
Novel Aptamer-Based Small-Molecule Drug Screening Assay to Identify Potential Sclerostin Inhibitors against Osteoporosis


 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Construction of the Chemical Library for Aptamer-Based Drug Screening
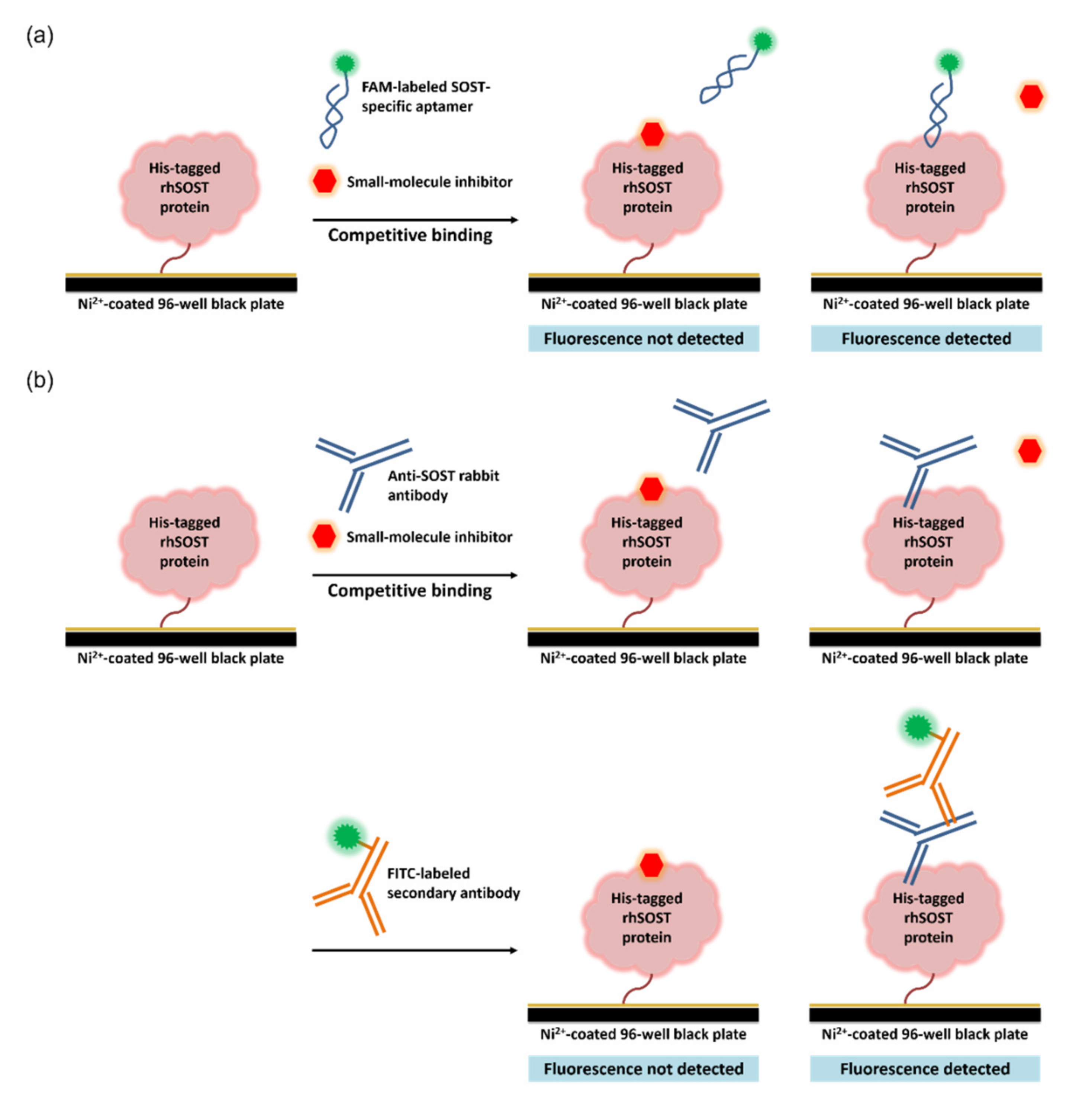
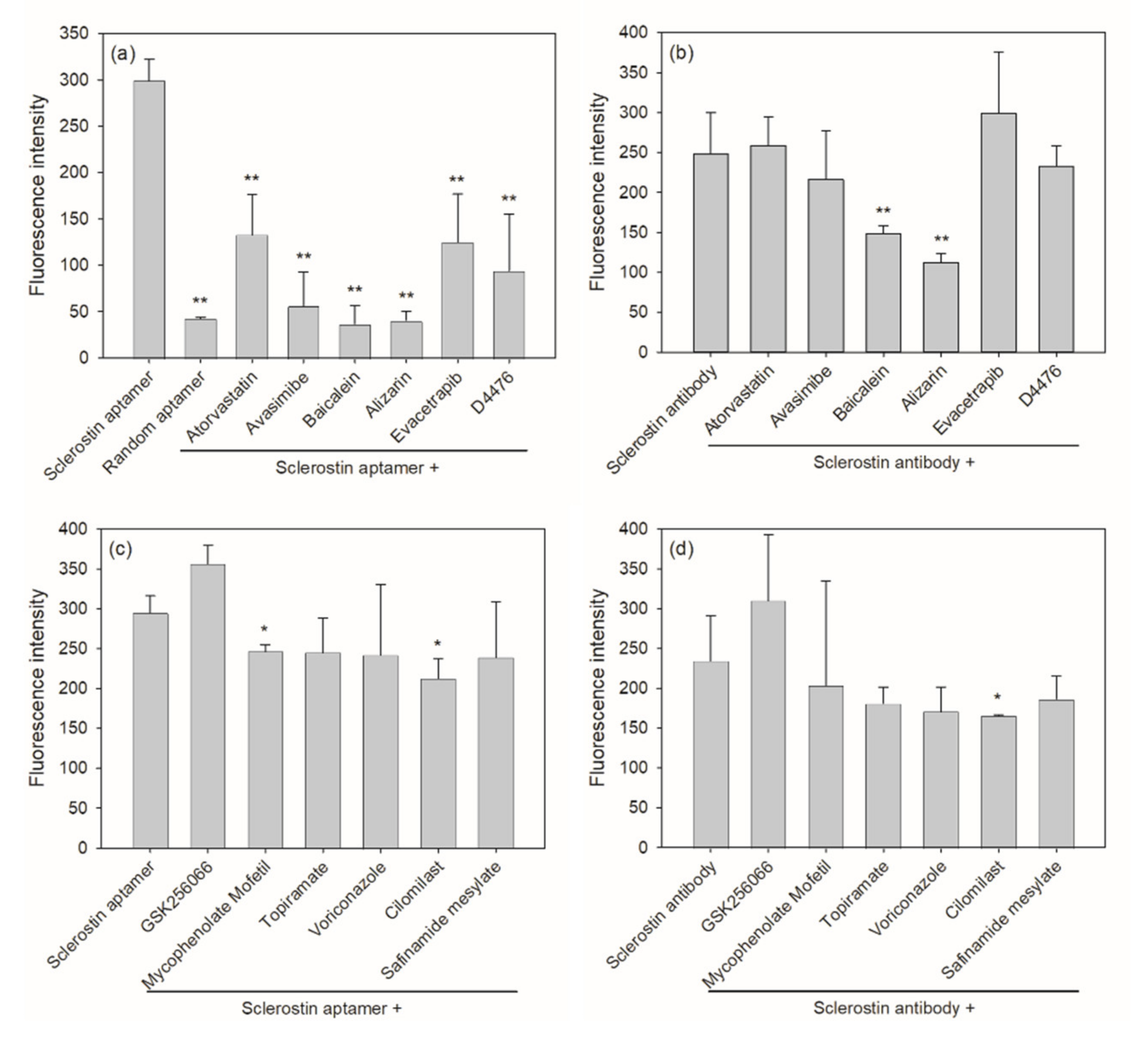
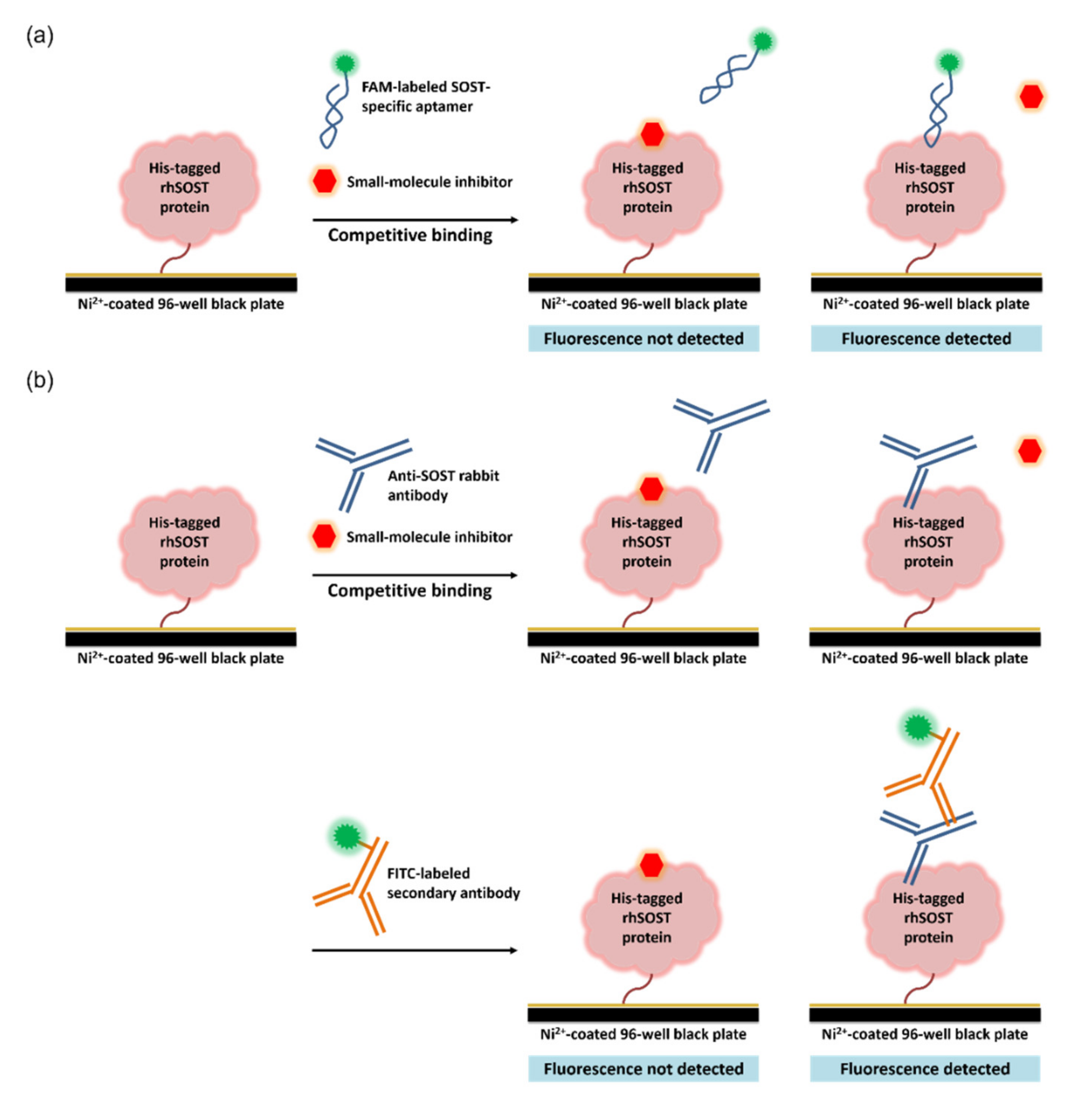
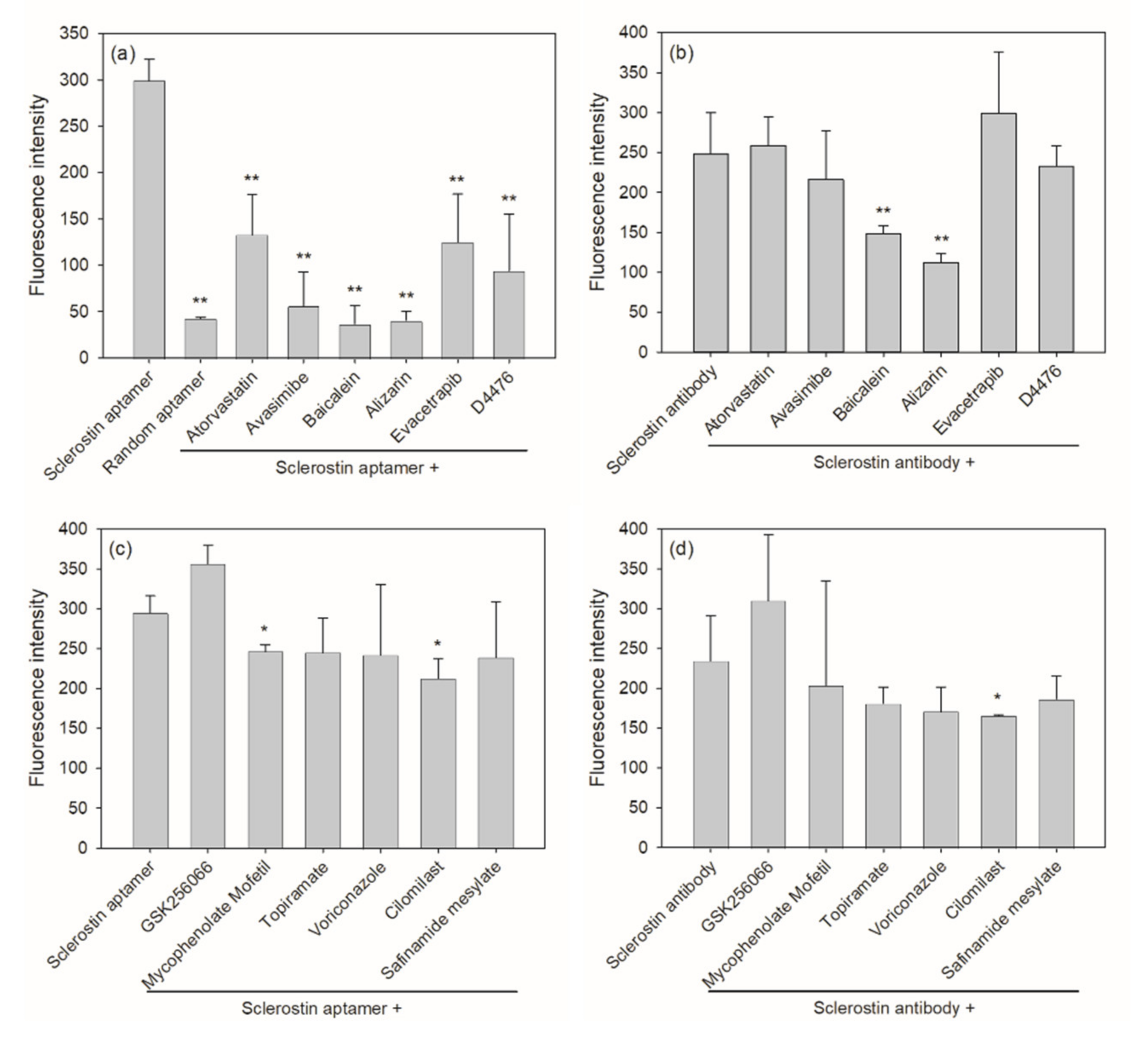
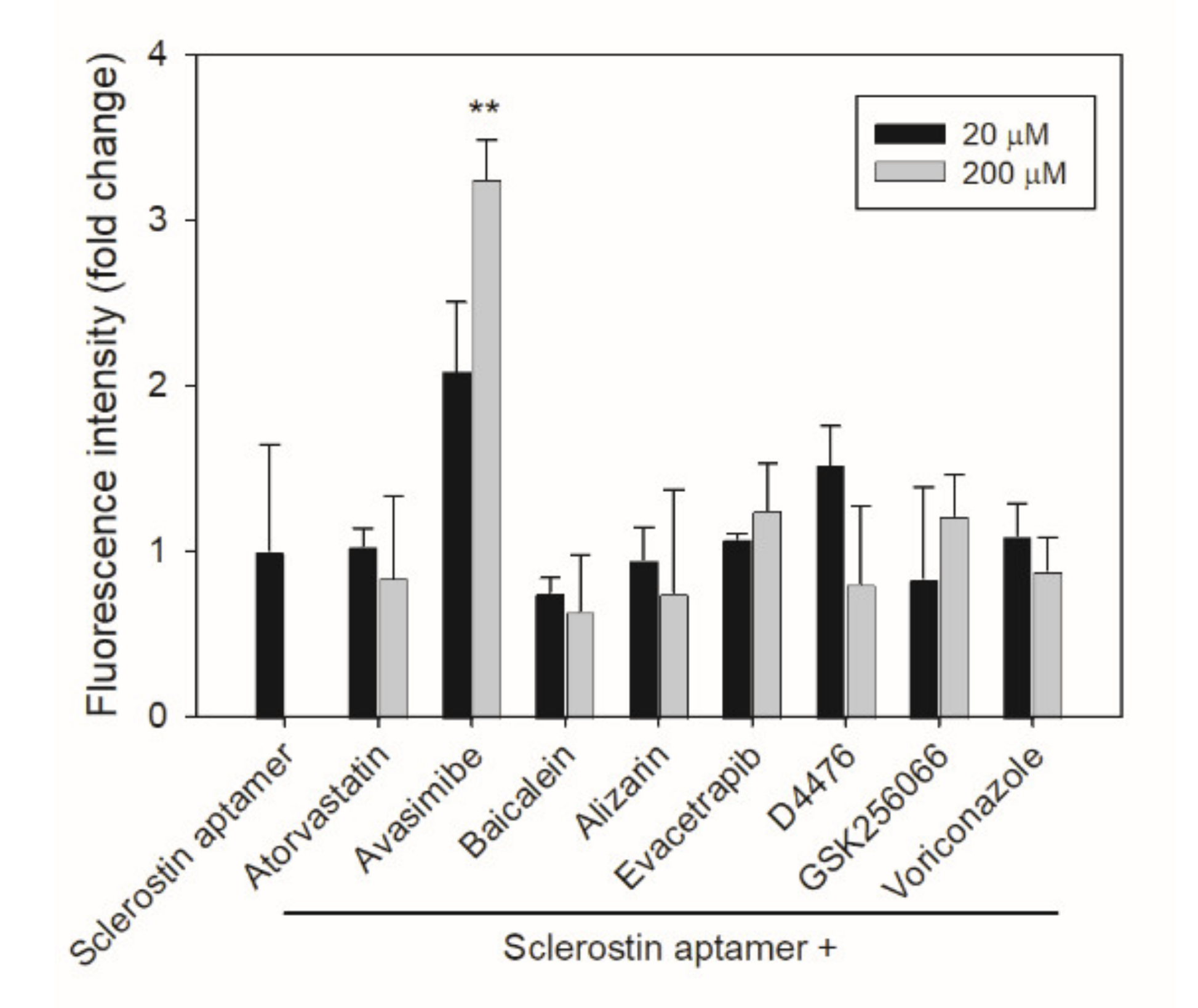
2.2. Aptamer-Based Competitive Drug Screening Assay
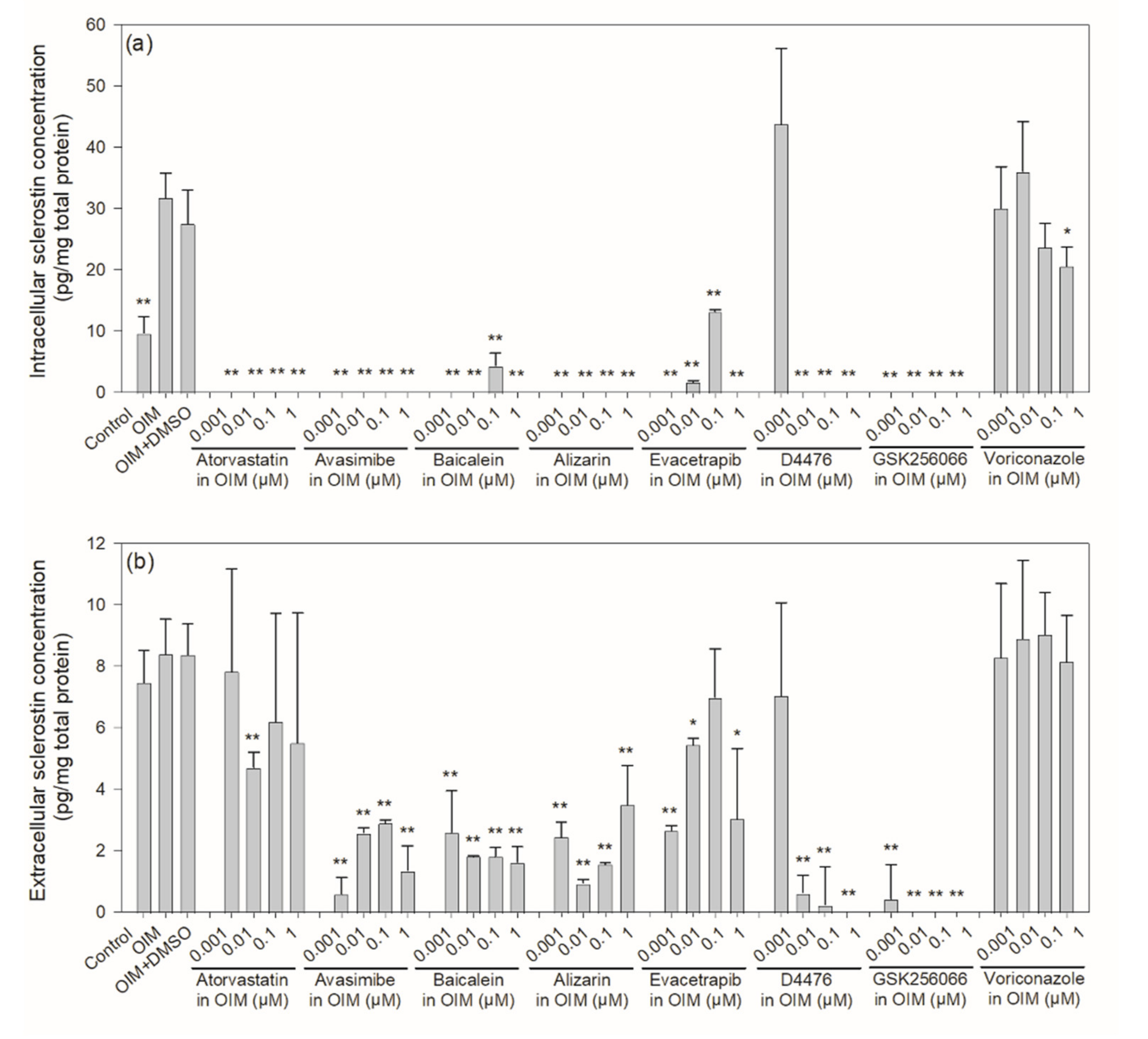
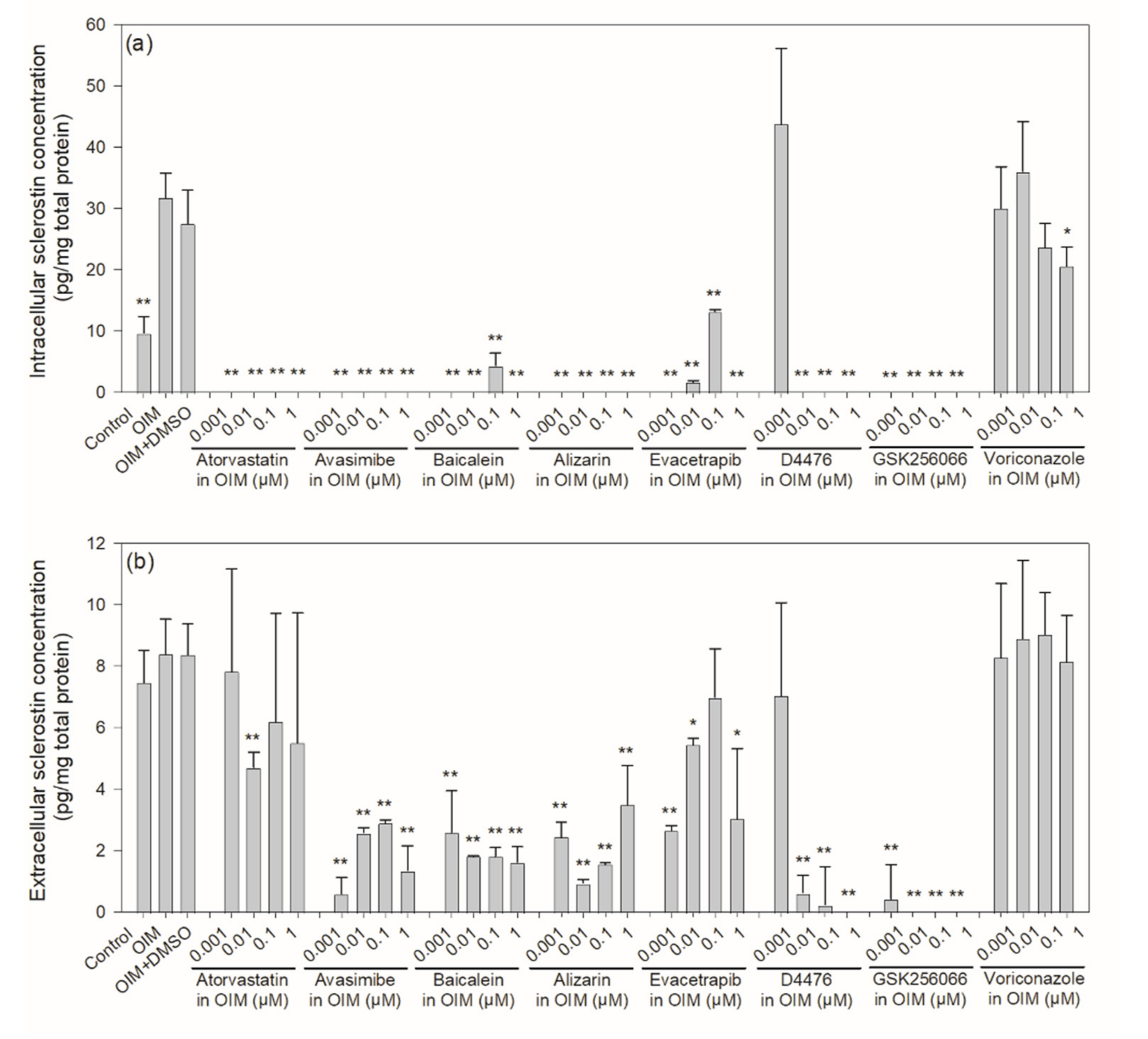
2.3. Intracellular and Extracellular Levels of Sclerostin in Mouse IDG-SW3 Cells
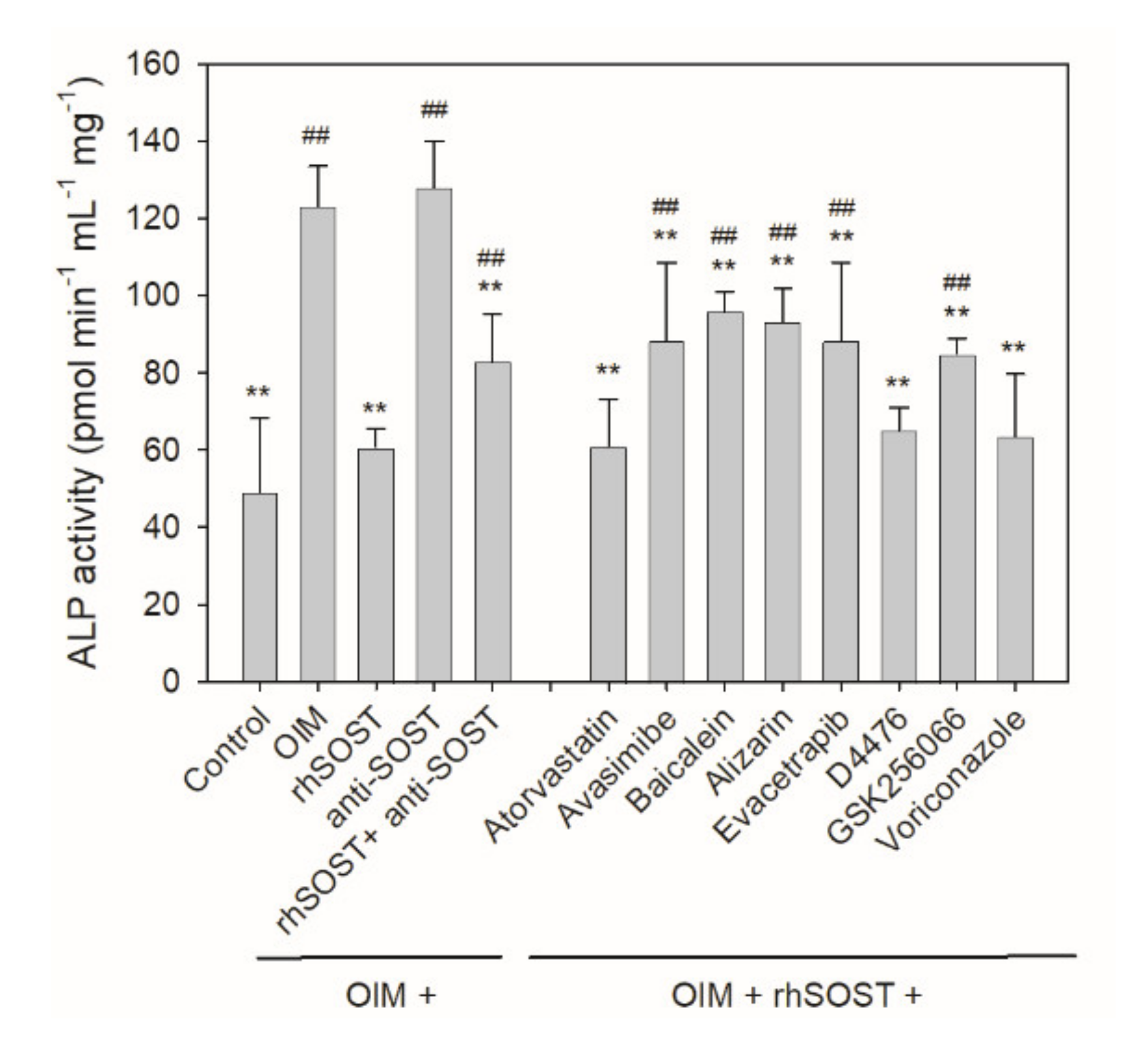
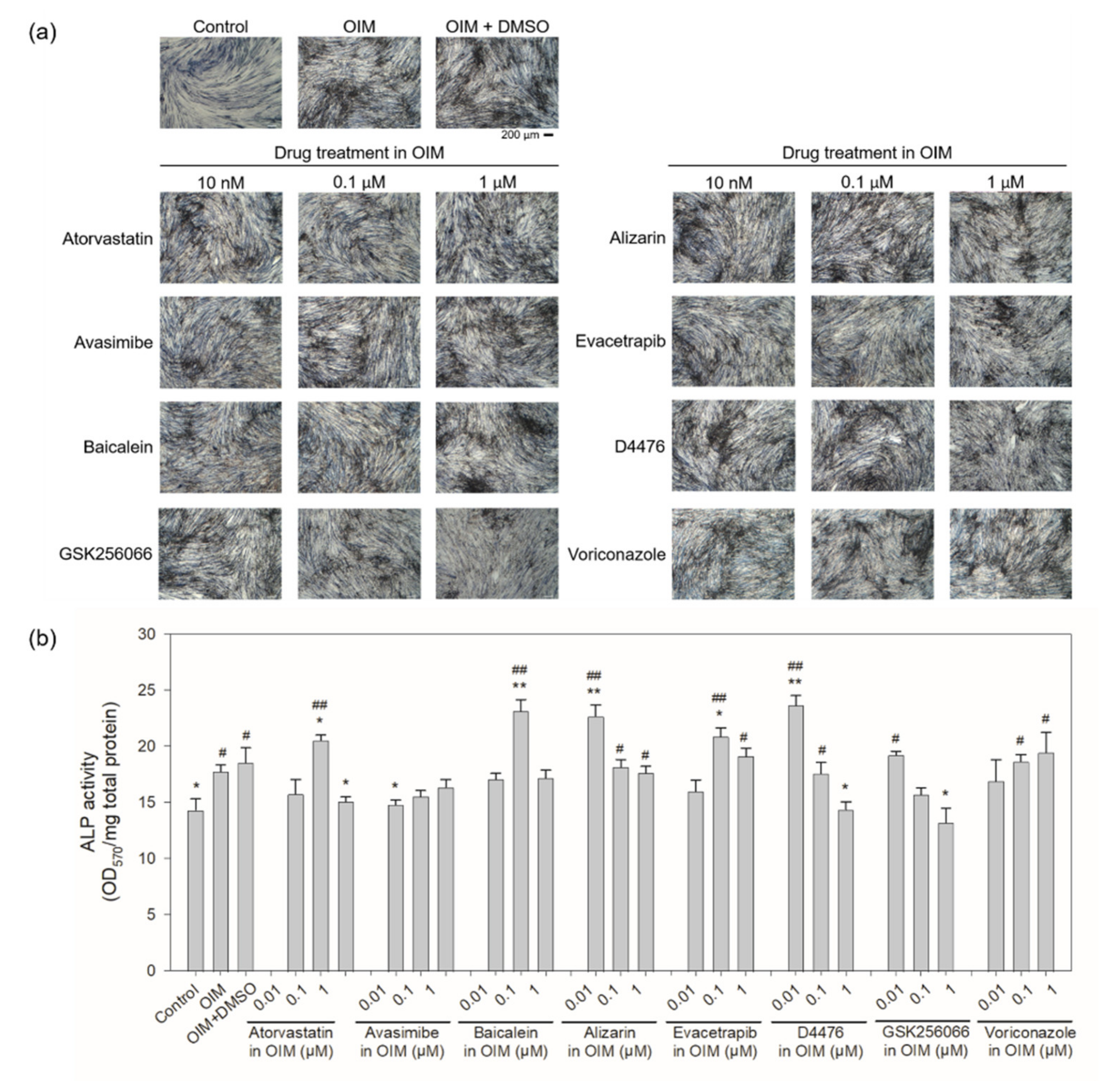
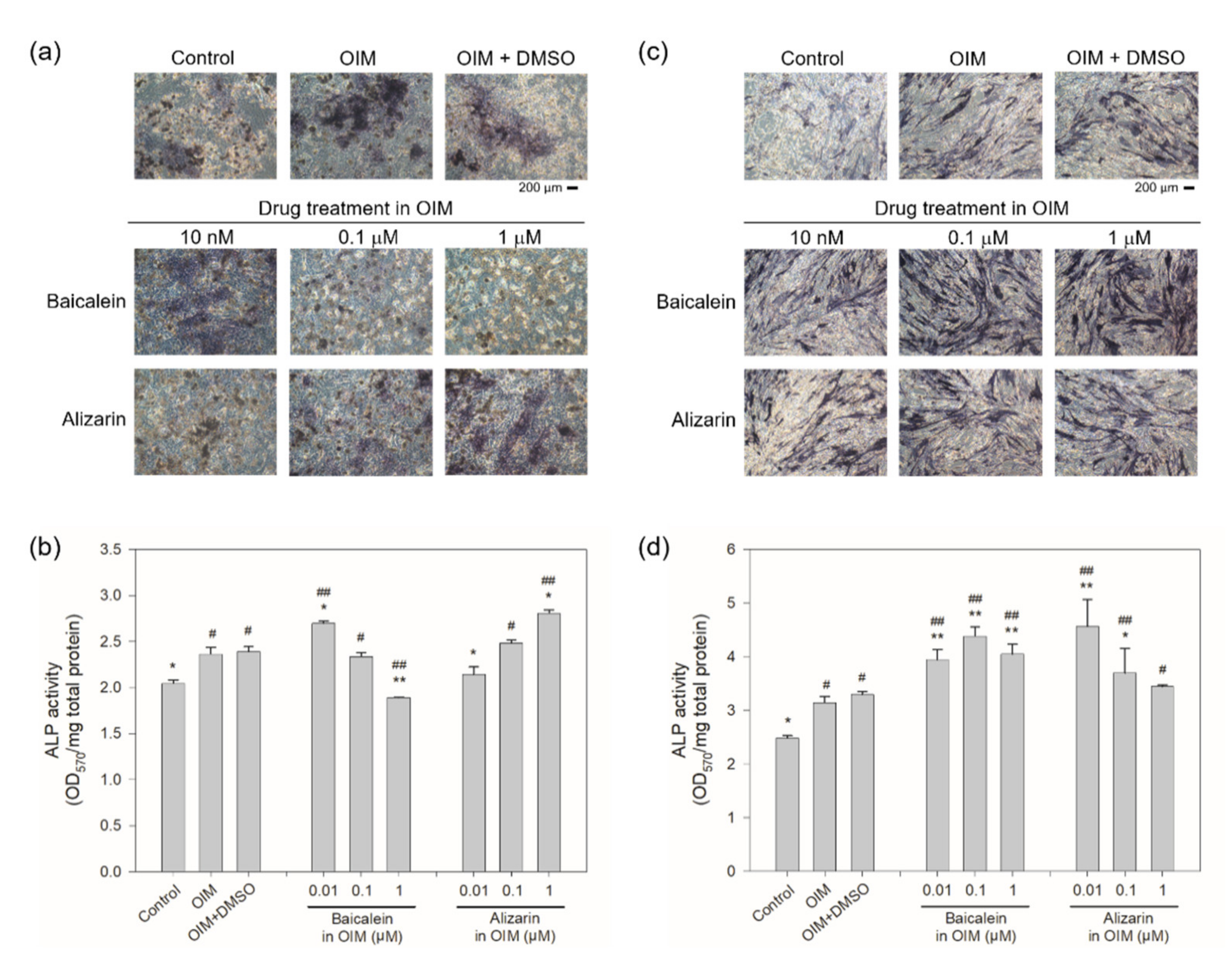
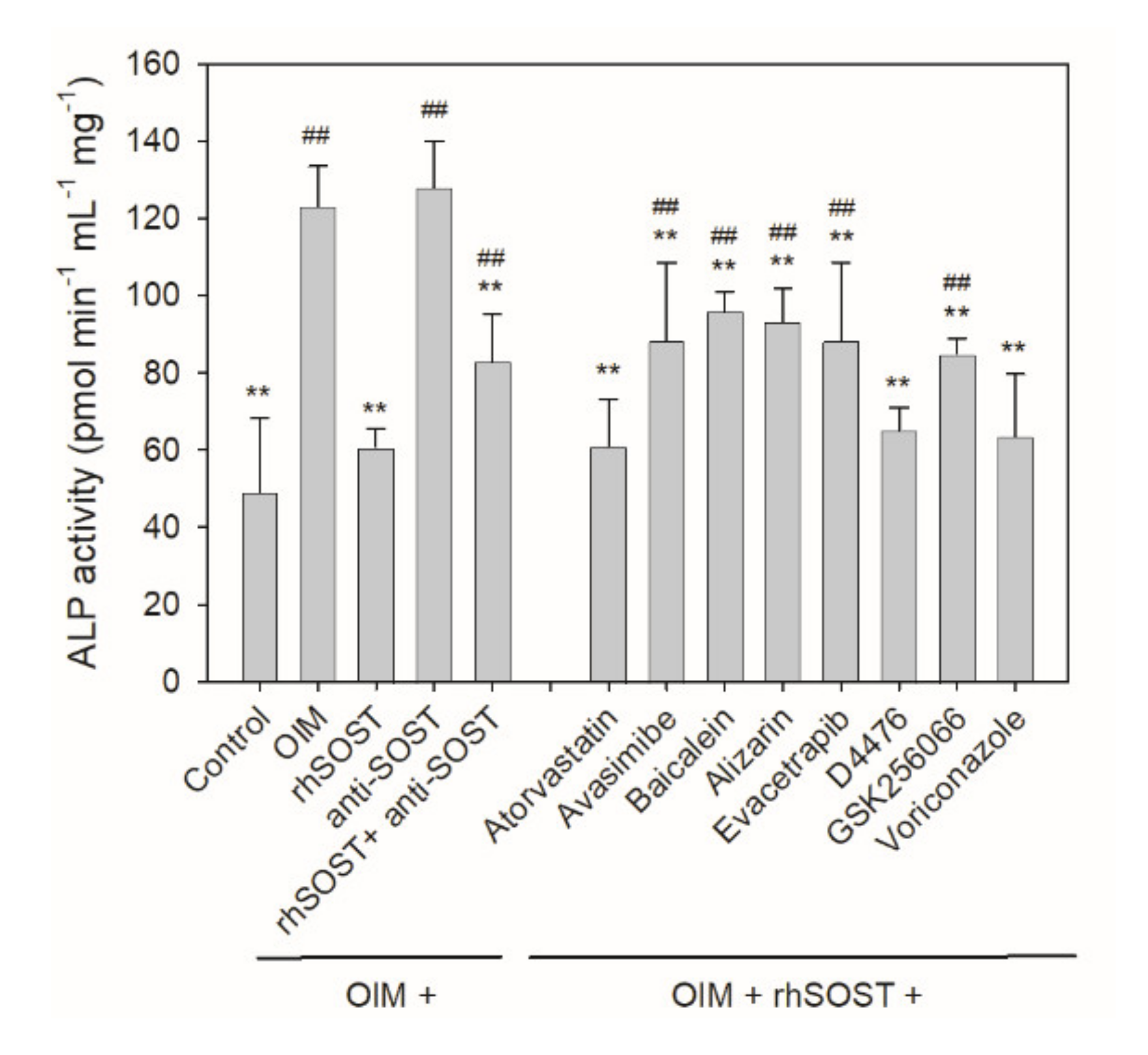
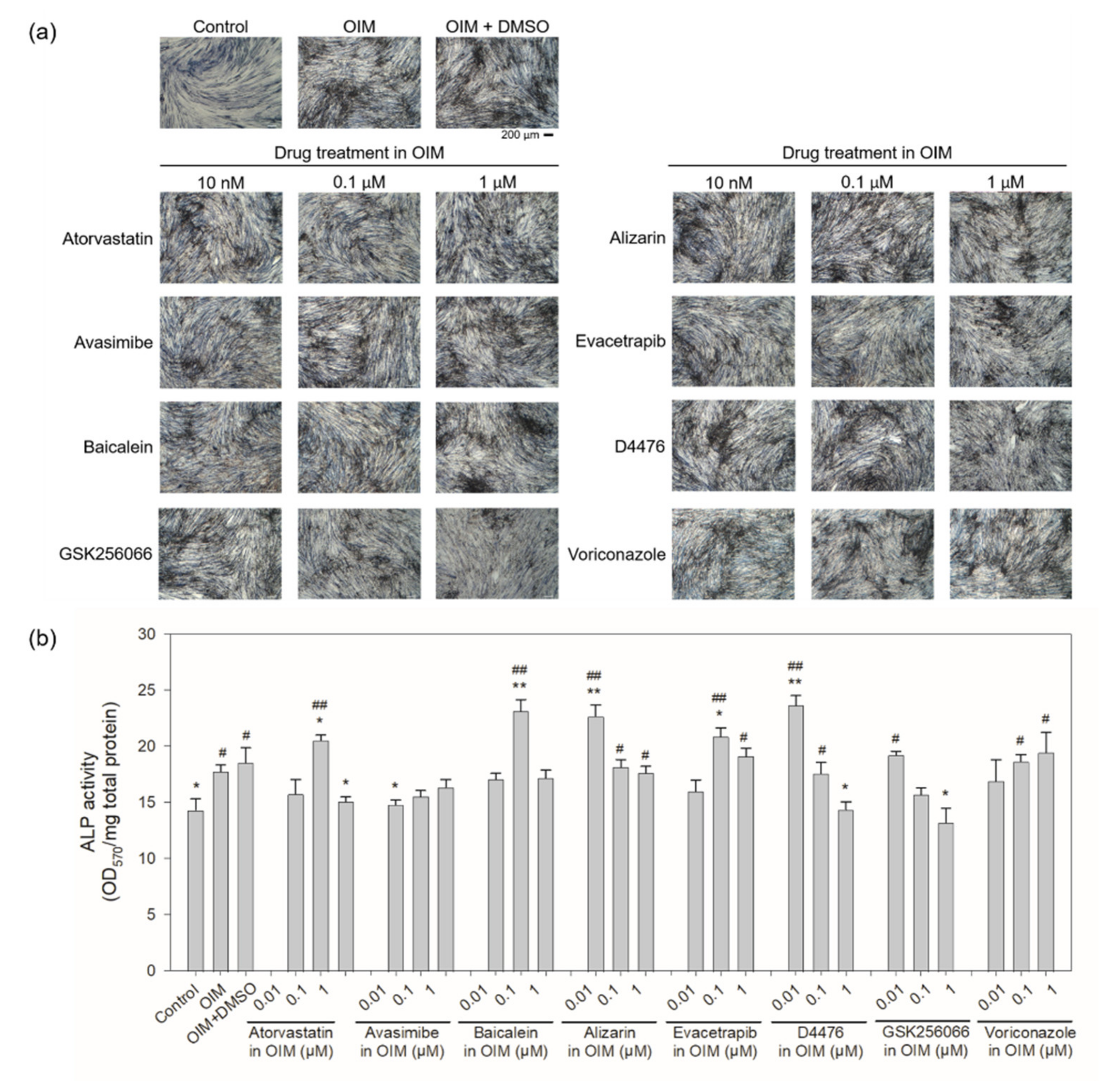
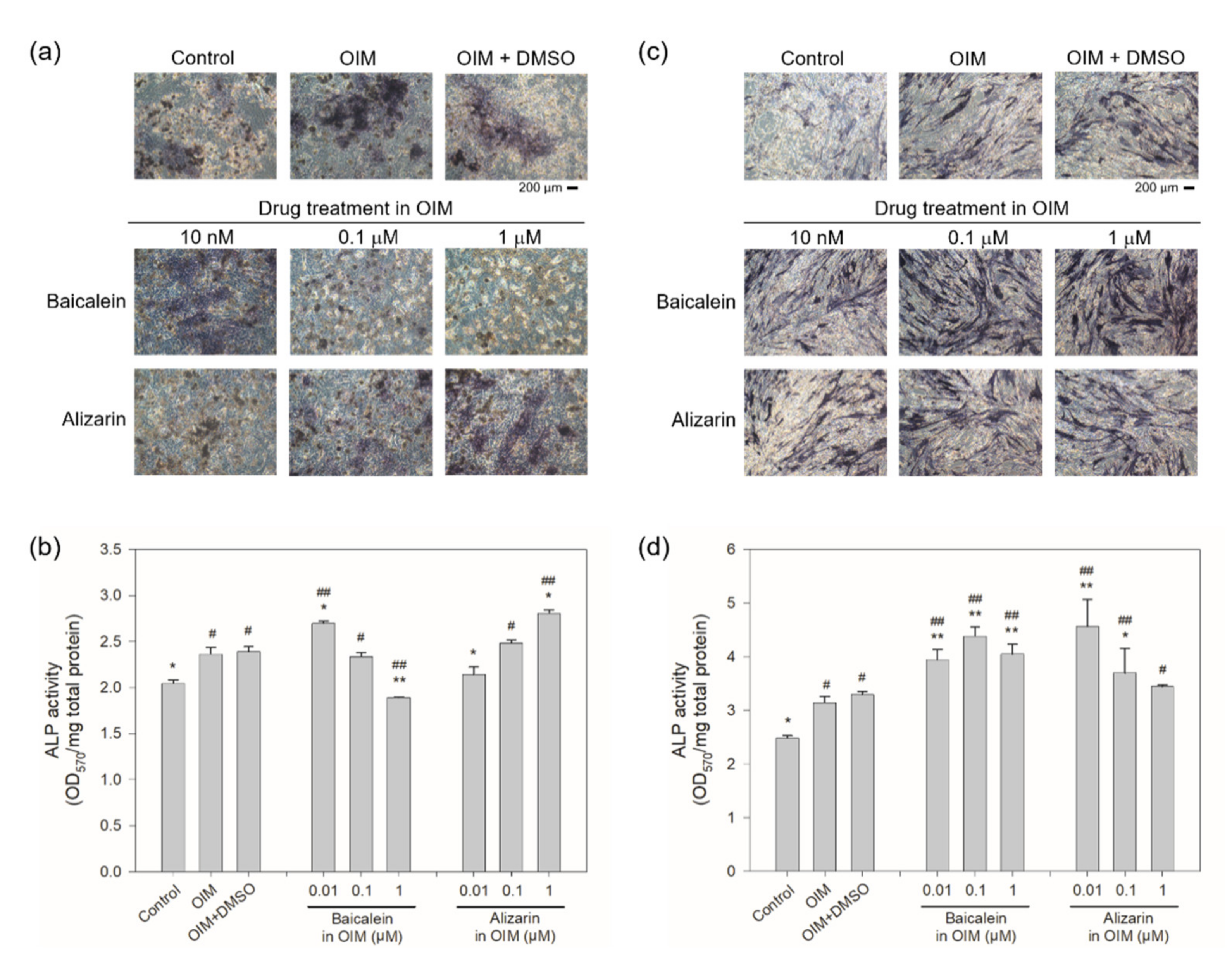
2.4. Cellular Rescue Assay and Osteogenic Activity of Potential Sclerostin-Binding Inhibitors
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Aptamer-Based Competitive Drug Screening Assay
4.3. Antibody-Based Competitive Drug Screening Assay
4.4. IDG-SW3 Cell Culture
4.5. hBMSC Culture
4.6. hFOB1.19 Cell Culture
4.7. SOST ELISA
4.8. Cellular Rescue Assay
