
Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease

 , and
, and
Abstract
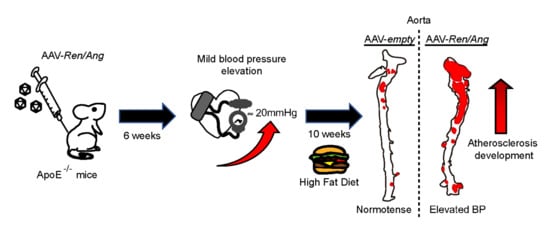
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Generation of a Model of Long-Term Elevated BP Using AAV-Ren/Ang Vectors
2.2. Elevation of BP Exacerbates Atherosclerosis in ApoE−/− Mice Fed a High-Fat Diet (HFD)
2.3. The Mechanical Effect of a Mild elevation in BP Induces Atherosclerosis Independently of the Activation of the RAAS Pathway
3. Discussion
3.1. Study limitations
3.2. Conclusions
4. Materials and Methods
4.1. Animal Experimental Design
4.2. Adeno-Associated Virus (AAV) Vector Production and Purification
4.3. Determination of AAV Vector Titter
4.4. Quantitative Real-Time PCR
4.5. Serum Analysis
4.6. Blood Pressure (BP) Measurements in Mice
4.7. Analysis of Atherosclerotic Lesions in ApoE-Deficient Mice
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kearney, P.M.; Whelton, M.; Reynolds, K.; Muntner, P.; Whelton, P.K.; He, J. Global burden of hypertension: Analysis of worldwide data. Lancet 2005, 365, 217–223. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, e127–e248. [Google Scholar]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Lewington, S.; Clarke, R.; Qizilbash, N.; Peto, R.; Collins, R.; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: A meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002, 360, 1903–1913. [Google Scholar]
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chang, Y.; Kim, S.; Sung, K.C.; Shin, H.; Ryu, S. Increased burden of coronary artery calcium from elevated blood pressure in low-risk young adults. Atherosclerosis 2019, 282, 188–195. [Google Scholar] [CrossRef]
- Whelton, S.P.; McEvoy, J.W.; Shaw, L.; Psaty, B.M.; Lima, J.A.C.; Budoff, M.; Nasir, K.; Szklo, M.; Blumenthal, R.S.; Blaha, M.J. Association of Normal Systolic Blood Pressure Level with Cardiovascular Disease in the Absence of Risk Factors. JAMA Cardiol. 2020, 5, 1011–1018. [Google Scholar] [CrossRef]
- Tocci, G.; Presta, V.; Figliuzzi, I.; Miceli, F.; Citoni, B.; Coluccia, R.; Paini, A.; Salvetti, M.; Ferrucci, A.; Muiesan, M.L.; et al. Reclassification of Hypertensive Outpatients According to New US Guidelines on High Blood Pressure. Am. J. Hypertens 2019, 32, 77–87. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Ruiz-Hurtado, G.; Barderas, M.G.; de la Cruz, J.J.; Lucia, A.; de la Sierra, A.; Gorostidi, M.; Vinyoles, E.; Segura, J.; Solis, J.; et al. Frequency and Prognosis of Treated Hypertensive Patients According to Prior and New Blood Pressure Goals. Hypertension 2019, 74, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.A.; Xu, J.; Peterson, L.E.; Arnold, R.M.; Diamond, J.A.; Schussheim, A.E. Impact of Cardiovascular Risk on the Relative Benefit and Harm of Intensive Treatment of Hypertension. J. Am. Coll. Cardiol. 2018, 71, 1601–1610. [Google Scholar] [CrossRef]
- Leong, X.F.; Ng, C.Y.; Jaarin, K. Animal Models in Cardiovascular Research: Hypertension and Atherosclerosis. Biomed. Res. Int. 2015, 2015, 528757. [Google Scholar] [CrossRef]
- Lerman, L.O.; Kurtz, T.W.; Touyz, R.M.; Ellison, D.H.; Chade, A.R.; Crowley, S.D.; Mattson, D.L.; Mullins, J.J.; Osborn, J.; Eirin, A.; et al. Animal Models of Hypertension: A Scientific Statement from the American Heart Association. Hypertension 2019, 73, e87–e120. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Roche-Molina, M.; Sanz-Rosa, D.; Cruz, F.M.; Garcia-Prieto, J.; Lopez, S.; Abia, R.; Muriana, F.J.; Fuster, V.; Ibanez, B.; Bernal, J.A. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 50–59. [Google Scholar] [CrossRef]
- Agabiti-Rosei, E.; Muiesan, M.L.; Rizzoni, D.; Zulli, R.; Calebich, S.; Castellano, M.; Cavaliere, G.; Nordio, G.; Pasini, F.L.; Zuccato, F. Cardiovascular structural changes and calcium antagonist therapy in patients with hypertension. J. Cardiovasc. Pharmacol. 1994, 24, S37–S43. [Google Scholar]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 2000, 105, 1605–1612. [Google Scholar] [CrossRef]
- Rateri, D.L.; Howatt, D.A.; Moorleghen, J.J.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Prolonged infusion of angiotensin II in apoE(-/-) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol. 2011, 179, 1542–1548. [Google Scholar] [CrossRef]
- Huang, B.S.; Ahmadi, S.; Ahmad, M.; White, R.A.; Leenen, F.H. Central neuronal activation and pressor responses induced by circulating ANG II: Role of the brain aldosterone-“ouabain” pathway. Am. J. Physiol. Heart. Circ. Physiol. 2010, 299, H422–H430. [Google Scholar] [CrossRef][Green Version]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [CrossRef]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Pizarro, G.; Jimenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- Hauswirth, W.W.; Lewin, A.S.; Zolotukhin, S.; Muzyczka, N. Production and purification of recombinant adeno-associated virus. Methods Enzymol. 2000, 316, 743–761. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Guerra, A.; Roche-Molina, M.; García-Quintáns, N.; Sánchez-Ramos, C.; Martín-Pérez, D.; Lytvyn, M.; de Nicolás-Hernández, J.; Rivera-Torres, J.; Arroyo, D.F.; Sanz-Rosa, D.; et al. Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease. Int. J. Mol. Sci. 2021, 22, 8448. https://doi.org/10.3390/ijms22168448
Gonzalez-Guerra A, Roche-Molina M, García-Quintáns N, Sánchez-Ramos C, Martín-Pérez D, Lytvyn M, de Nicolás-Hernández J, Rivera-Torres J, Arroyo DF, Sanz-Rosa D, et al. Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease. International Journal of Molecular Sciences. 2021; 22(16):8448. https://doi.org/10.3390/ijms22168448
Chicago/Turabian StyleGonzalez-Guerra, Andrés, Marta Roche-Molina, Nieves García-Quintáns, Cristina Sánchez-Ramos, Daniel Martín-Pérez, Mariya Lytvyn, Javier de Nicolás-Hernández, José Rivera-Torres, Diego F. Arroyo, David Sanz-Rosa, and et al. 2021. "Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease" International Journal of Molecular Sciences 22, no. 16: 8448. https://doi.org/10.3390/ijms22168448
APA StyleGonzalez-Guerra, A., Roche-Molina, M., García-Quintáns, N., Sánchez-Ramos, C., Martín-Pérez, D., Lytvyn, M., de Nicolás-Hernández, J., Rivera-Torres, J., Arroyo, D. F., Sanz-Rosa, D., & Bernal, J. A. (2021). Sustained Elevated Blood Pressure Accelerates Atherosclerosis Development in a Preclinical Model of Disease. International Journal of Molecular Sciences, 22(16), 8448. https://doi.org/10.3390/ijms22168448