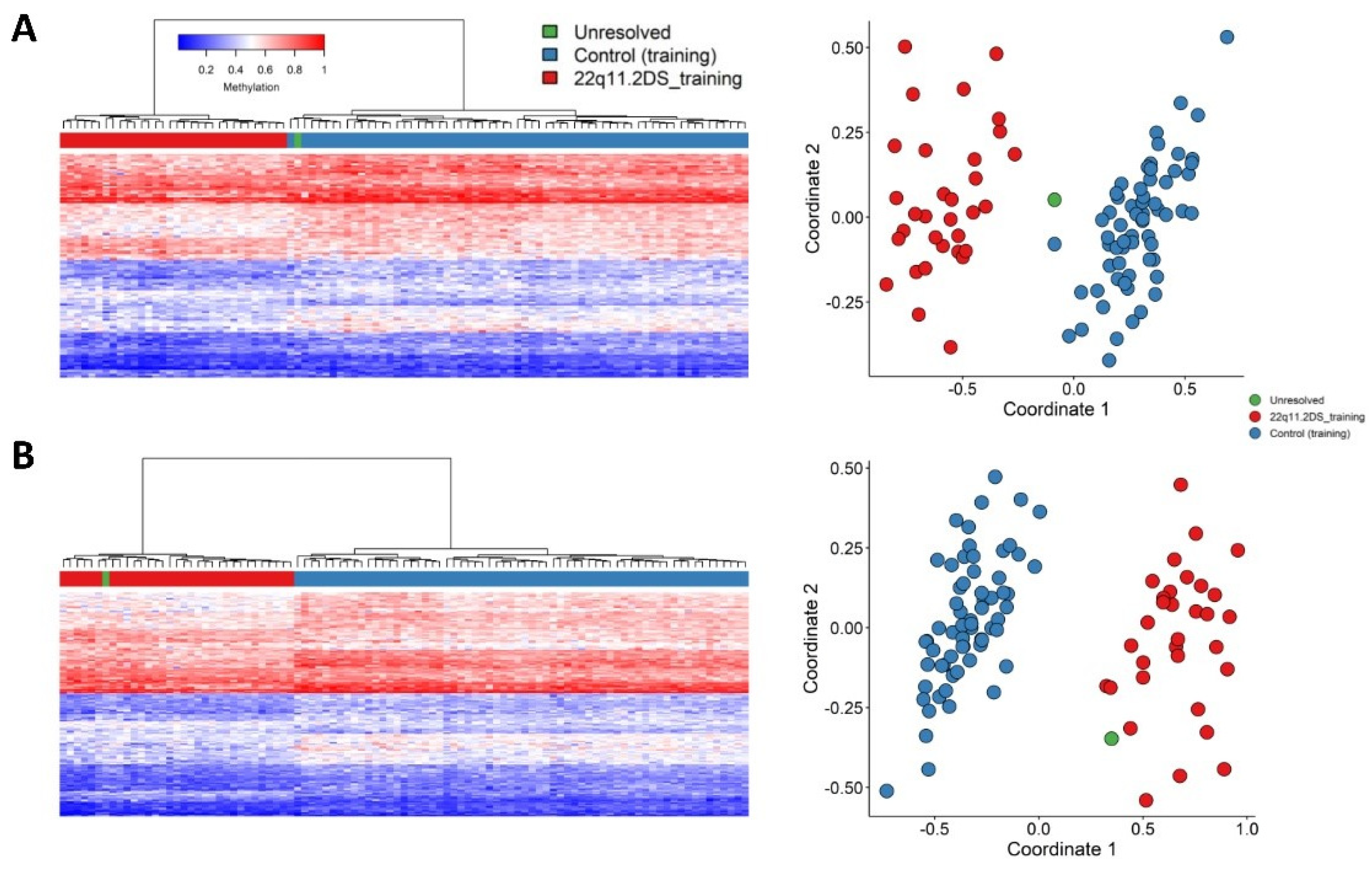
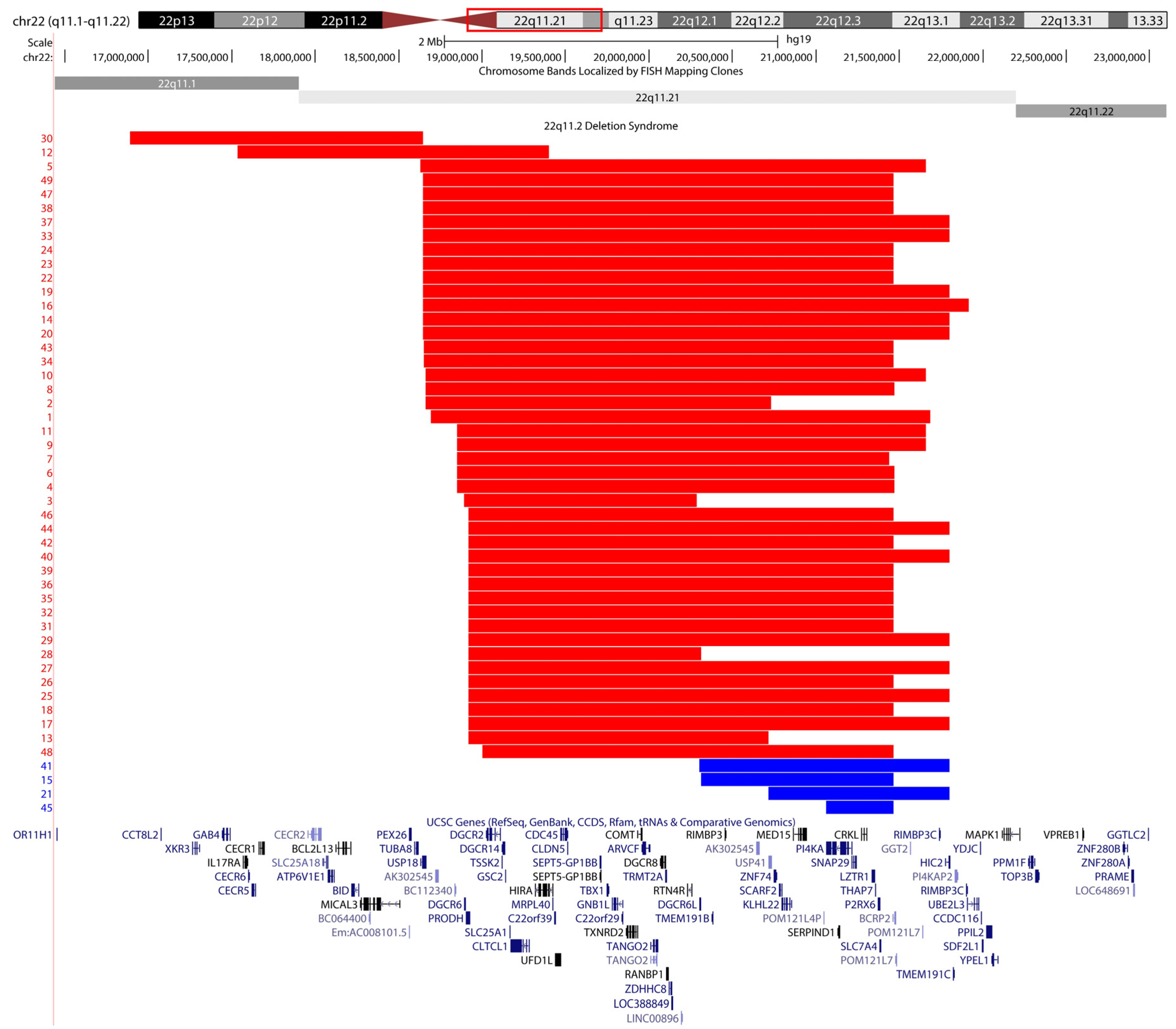
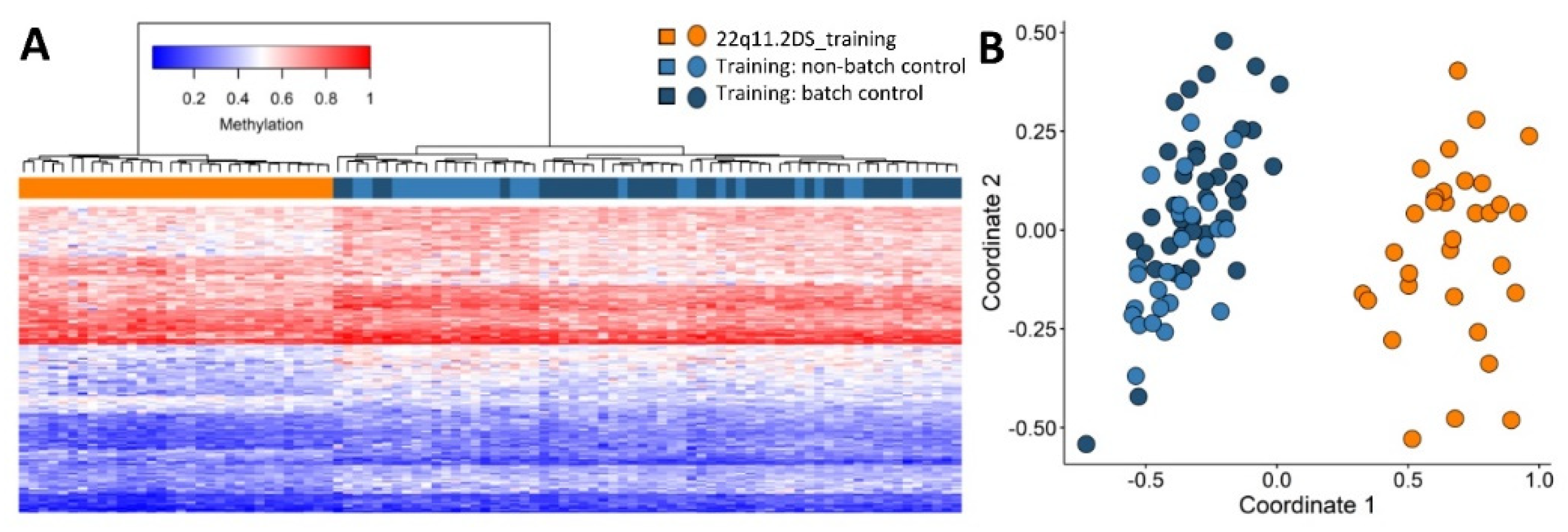
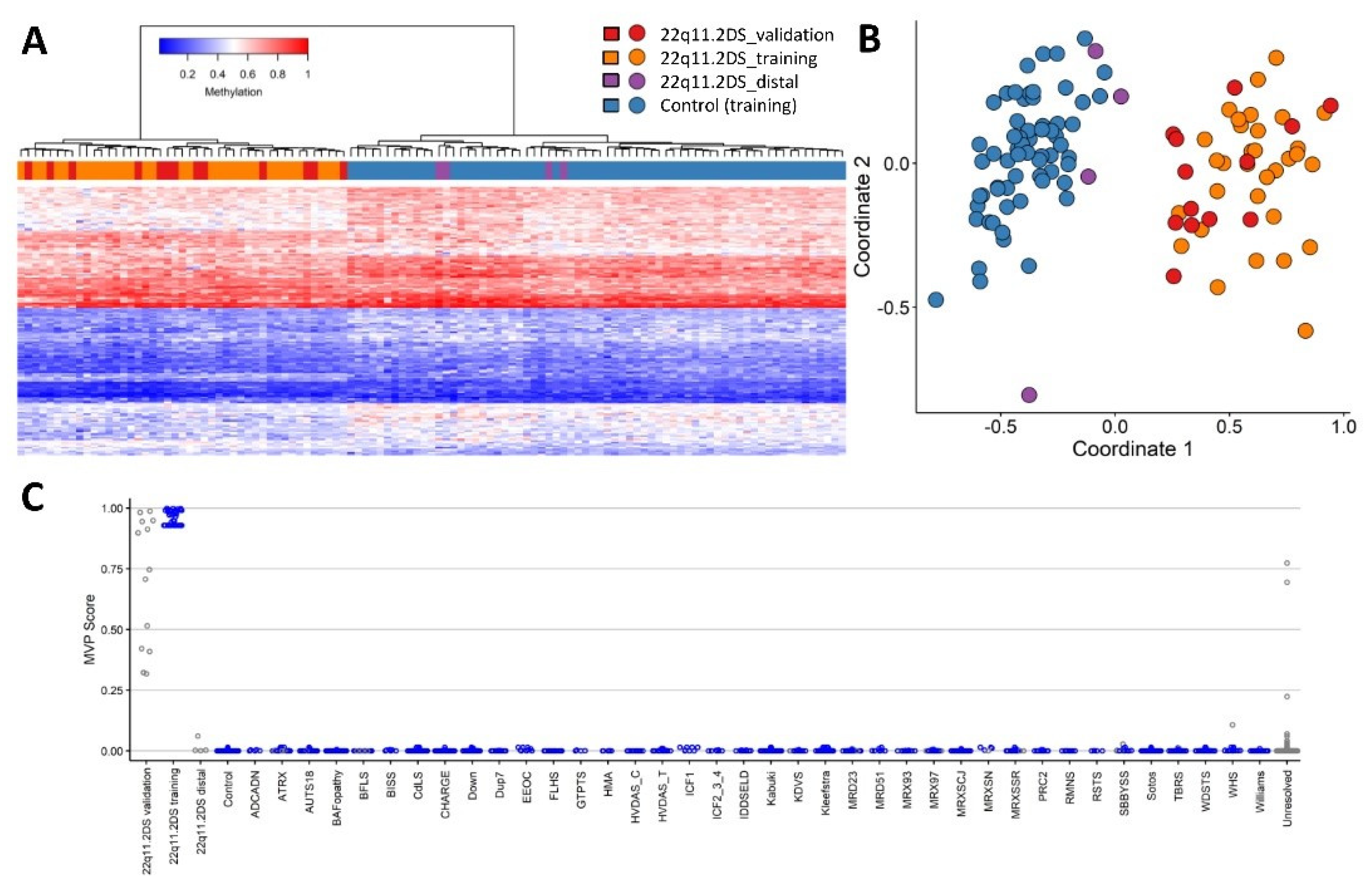
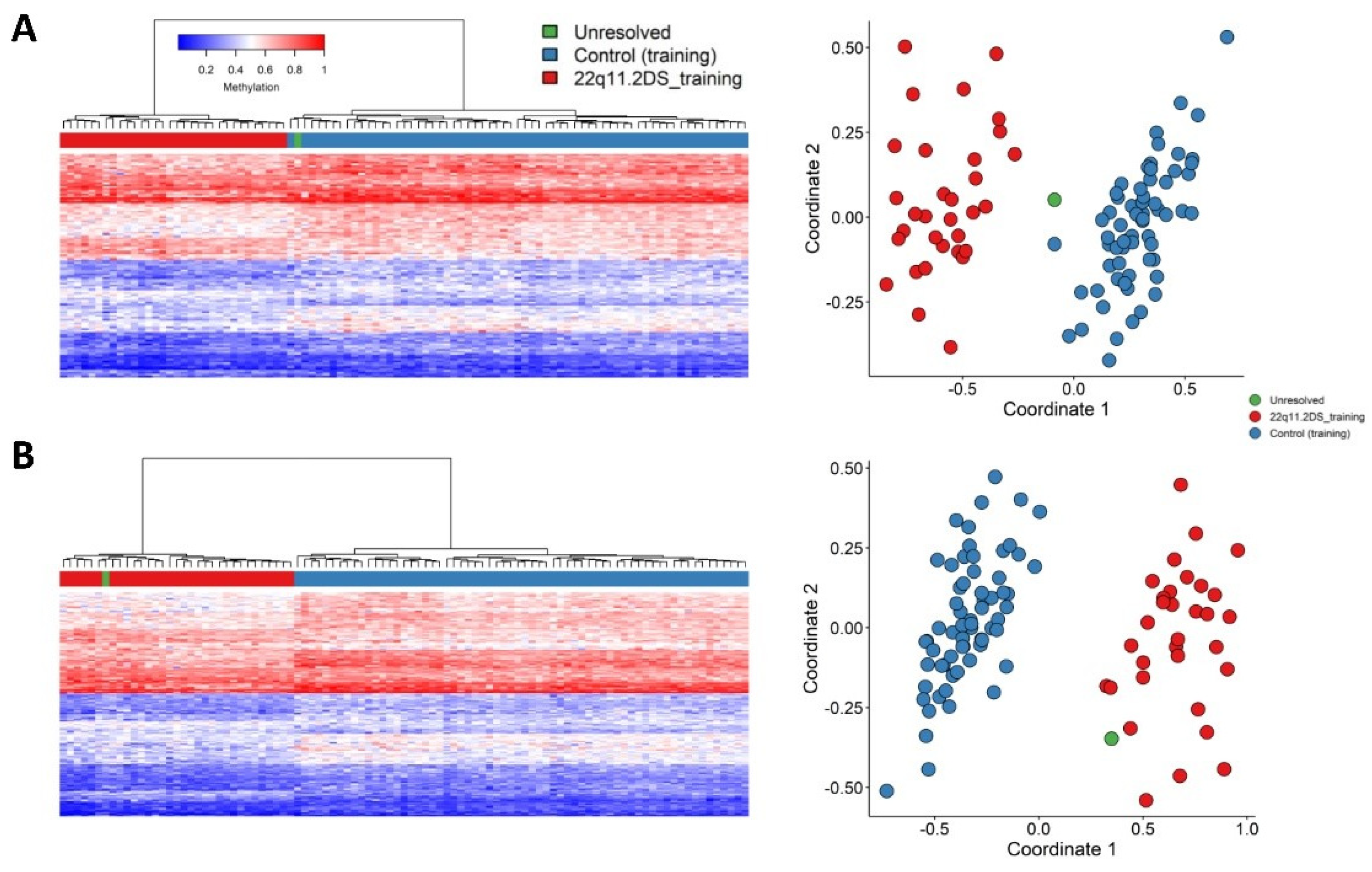
2.1. Screening Unresolved Cases
2.2. Differentially Methylated Regions
3. Discussion
4. Material and Methods
4.1. Study Cohort
4.2. DNA Methylation Experiment
4.3. Probe Selection, Clustering and Dimension Reduction
4.4. Cross Validation and Batch Structure
4.5. Classifier Model and Differentially Methylated Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald-Mcginn, D.M.; Sullivan, K.E. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 2011, 90, 1–18. [Google Scholar] [CrossRef]
- Gong, W.; Emanuel, B.S.; Collins, J.; Kim, D.H.; Wang, Z.; Chen, F.; Zhang, G.; Roe, B.; Budarf, M.L. A transcription map of the DiGeorge and velo-cardio-facial syndrome minimal critical region on 22q11. Hum. Mol. Genet. 1996, 5, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; Mcginn, D.E.; Mcginn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Harold, I.; Gaynor, J.W.; et al. The children’s hospital of Philadelphia. Am. J. Med. Genet. 2018, 176, 2058–2069. [Google Scholar] [CrossRef]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22Q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staple, L.; Andrews, T.; McDonald-McGinn, D.; Zackai, E.; Sullivan, K.E. Allergies in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) and patients with chronic granulomatous disease. Pediatr. Allergy Immunol. 2005, 16, 226–230. [Google Scholar] [CrossRef]
