
Autophagy and Metabolism in Normal and Malignant Hematopoiesis


Abstract
:1. Introduction
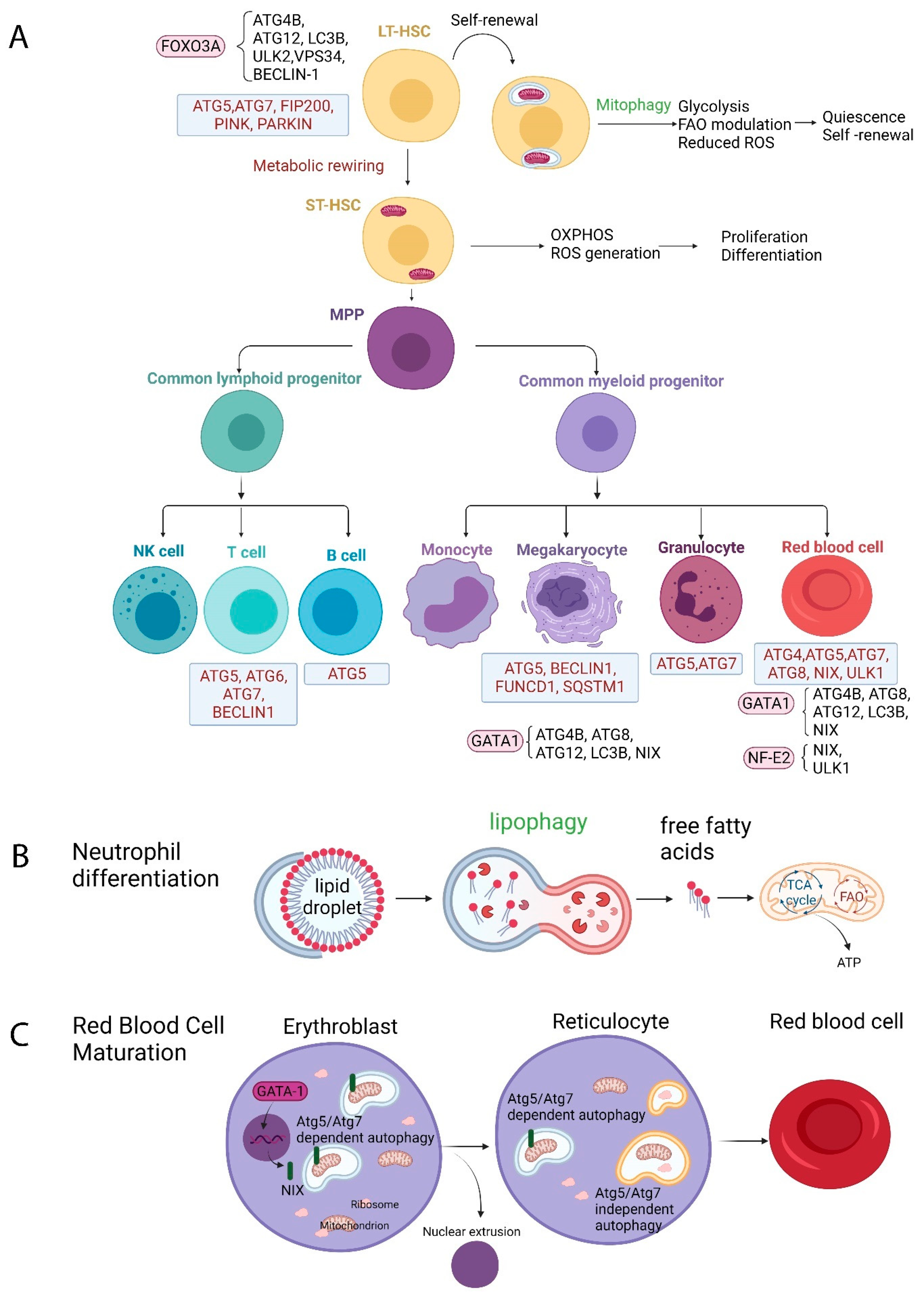
2. The Role of Autophagy in Normal Hematopoiesis
2.1. Autophagy and HSCs
2.2. Autophagy and Erythropoiesis
2.3. Autophagy and Granulopoiesis
2.4. Autophagy and Megakaryopoiesis
2.5. Autophagy and Lymphopoiesis
2.5.1. T Lymphocytes
2.5.2. B Lymphocytes
3. Autophagy in Malignant Hematopoiesis
3.1. Autophagy in Acute Myeloid Leukemia (AML)
3.2. Autophagy in Myelodysplastic Syndromes (MDS)
3.3. Autophagy in Chronic Myeloid Leukemia (CML)
3.4. Autophagy in Acute Lymphoblastic Leukemia (ALL)
3.5. Autophagy in Chronic Lymphocytic Leukemia (CLL) and Lymphoma
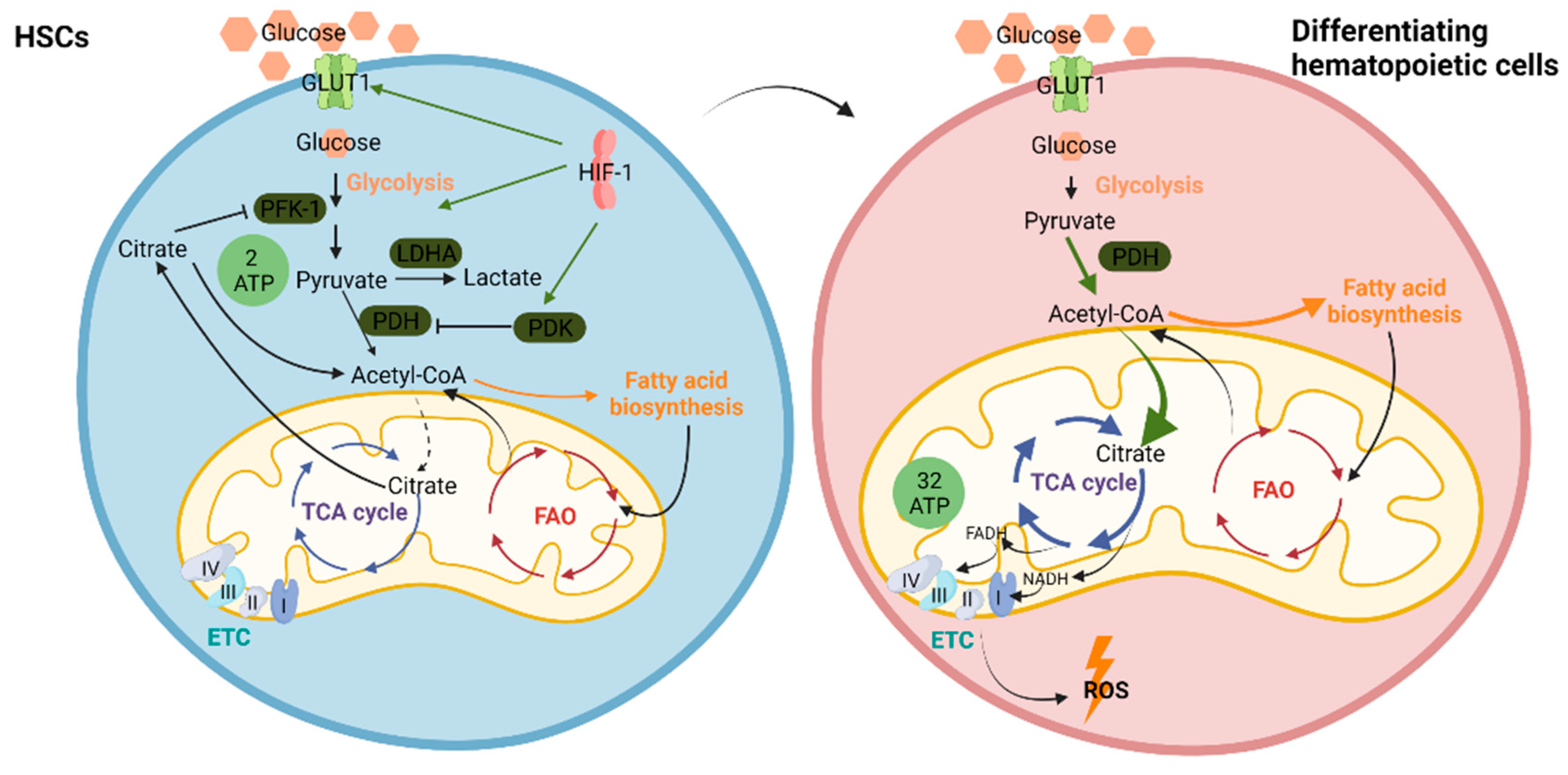
4. Metabolism in Normal Hematopoiesis
4.1. Regulation of HSC Metabolism–Glycolysis Versus OXPHOS
4.2. Fatty Acid Oxidation (FAO) in HSCs
5. Metabolism in Malignant Hematopoiesis
5.1. Glucose Metabolism
5.2. Glutamine Metabolism
5.3. Fatty Acid Metabolism
5.4. Mitochondrial Metabolism
6. Links between Autophagy and Metabolism
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ito, K.; Bonora, M.; Ito, K. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 2019, 109, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Kohli, L.; Passegué, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Chen, W.L.; Wang, J.H.; Li, N.; Li, J.M.; Mi, J.Q.; Zhang, W.N.; Li, Y.; Wu, S.F.; et al. Rapid diagnosis and prognosis of de novo acute myeloid leukemia by serum metabonomic analysis. J. Proteome Res. 2013, 12, 4393–4401. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warr, M.R.; Kohli, L.; Passegué, E. Born to survive: Autophagy in hematopoietic stem cell maintenance. Cell Cycle 2013, 12, 1979–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-A.; Sanalkumar, R.; O’Geen, H.; Linnemann, A.K.; Chang, C.-J.; Bouhassira, E.E.; Farnham, P.J.; Keles, S.; Bresnick, E.H. Autophagy driven by a master regulator of hematopoiesis. Mol. Cell. Biol. 2012, 32, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Karagianni, P.; Giannouli, S.; Voulgarelis, M. From the (Epi)Genome to Metabolism and Vice Versa; Examples from Hematologic Malignancy. Int. J. Mol. Sci. 2021, 22, 6321. [Google Scholar] [CrossRef]
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An evolving paradigm for stem cell biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef]
- Phadwal, K.; Watson, A.S.; Simon, A.K. Tightrope act: Autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol. Life Sci. 2013, 70, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vos, K.E.; Gomez-Puerto, C.; Coffer, P.J. Regulation of autophagy by Forkhead box (FOX) O transcription factors. Adv. Biol. Regul. 2012, 52, 122–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.-U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.W.; et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.; Ferguson, D.J.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Aerbajinai, W.; Giattina, M.; Lee, Y.T.; Raffeld, M.; Miller, J.L. The proapoptotic factor Nix is coexpressed with Bcl-xL during terminal erythroid differentiation. Blood 2003, 102, 712–717. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Arakawa, S.; Nishida, Y.; Yamaguchi, H.; Ishii, E.; Shimizu, S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 2014, 5, 4004. [Google Scholar] [CrossRef] [PubMed]