4.9. Alkaline Phosphatase Activity Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estell, E.G.; Rosen, C.J. Emerging insights into the comparative effectiveness of anabolic therapies for osteoporosis. Nat. Rev. Endocrinol. 2021, 17, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Neer, R.M.; Arnaud, C.D.; Zanchetta, J.R.; Prince, R.; Gaich, G.A.; Reginster, J.-Y.; Hodsman, A.B.; Eriksen, E.F.; Ish-Shalom, S.; Genant, H.K.; et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. New Engl. J. Med. 2001, 344, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Vahle, J.L.; Long, G.G.; Sandusky, G.; Westmore, M.; Ma, Y.L.; Sato, M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1-34)] are dependent on duration of treatment and dose. Toxicol. Pathol. 2004, 32, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J. Biol. Chem. 2015, 290, 16744–16758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suen, P.K.; Ling, Q. Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: A general review. J. Orthop. Translat. 2016, 4, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.-S.; Choi, S.-H.; Choi, E.H.; Kim, K.-M.; Chu, P.K. Enhanced osteogenic differentiation of human mesenchymal stem cells on amine-functionalized titanium using humidified ammonia supplied nonthermal atmospheric pressure plasma. Int. J. Mol. Sci. 2020, 21, 6085. [Google Scholar] [CrossRef]