- Fung, W.L.A.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.C.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Boot, E.; Butcher, N.J.; Udow, S.; Marras, C.; Mok, K.Y.; Kaneko, S.; Barrett, M.J.; Prontera, P.; Berman, B.D.; Masellis, M.; et al. Typical features of Parkinson disease and diagnostic challenges with microdeletion 22q11.2. Neurology 2018, 90, E2059–E2067. [Google Scholar] [CrossRef] [Green Version]
- Carmel, M.; Michaelovsky, E.; Weinberger, R.; Frisch, A.; Mekori-Domachevsky, E.; Gothelf, D.; Weizman, A. Differential methylation of imprinting genes and MHC locus in 22q11.2 deletion syndrome-related schizophrenia spectrum disorders. World J. Biol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Crowley, B.; Ruffner, M.; McDonald McGinn, D.M.; Sullivan, K.E. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2018, 176, 2082–2086. [Google Scholar] [CrossRef]
- Meechan, D.W.; Maynard, T.M.; Gopalakrishna, D.; Wu, Y.; LaMantia, A.S. When half is not enough: Gene expression and dosage in the 22q11 Deletion syndrome. Gene Expr. 2007, 13, 299–310. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Rodenhiser, D.I.; Ainsworth, P.J.; Paré, G.; Sadikovic, B. DNA methylation analysis in constitutional disorders: Clinical implications of the epigenome. Crit. Rev. Clin. Lab. Sci. 2016, 53, 147–165. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; Dubourg, C.