- Barde, I.; Rauwel, B.; Marin-Florez, R.M.; Corsinotti, A.; Laurenti, E.; Verp, S.; Offner, S.; Marquis, J.; Kapopoulou, A.; Vanicek, J.; et al. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 2013, 340, 350–353. [Google Scholar] [CrossRef] [Green Version]
- Gothwal, M.; Wehrle, J.; Aumann, K.; Zimmermann, V.; Gründer, A.; Pahl, H.L. A novel role for nuclear factor-erythroid 2 in erythroid maturation by modulation of mitochondrial autophagy. Haematologica 2016, 101, 1054–1064. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e465. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Wei, Q.; Shin, J.N.; Fattah, E.A.; Bonilla, D.L.; Xiang, Q.; Eissa, N.T. Autophagy Is Required for Neutrophil-Mediated Inflammation. Cell Rep. 2015, 12, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Iula, L.; Keitelman, I.A.; Sabbione, F.; Fuentes, F.; Guzman, M.; Galletti, J.G.; Gerber, P.P.; Ostrowski, M.; Geffner, J.R.; Jancic, C.C.; et al. Autophagy Mediates Interleukin-1β Secretion in Human Neutrophils. Front. Immunol. 2018, 9, 269. [Google Scholar] [CrossRef] [Green Version]
- Colosetti, P.; Puissant, A.; Robert, G.; Luciano, F.; Jacquel, A.; Gounon, P.; Cassuto, J.P.; Auberger, P. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 2009, 5, 1092–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Cai, J.; Zhang, S.; Yuan, N.; Li, X.; Fang, Y.; Song, L.; Shang, M.; Liu, S.; Zhao, W.; et al. Loss of autophagy leads to failure in megakaryopoiesis, megakaryocyte differentiation, and thrombopoiesis in mice. Exp. Hematol. 2015, 43, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Chang, C.; Luo, D.; Su, H.; Yu, S.; Hua, W.; Chen, Z.; Hu, H.; Liu, W. Dissection of autophagy in human platelets. Autophagy 2014, 10, 642–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ren, H.; Xu, C.; Zhu, C.; Wu, H.; Liu, D.; Wang, J.; Liu, L.; Li, W.; Ma, Q.; et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 2016, 5, e21407. [Google Scholar] [CrossRef]
- Pua, H.H.; Dzhagalov, I.; Chuck, M.; Mizushima, N.; He, Y.W. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007, 204, 25–31. [Google Scholar] [CrossRef]
- Stephenson, L.M.; Miller, B.C.; Ng, A.; Eisenberg, J.; Zhao, Z.; Cadwell, K.; Graham, D.B.; Mizushima, N.N.; Xavier, R.; Virgin, H.W.; et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009, 5, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055. [Google Scholar] [CrossRef]
- Farfariello, V.; Amantini, C.; Santoni, G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J. Leukoc. Biol. 2012, 92, 421–431. [Google Scholar] [CrossRef]
- Arsov, I.; Adebayo, A.; Kucerova-Levisohn, M.; Haye, J.; MacNeil, M.; Papavasiliou, F.N.; Yue, Z.; Ortiz, B.D. A role for autophagic protein beclin 1 early in lymphocyte development. J. Immunol. 2011, 186, 2201–2209. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Zhao, Z.; Stephenson, L.M.; Cadwell, K.; Pua, H.H.; Lee, H.K.; Mizushima, N.N.; Iwasaki, A.; He, Y.W.; Swat, W.; et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008, 4, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.L.; Kuballa, P.; Khor, B.; Zhang, M.; Shi, H.N.; Virgin, H.W.; Xavier, R.J. ATG5 regulates plasma cell differentiation. Autophagy 2013, 9, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Hong, M.J.; Sun, H.; Wang, L.; Shi, X.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 2014, 20, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Jordan, C.T. The leukemic stem cell. Best Pract. Res. Clin. Haematol. 2007, 20, 13–18. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; de Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2017, 31, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Payton, J.E.; Ries, R.E.; Shannon, W.D.; Deshmukh, H.; Zhao, Y.; Baty, J.; Heath, S.; Westervelt, P.; Watson, M.A.; et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc. Natl. Acad. Sci. USA 2009, 106, 12950–12955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxid. Med. Cell Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N., Jr.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.-G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016, 7, e2361. [Google Scholar] [CrossRef]
- Amy, H.P.; Lucie Leveque-El, M.; Therese, V.; Claudia, B.; Joanne, S.; Sebastien, J.; Geoffrey, R.H.; Kelli, P.A.M.; Steven, W.L. Acute myeloid leukemia stem cell function is preserved in the absence of autophagy. Haematologica 2017, 102, e344–e347. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.-L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Bühler, C.; Marcantonio, D.D.; Martinez, E.; Göllner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). Classification of Tumours of Haematopoietic and Lymphoid Tissues, WHO Classification of Tumours, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; Volume 2. [Google Scholar]
- Visconte, V.; Przychodzen, B.P.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.; Hirsch, C.M.; Carraway, H.E.; Parker, Y.; Lindner, D.; et al. Genetic and Epigenetic Defects in the Autophagy Machinery in Myelodysplastic Syndromes. Blood 2016, 128, 4301. [Google Scholar] [CrossRef]
- Houwerzijl, E.J.; Pol, H.W.; Blom, N.R.; van der Want, J.J.L.; Wolf, J.d.; Vellenga, E. Erythroid precursors from patients with low-risk myelodysplasia demonstrate ultrastructural features of enhanced autophagy of mitochondria. Leukemia 2009, 23, 886–891. [Google Scholar] [CrossRef]
- Van de Loosdrecht, A.A.; Brada, S.J.; Blom, N.R.; Hendriks, D.W.; Smit, J.W.; van den Berg, E.; de Wolf, J.T.; Vellenga, E. Mitochondrial disruption and limited apoptosis of erythroblasts are associated with high risk myelodysplasia. An ultrastructural analysis. Leuk. Res. 2001, 25, 385–393. [Google Scholar] [CrossRef]
- Stergiou, I.E.; Kambas, K.; Poulaki, A.; Giannouli, S.; Katsila, T.; Dimitrakopoulou, A.; Vidali, V.; Mouchtouris, V.; Kloukina, I.; Xingi, E.; et al. Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology. Int. J. Mol. Sci. 2021, 22, 4099. [Google Scholar] [CrossRef]
- Lazarini, M.; Machado-Neto, J.A.; Duarte, A.d.S.S.; Pericole, F.V.; Vieira, K.P.; Niemann, F.S.; Alvarez, M.; Traina, F.; Saad, S.T.O. BNIP3L in myelodysplastic syndromes and acute myeloid leukemia: Impact on disease outcome and cellular response to decitabine. Haematologica 2016, 101, e445–e448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Yang, L.; Guo, L.; Cui, N.; Zhang, G.; Liu, C.; Xing, L.; Shao, Z.; Wang, H. Impaired Mitophagy of Nucleated Erythroid Cells Leads to Anemia in Patients with Myelodysplastic Syndromes. Oxidative Med. Cell. Longev. 2018, 2018, 6328051. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Ma, L.; Sun, J.E.; Zhu, L.J.; Green, M.R. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011, 118, 2840–2848. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Crowley, L.C.; Elzinga, B.M.; O’Sullivan, G.C.; McKenna, S.L. Autophagy induction by Bcr-Abl-expressing cells facilitates their recovery from a targeted or nontargeted treatment. Am. J. Hematol. 2011, 86, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Rothe, K.; Yen, R.; Fruhstorfer, C.; Maetzig, T.; Chen, M.; Forrest, D.L.; Humphries, R.K.; Jiang, X. A novel AHI-1-BCR-ABL-DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia 2017, 31, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef]