- Clarke, B.L. Anti-sclerostin antibodies: Utility in treatment of osteoporosis. Maturitas 2014, 78, 199–204. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [Green Version]
- Atkins, G.J.; Rowe, P.S.; Lim, H.P.; Welldon, K.J.; Ormsby, R.; Wijenayaka, A.R.; Zelenchuk, L.; Evdokiou, A.; Findlay, D.M. Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J. Bone Miner. Res. 2011, 26, 1425–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Turcott, E.; Skonier, J.E.; Winkler, D.G.; Latham, J.A. Sclerostin promotes the apoptosis of human osteoblastic cells: A novel regulation of bone formation. Bone 2004, 35, 828–835. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Kramer, I.; Loots, G.G.; Studer, A.; Keller, H.; Kneissel, M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J. Bone Miner. Res. 2010, 25, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.; Scott, L.J. Romosozumab: A review in postmenopausal osteoporosis. Drug Aging 2020, 37, 845–855. [Google Scholar] [CrossRef]
- Kerschan-Schindl, K. Romosozumab: A novel bone anabolic treatment option for osteoporosis? Wien. Med. Wochenschr. 2020, 170, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab treatment in postmenopausal women with osteoporosis. New Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. New Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fixen, C.; Tunoa, J. Romosozumab: A review of efficacy, safety, and cardiovascular risk. Curr. Osteoporos. Rep. 2021. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, P.; Girod, A.; Blind, M. Aptamers as tools for target prioritization and lead identification. Drug Discov. Today 2002, 7, 1221–1228. [Google Scholar] [CrossRef]
- Green, L.S.; Bell, C.; Janjic, N. Aptamers as reagents for high-throughput screening. BioTechniques 2001, 30, 1094–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartig, J.S.; Najafi-Shoushtari, S.H.; Grune, I.; Yan, A.; Ellington, A.D.; Famulok, M. Protein-dependent ribozymes report molecular interactions in real time. Nat. Biotechnol. 2002, 20, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Proske, D.; Blank, M.; Buhmann, R.; Resch, A. Aptamers--basic research, drug development, and clinical applications. Appl. Microbiol. Biotechnol. 2005, 69, 367–374. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Benoit, D.S.W.; Schwartz, M.P.; Durney, A.R.; Anseth, K.S. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat. Mater. 2008, 7, 816–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.E.J.; Kawazoe, N.; Chen, G. Gold nanoparticles with different charge and moiety induce differential cell response on mesenchymal stem cell osteogenesis. Biomaterials 2015, 54, 226–236. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, N.; Shi, H.; Liu, J.; Shi, L.; Zhang, B.; Wang, H.; Ji, J.; Chu, P.K. Upregulation of BMSCs osteogenesis by positively-charged tertiary amines on polymeric implants via charge/iNOS signaling pathway. Sci. Rep. 2015, 5, 9369. [Google Scholar] [CrossRef] [Green Version]
- Shum, K.T.; Chan, C.; Leung, C.M.; Tanner, J.A. Identification of a DNA aptamer that inhibits sclerostin’s antagonistic effect on Wnt signalling. Biochem. J. 2011, 434, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.M.; Rosser, J.; Dusevich, V.; Kalajzic, I.; Bonewald, L.F. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J. Bone Miner. Res. 2011, 26, 2634–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, N.R.; Williams, K.M.; Joca, H.C.; Torre, O.M.; Lyons, J.S.; Leser, J.M.; Srikanth, M.P.; Hughes, M.; Khairallah, R.J.; Feldman, R.A.; et al. Disparate bone anabolic cues activate bone formation by regulating the rapid lysosomal degradation of sclerostin protein. eLife 2021, 10, e64393. [Google Scholar] [CrossRef]
- Veverka, V.; Henry, A.J.; Slocombe, P.M.; Ventom, A.; Mulloy, B.; Muskett, F.W.; Muzylak, M.; Greenslade, K.; Moore, A.; Zhang, L.; et al. Characterization of the structural features and interactions of sclerostin. J. Biol. Chem. 2009, 284, 10890–10900. [Google Scholar] [CrossRef] [Green Version]