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Ciolfi, A.; Aref-Eshghi, E.; Pizzi, S.; Pedace, L.; Miele, E.; Kerkhof, J.; Flex, E.; Martinelli, S.; Radio, F.C.; Ruivenkamp, C.A.L.; et al. Frameshift mutations at the C-terminus of HIST1H1E result in a specific DNA hypomethylation signature. Clin. Epigenetics 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bend, E.G.; Aref-Eshghi, E.; Everman, D.B.; Rogers, R.C.; Cathey, S.S.; Prijoles, E.J.; Lyons, M.J.; Davis, H.; Clarkson, K.; Gripp, K.W.; et al. Gene domain-specific DNA methylation episignatures highlight distinct molecular entities of ADNP syndrome. Clin. Epigenetics 2019, 11, 1–17. [Google Scholar] [CrossRef]
- Krzyzewska, I.M.; Maas, S.M.; Henneman, P.; Lip, K.V.D.; Venema, A.; Baranano, K.; Chassevent, A.; Aref-Eshghi, E.; Van Essen, A.J.; Fukuda, T.; et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin. Epigenetics 2019, 11, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref-Eshghi, E.; Bend, E.G.; Colaiacovo, S.; Caudle, M.; Chakrabarti, R.; Napier, M.; Brick, L.; Brady, L.; Carere, D.A.; Levy, M.A.; et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet. 2019, 104, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Aref-Eshghi, E.; Bend, E.G.; Hood, R.L.; Schenkel, L.C.; Carere, D.A.; Chakrabarti, R.; Nagamani, S.C.S.; Cheung, S.W.; Campeau, P.M.; Prasad, C.; et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin–Siris and Nicolaides–Baraitser syndromes. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Aref-Eshghi, E.; Skinner, C.; Ainsworth, P.; Lin, H.; Paré, G.; Rodenhiser, D.I.; Schwartz, C.; Sadikovic, B. Peripheral blood epi-signature of Claes-Jensen syndrome enables sensitive and specific identification of patients