- Drullion, C.; Trégoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.; Auberger, P.; Mahon, F.X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef] [PubMed]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Ianniciello, A.; Dumas, P.-Y.; Drullion, C.; Guitart, A.; Villacreces, A.; Peytour, Y.; Chevaleyre, J.; de la Grange, P.B.; Vigon, I.; Desplat, V.; et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 2017, 8, 96984–96992. [Google Scholar] [CrossRef] [Green Version]
- Golub, T.R.; Barker, G.F.; Bohlander, S.K.; Hiebert, S.W.; Ward, D.C.; Bray-Ward, P.; Morgan, E.; Raimondi, S.C.; Rowley, J.D.; Gilliland, D.G. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 1995, 92, 4917–4921. [Google Scholar] [CrossRef] [Green Version]
- Polak, R.; Bierings, M.B.; van der Leije, C.S.; Sanders, M.A.; Roovers, O.; Marchante, J.R.M.; Boer, J.M.; Cornelissen, J.J.; Pieters, R.; den Boer, M.L.; et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica 2019, 104, 738–748. [Google Scholar] [CrossRef]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.N.; Liu, Q.Q.; Zhang, S.P.; Yuan, N.; Cao, Y.; Cai, J.Y.; Lin, W.W.; Xu, F.; Wang, Z.J.; Chen, B.; et al. Alternative messenger RNA splicing of autophagic gene Beclin 1 in human B-cell acute lymphoblastic leukemia cells. Asian Pac. J. Cancer Prev. 2014, 15, 2153–2158. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xie, M.; Kang, R.; Livesey, K.M.; Cao, L.; Tang, D. HMGB1 is a therapeutic target for leukemia. Am. J. Blood Res. 2012, 2, 36–43. [Google Scholar]
- Laane, E.; Tamm, K.P.; Buentke, E.; Ito, K.; Kharaziha, P.; Oscarsson, J.; Corcoran, M.; Björklund, A.C.; Hultenby, K.; Lundin, J.; et al. Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ. 2009, 16, 1018–1029. [Google Scholar] [CrossRef]
- Heidari, N.; Hicks, M.A.; Harada, H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 2010, 1, e76. [Google Scholar] [CrossRef] [Green Version]
- Neri, L.M.; Cani, A.; Martelli, A.M.; Simioni, C.; Junghanss, C.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; McCubrey, J.A.; et al. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia 2014, 28, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered expression of autophagy-related genes might contribute to glucocorticoid resistance in precursor B-cell-type acute lymphoblastic leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef]
- Kumar, K.; Kaur, J.; Walia, S.; Pathak, T.; Aggarwal, D. L-asparaginase: An effective agent in the treatment of acute lymphoblastic leukemia. Leuk. Lymphoma 2014, 55, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyczynski, M.; Vesterlund, M.; Björklund, A.C.; Zachariadis, V.; Janssen, J.; Gallart-Ayala, H.; Daskalaki, E.; Wheelock, C.E.; Lehtiö, J.; Grandér, D.; et al. Metabolic reprogramming of acute lymphoblastic leukemia cells in response to glucocorticoid treatment. Cell Death Dis. 2018, 9, 846. [Google Scholar] [CrossRef] [Green Version]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.C.