- Weidauer, S.E.; Schmieder, P.; Beerbaum, M.; Schmitz, W.; Oschkinat, H.; Mueller, T.D. NMR structure of the Wnt modulator protein Sclerostin. Biochem. Biophys. Res. Commun. 2009, 380, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiang, S.; Wei, X.; Yadav, R.I.; Han, M.; Zheng, W.; Zhao, L.; Shi, Y.; Cao, Y. Icariin promotes the osteogenesis of bone marrow mesenchymal stem cells through regulating sclerostin and activating the Wnt/β-catenin signaling pathway. BioMed Res. Int. 2021, 2021, 6666836. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Zhang, W.; Li, X.-L. Effects of SOST gene silencing on proliferation, apoptosis, invasion, and migration of human osteosarcoma cells through the Wnt/β-catenin signaling pathway. Calcif. Tissue Int. 2017, 100, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfield, H.; Rosen, C.J.; Reagan, M.R. Connecting bone and fat: The potential role for sclerostin. Curr. Mol. Biol. Rep. 2017, 3, 114–121. [Google Scholar] [CrossRef]
- Ukita, M.; Yamaguchi, T.; Ohata, N.; Tamura, M. Sclerostin enhances adipocyte differentiation in 3T3-L1 cells. J. Cell. Biochem. 