and healthy carriers with pathogenic mutations in KDM5C. Clin. Epigenetics 2018, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Cigana Schenkel, L.; Huang, L.; Smith, A.; Pare, G.; Ainsworth, P.; Care4Rare Canada Consortium; Boycott, K.M.; Warman-Chardon, J.; Sadikovic, B. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Clin. Epigenetics 2016, 8, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Hood, R.L.; Schenkel, L.C.; Nikkel, S.M.; Ainsworth, P.J.; Pare, G.; Boycott, K.M.; Bulman, D.E.; Sadikovic, B. The defining DNA methylation signature of Floating-Harbor Syndrome. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Aref-Eshghi, E.; Levy, M.A.; Rodenhiser, D. DNA methylation signatures in mendelian developmental disorders as a diagnostic bridge between genotype and phenotype. Epigenomics 2019, 11, 563–575. [Google Scholar] [CrossRef]
- Sadikovic, B.; Levi, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 1–10. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; V Lord, R.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Strong, E.; Butcher, D.T.; Singhania, R.; Mervis, C.B.; Morris, C.A.; De Carvalho, D.; Weksberg, R.; Osborne, L.R. Symmetrical Dose-Dependent DNA-Methylation Profiles in Children with Deletion or Duplication of 7q11.23. Am. J. Hum. Genet. 2015, 97, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, L.C.; Aref-Eshghi, E.; Rooney, K.; Kerkhof, J.; Levy, M.A.; McConkey, H.; Rogers, R.C.; Phelan, K.; Sarasua, S.M.; Jain, L.; et al. DNA methylation epi-signature is associated with two molecularly and phenotypically distinct clinical subtypes of Phelan-McDermid syndrome. Clin. Epigenetics 2021, 13, 1–17. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Rodenhiser, D.I.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Hood, R.L.; Bulman, D.E.; Kernohan, K.D.; et al. Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am. J. Hum. Genet. 2018, 102, 156–174. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Zink, S.; Zweier, C.; Thiel, C.T.; Koch, A.; Rauch, R.; Lascorz, J.; Hüffmeier, U.; Weyand, M.; Singer, H.; et al. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J. Med. Genet. 2005, 42, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Deneen, B.; Ho, R.; Lukaszewicz, A.; Hochstim, C.J.; Gronostajski, R.M.; Anderson, D.J. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 2006, 52, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef]
- Jeanne, M.; Vuillaume, M.L.; Ung, D.C.; Vancollie, V.E.; Wagner, C.; Collins, S.C.; Vonwill, S.; Haye, D.; Chelloug, N.; Pfundt, R.; et al. Haploinsufficiency of the HIRA gene located in the 22q11 deletion syndrome region is associated with abnormal neurodevelopment and impaired dendritic outgrowth. Hum. Genet. 2021, 140, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, M.A.; Warzecha, C.; Bachu, M.; Dey, A.; Wu, T.; Adams, P.D.; Macfarlan, T.; Love, P.; Ozato, K. HIRA, a DiGeorge Syndrome Candidate Gene, Confers Proper Chromatin Accessibility on HSCs and Supports All Stages of Hematopoiesis. Cell Rep. 2020, 30, 2136–2149e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chieffo, C.; Garvey, N.; Gong, W.; Roe, B.; Zhang, G.; Silver, L.; Emanuel, B.S.; Budarf, M.L. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 1997, 43, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.I.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Shifman, S.; Bronstein, M.; Sternfeld, M.; Pisanté-Shalom, A.; Lev-Lehman, E.; Weizman, A.; Reznik, I.; Spivak, B.; Grisaru, N.; Karp, L.; et al. A highly significant association between a COMT haplotype and schizophrenia. Am. J. Hum. Genet. 2002, 71, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shprintzen, R.J.; Goldberg, R.; Golding-Kushner, K.J.; Marion, R.W. Late-onset psychosis in the velo-cardio-facial syndrome (5). Am. J. Med. Genet. 1992, 42, 141–142. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics 2017, 12, 923–933. [Google Scholar] [CrossRef]
- Prontera, P.; Napolioni, V.; Ottaviani, V.; Rogaia, D.; Fusco, C.; Augello, B.; Serino, D.; Parisi, V.; Bernardini, L.; Merla, G.; et al. DPP6 gene disruption in a family with Gilles de la Tourette syndrome. Neurogenetics 2014, 15, 237–242. [Google Scholar] [CrossRef]
- Prontera, P.; Ottaviani, V.; Rogaia, D.; Isidori, I.; Mencarelli, A.; Malerba, N.; Cocciadiferro, D.; Rolph, P.; Stangoni, G.; Vulto-van Silfhout, A.; et al. A novel MED12 mutation: Evidence for a fourth phenotype. Am. J. Med. Genet. Part A 2016, 170, 2377–2382. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smola, A.; Bartlett, P.; Schuurmans, D. Advances in Large Margin Classifiers; MIT Press: Cambridge, UK, 2000. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of CpGs | Methylation Difference (%) | Number of DMRs |