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gade, P.; Kimball, A.S.; DiNardo, A.C.; Gangwal, P.; Ross, D.D.; Boswell, H.S.; Keay, S.K.; Kalvakolanu, D.V. Death-associated Protein Kinase-1 Expression and Autophagy in Chronic Lymphocytic Leukemia Are Dependent on Activating Transcription Factor-6 and CCAAT/Enhancer-binding Protein-β. J. Biol. Chem. 2016, 291, 22030–22042. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bologna, C.; Buonincontri, R.; Serra, S.; Vaisitti, T.; Audrito, V.; Brusa, D.; Pagnani, A.; Coscia, M.; D’Arena, G.; Mereu, E.; et al. SLAMF1 regulation of chemotaxis and autophagy determines CLL patient response. J. Clin. Investig. 2016, 126, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar] [PubMed]
- Huang, J.J.; Zhu, Y.J.; Lin, T.Y.; Jiang, W.Q.; Huang, H.Q.; Li, Z.M. Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Hum. Pathol. 2011, 42, 1459–1466. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Bertolo, C.; Roa, S.; Sagardoy, A.; Mena-Varas, M.; Robles, E.F.; Martinez-Ferrandis, J.I.; Sagaert, X.; Tousseyn, T.; Orta, A.; Lossos, I.S.; et al. LITAF, a BCL6 target gene, regulates autophagy in mature B-cell lymphomas. Br. J. Haematol. 2013, 162, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhou, X.; Zhang, Y.; Yang, J.; Xu, Y.; Zhao, Y.; Wang, X. CUL4B regulates autophagy via JNK signaling in diffuse large B-cell lymphoma. Cell Cycle 2019, 18, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; He, M.; Cheng, F.; Bai, R.; da Silva, S.R.; Aguiar, R.C.T.; Gao, S.-J. Tenovin-6 inhibits proliferation and survival of diffuse large B-cell lymphoma cells by blocking autophagy. Oncotarget 2017, 8, 14912–14924. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.; Marzec, J.; Clear, A.; Petty, R.D.; Coutinho, R.; Matthews, J.; Wilson, A.; Iqbal, S.; Calaminici, M.; Gribben, J.G.; et al. Dysregulation of autophagy in human follicular lymphoma is independent of overexpression of BCL-2. Oncotarget 2014, 5, 11653–11668. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Gatica, D.; Ying, Z.X.; Peterson, L.F.; Kim, P.; Bernard, D.; Saiya-Cork, K.; Wang, S.; Kaminski, M.S.; Chang, A.E.; et al. Follicular lymphoma-associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J. Clin. Investig. 2019, 129, 1626–1640. [Google Scholar] [CrossRef]
- Heine, S.; Kleih, M.; Giménez, N.; Böpple, K.; Ott, G.; Colomer, D.; Aulitzky, W.E.; van der Kuip, H.; Silkenstedt, E. Cyclin D1-CDK4 activity drives sensitivity to bortezomib in mantle cell lymphoma by blocking autophagy-mediated proteolysis of NOXA. J. Hematol. Oncol. 2018, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.; Miranda, R.N.; Medeiros, L.J.; McCarty, N. TG2 and NF-κB Signaling Coordinates the Survival of Mantle Cell Lymphoma Cells via IL6-Mediated Autophagy. Cancer Res. 2016, 76, 6410–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Teo, A.E.; McCarty, N. ROS-Induced CXCR4 Signaling Regulates Mantle Cell Lymphoma (MCL) Cell Survival and Drug Resistance in the Bone Marrow Microenvironment via Autophagy. Clin. Cancer Res. 2016, 22, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Zon, L.I. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 2008, 453, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Cooper, D.D.; Hayes, S.F.; Spangrude, G.J. Rhodamine-123 staining in hematopoietic stem cells of young mice indicates mitochondrial activation rather than dye efflux. Blood 1998, 91, 4106–4117. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Parmar, K.; Mauch, P.; Vergilio, J.A.; Sackstein, R.; Down, J.D. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 5431–5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, J.J.; Bui, T.; Gruber, M.; Gordan, J.D.; DeBerardinis, R.J.; Covello, K.L.; Simon, M.C.; Thompson, C.B. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007, 21, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- De Almeida, M.J.; Luchsinger, L.L.; Corrigan, D.J.; Williams, L.J.; Snoeck, H.W. Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell 2017, 21, 725–729.e724. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, L.L.; de Almeida, M.J.; Corrigan, D.J.; Mumau, M.; Snoeck, H.W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 2016, 529, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrin, M.; Widjaja, A.; Liu, Y.; Luo, H.; Chen, D. The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell 2018, 17, e12756. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimmelé, P.; Liang, R.; Bigarella, C.L.; Kocabas, F.; Xie, J.; Serasinghe, M.N.; Chipuk, J.; Sadek, H.; Zhang, C.C.; Ghaffari, S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015, 16, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigarella, C.L.; Li, J.; Rimmelé, P.; Liang, R.; Sobol, R.W.; Ghaffari, S. FOXO3 Transcription Factor Is Essential for Protecting Hematopoietic Stem and Progenitor Cells from Oxidative DNA Damage. J. Biol. Chem. 2017, 292, 3005–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Li, L.; Zou, P.; Wang, J.; Shao, L.; Zhou, D.; Liu, L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation 2014, 97, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Ni, M.; Cao, H.; Signer, R.A.J.; Li, D.; Li, M.; Gu, Z.; Hu, Z.; Dickerson, K.E.; et al. Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat. Cell Biol. 2017, 19, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girão, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; van der Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e724. [Google Scholar] [CrossRef] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoechel, B.; Aster, J.C. Metabolic Mechanisms of Drug Resistance in Leukemia. Cell Metab. 2015, 22, 759–760. [Google Scholar] [CrossRef] [Green Version]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef] [Green Version]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Perea, G.; Domingo, A.; Villamor, N.; Palacios, C.; Juncà, J.; Torres, P.; Llorente, A.; Fernández, C.; Tormo, M.; de Llano, M.P.O.; et al. Adverse prognostic impact of CD36 and CD2 expression in adult de novo acute myeloid leukemia patients. Leuk. Res. 2005, 29, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Tucci, J.; Sheng, X.; Mittelman, S.D. Abstract 4339: Acute lymphoblastic leukemia cells stimulate adipocyte lipolysis and utilize adipocyte-derived free-fatty acids for proliferation. Cancer Res. 2014, 74, 4339. [Google Scholar] [CrossRef]
- Tucci, J.; Chen, T.; Margulis, K.; Orgel, E.; Paszkiewicz, R.L.; Cohen, M.D.; Oberley, M.J.; Wahhab, R.; Jones, A.E.; Divakaruni, A.S.; et al. Adipocytes Provide Fatty Acids to Acute Lymphoblastic Leukemia Cells. Front. Oncol. 2021, 11, 665763. [Google Scholar] [CrossRef]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.M.; Li, P.; Liu, Z.; Wu, J.Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hurren, R.; MacLean, N.; Gronda, M.; Jitkova, Y.; Sukhai, M.A.; Minden, M.D.; Schimmer, A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis 2015, 20, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine 2016, 14, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- de Molina, A.R.; Gallego-Ortega, D.; Sarmentero-Estrada, J.; Lagares, D.; Del Pulgar, T.G.; Bandrés, E.; García-Foncillas, J.; Lacal, J.C. Choline kinase as a link connecting phospholipid metabolism and cell cycle regulation: Implications in cancer therapy. Int. J. Biochem. Cell Biol. 2008, 40, 1753–1763. [Google Scholar] [CrossRef]