|---|---|---|
| 5 | 10 | 0 |
| 5 | 5 | 329 |
| 3 | 10 | 5 |
| 3 | 5 | 1064 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rooney, K.; Levy, M.A.; Haghshenas, S.; Kerkhof, J.; Rogaia, D.; Tedesco, M.G.; Imperatore, V.; Mencarelli, A.; Squeo, G.M.; Di Venere, E.; et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. Int. J. Mol. Sci. 2021, 22, 8611. https://doi.org/10.3390/ijms22168611
Rooney K, Levy MA, Haghshenas S, Kerkhof J, Rogaia D, Tedesco MG, Imperatore V, Mencarelli A, Squeo GM, Di Venere E, et al. Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. International Journal of Molecular Sciences. 2021; 22(16):8611. https://doi.org/10.3390/ijms22168611
Chicago/Turabian StyleRooney, Kathleen, Michael A. Levy, Sadegheh Haghshenas, Jennifer Kerkhof, Daniela Rogaia, Maria Giovanna Tedesco, Valentina Imperatore, Amedea Mencarelli, Gabriella Maria Squeo, Eleonora Di Venere, and et al. 2021. "Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome" International Journal of Molecular Sciences 22, no. 16: 8611. https://doi.org/10.3390/ijms22168611
APA StyleRooney, K., Levy, M. A., Haghshenas, S., Kerkhof, J., Rogaia, D., Tedesco, M. G., Imperatore, V., Mencarelli, A., Squeo, G. M., Di Venere, E., Di Cara, G., Verrotti, A., Merla, G., Tedder, M. L., DuPont, B. R., Sadikovic, B., & Prontera, P. (2021). Identification of a DNA Methylation Episignature in the 22q11.2 Deletion Syndrome. International Journal of Molecular Sciences, 22(16), 8611. https://doi.org/10.3390/ijms22168611