- Lo Presti, C.; Fauvelle, F.; Jacob, M.-C.; Mondet, J.; Mossuz, P. The metabolic reprogramming in acute myeloid leukemia patients depends on their genotype and is a prognostic marker. Blood Adv. 2021, 5, 156–166. [Google Scholar] [CrossRef]
- Xu, M.; Seneviratne, A.K.; Schimmer, A.D. Phospholipid metabolism regulates AML growth and stemness. Aging 2019, 11, 3895–3897. [Google Scholar] [CrossRef] [PubMed]
- Stefanko, A.; Thiede, C.; Ehninger, G.; Simons, K.; Grzybek, M. Lipidomic approach for stratification of acute myeloid leukemia patients. PLoS ONE 2017, 12, e0168781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megías-Vericat, J.E.; Montesinos, P.; Herrero, M.J.; Moscardó, F.; Bosó, V.; Rojas, L.; Martínez-Cuadrón, D.; Rodríguez-Veiga, R.; Sendra, L.; Cervera, J.; et al. Impact of NADPH oxidase functional polymorphisms in acute myeloid leukemia induction chemotherapy. Pharmacogenom. J. 2018, 18, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Julie, M.; Caroline Lo, P.; Catherine, G.; Kristina, S.; Clara, M.; Sylvain, C.; Sophie, P.; Martin, C.; Claude-Eric, B.; Lysiane, M.; et al. Adult patients with de novo acute myeloid leukemia show a functional deregulation of redox balance at diagnosis which is correlated with molecular subtypes and overall survival. Haematologica 2019, 104, e393–e397. [Google Scholar] [CrossRef] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.; Yan, S.; Vestal, C.G. Oxidative stress, bone marrow failure, and genome instability in hematopoietic stem cells. Int. J. Mol. Sci. 2015, 16, 2366–2385. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Logan, T.D.; Hochberg, M.L.; Shelat, S.G.; Yu, X.; Wilding, G.E.; Tan, W.; Kujoth, G.C.; Prolla, T.A.; Selak, M.A.; et al. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 2009, 114, 4045–4053. [Google Scholar] [CrossRef] [Green Version]
- Gattermann, N. From sideroblastic anemia to the role of mitochondrial DNA mutations in myelodysplastic syndromes. Leuk. Res. 2000, 24, 141–151. [Google Scholar] [CrossRef]
- Linnartz, B.; Anglmayer, R.; Zanssen, S. Comprehensive scanning of somatic mitochondrial DNA alterations in acute leukemia developing from myelodysplastic syndromes. Cancer Res. 2004, 64, 1966–1971. [Google Scholar] [CrossRef] [Green Version]
- Wulfert, M.; Küpper, A.C.; Tapprich, C.; Bottomley, S.S.; Bowen, D.; Germing, U.; Haas, R.; Gattermann, N. Analysis of mitochondrial DNA in 104 patients with myelodysplastic syndromes. Exp. Hematol. 2008, 36, 577–586. [Google Scholar] [CrossRef]
- Stevens, B.M.; Khan, N.; D’Alessandro, A.; Nemkov, T.; Winters, A.; Jones, C.L.; Zhang, W.; Pollyea, D.A.; Jordan, C.T. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat. Commun. 2018, 9, 3694. [Google Scholar] [CrossRef] [PubMed]
- Poulaki, A.; Katsila, T.; Stergiou, I.E.; Giannouli, S.; Gόmez-Tamayo, J.C.; Piperaki, E.T.; Kambas, K.; Dimitrakopoulou, A.; Patrinos, G.P.; Tzioufas, A.G.; et al. Bioenergetic Profiling of the Differentiating Human MDS Myeloid Lineage with Low and High Bone Marrow Blast Counts. Cancers 2020, 12, 3520. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting mitochondrial respiration selectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am. J. Cancer Res. 2017, 7, 2395–2405. [Google Scholar]
- Zhong, W.; Yi, Q.; Xu, B.; Li, S.; Wang, T.; Liu, F.; Zhu, B.; Hoffmann, P.R.; Ji, G.; Lei, P.; et al. ORP4L is essential for T-cell acute lymphoblastic leukemia cell survival. Nat. Commun. 2016, 7, 12702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Giessl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Onodera, J.; Ohsumi, Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J. Biol. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.W.; Onodera, J.; Ohsumi, Y. Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS ONE 2011, 6, e17412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Van der Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Gan, B.; Sahin, E.; Jiang, S.; Sanchez-Aguilera, A.; Scott, K.L.; Chin, L.; Williams, D.A.; Kwiatkowski, D.J.; DePinho, R.A. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc. Natl. Acad. Sci. USA 2008, 105, 19384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Kundu, M. Mitophagy in hematopoietic stem cells. Autophagy 2013, 9, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Ko, J.; Park, Y.S.; Park, J.; Hwang, J.; Koh, H.C. Clearance of Damaged Mitochondria Through PINK1 Stabilization by JNK and ERK MAPK Signaling in Chlorpyrifos-Treated Neuroblastoma Cells. Mol. Neurobiol. 2017, 54, 1844–1857. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Tan, Z.; Shi, F.; Tang, M.; Xie, L.; Zhao, L.; Li, Y.; Hu, J.; Zhou, M.; Bode, A.; et al. PGC1α/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. 2019, 110, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Autophagy Aberrations | Leukemia Model | Metabolic Effect | |
|---|---|---|---|
| ATG5 deletion | Myeloproliferation (mouse models) | [46,50] | |
| ATG7 deletion | Myeloproliferation (mouse models) | Accumulation of mitochondria Increased ROS | [45,50] |
| Low transcript levels of ULK1, FIP200, ATG14, ATG7, ATG3, ATG4B, ATG4D | Human AML blasts | [49] | |
| Loss of ATG10, ATG12, GABARAPL2, MAP1LC3B, GABARAP | AML with deletions of 5q, 16q, 17p | [46] | |
| AMPK deletion (downregulation of autophagy) | MLL-AF9-induced murine AML model | Increased oxidative stress | [56] |
| FIS deletion (mitophagy attenuation) | Human LSCs | Reduced mitochondrial activity | [57] |
| Loss of p62 (mitophagy impairment) | Murine myeloid leukemia | Increased ROS | [58] |
| AKT/mTORC1 activation (downregulation of autophagy) | AML cell lines | Glycolysis enhancement | [138] |
| Downregulation of ATF4 (autophagy inhibition) | FLT3-ITD-mutated AML cell lines | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stergiou, I.E.; Kapsogeorgou, E.K. Autophagy and Metabolism in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2021, 22, 8540. https://doi.org/10.3390/ijms22168540
Stergiou IE, Kapsogeorgou EK. Autophagy and Metabolism in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences. 2021; 22(16):8540. https://doi.org/10.3390/ijms22168540
Chicago/Turabian StyleStergiou, Ioanna E., and Efstathia K. Kapsogeorgou. 2021. "Autophagy and Metabolism in Normal and Malignant Hematopoiesis" International Journal of Molecular Sciences 22, no. 16: 8540. https://doi.org/10.3390/ijms22168540
APA StyleStergiou, I. E., & Kapsogeorgou, E. K. (2021). Autophagy and Metabolism in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences, 22(16), 8540. https://doi.org/10.3390/ijms22168540