2016, 117, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfield, H.; Falank, C.; Harris, E.; Demambro, V.; McDonald, M.; Pettitt, J.A.; Mohanty, S.T.; Croucher, P.; Kramer, I.; Kneissel, M.; et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J. Cell. Physiol. 2018, 233, 1156–1167. [Google Scholar] [CrossRef]
- Kim, S.P.; Frey, J.L.; Li, Z.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, S.B.; Jain, G.K. Statins and osteoporosis: New role for old drugs. J. Pharm. Pharmacol. 2006, 58, 3–18. [Google Scholar] [CrossRef]
- Hong, W.; Wei, Z.; Qiu, Z.; Li, Z.; Fu, C.; Ye, Z.; Xu, X. Atorvastatin promotes bone formation in aged apoE–/– mice through the Sirt1–Runx2 axis. J. Orthop. Surg. Res. 2020, 15, 303. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, C.Y.; Li, J.; Cheng, J.X.; Jang, M.; Kim, K.H. In vitro exploration of ACAT contributions to lipid droplet formation during adipogenesis. J. Lipid Res. 2018, 59, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Chen, Y.; Huang, Y.P.; Hsu, Y.C.; Chiang, L.H.; Chen, T.Y.; Wang, G.J. Exogenous polyamines promote osteogenic differentiation by reciprocally regulating osteogenic and adipogenic gene expression. J. Cell. Biochem. 2013, 114, 2718–2728. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Hu, B.B.; Shi, X.L.; Ren, M.M.; Yu, W.B.; Cen, S.D.; Hu, R.D.; Deng, H. Baicalein enhances the osteogenic differentiation of human periodontal ligament cells by activating the Wnt/beta-catenin signaling pathway. Arch. Oral Biol. 2017, 78, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saul, D.; Weber, M.; Zimmermann, M.H.; Kosinsky, R.L.; Hoffmann, D.B.; Menger, B.; Taudien, S.; Lehmann, W.; Komrakova, M.; Sehmisch, S. Effect of the lipoxygenase inhibitor baicalein on bone tissue and bone healing in ovariectomized rats. Nutr. Metab. 2019, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yuan, P.W.; Hao, Y.Q.; Lu, Z.M. Emodin enhances osteogenesis and inhibits adipogenesis. BMC Compl. Altern. Med. 2014, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Pengjam, Y.; Madhyastha, H.; Madhyastha, R.; Yamaguchi, Y.; Nakajima, Y.; Maruyama, M. Anthraquinone glycoside aloin induces osteogenic initiation of MC3T3-E1 cells: Involvement of MAPK mediated Wnt and Bmp signaling. Biomol. Ther. 2016, 24, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Armitage, J.; Holmes, M.V.; Preiss, D. Cholesteryl ester transfer protein inhibition for preventing cardiovascular events: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 2004, 23, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | M.W. | CAS Number | Chemical Structure 2 | Target |
|---|---|---|---|---|
| Atorvastatin calcium 1 | 1155.34 | 134523-03-8 | C66H68CaF2N4O10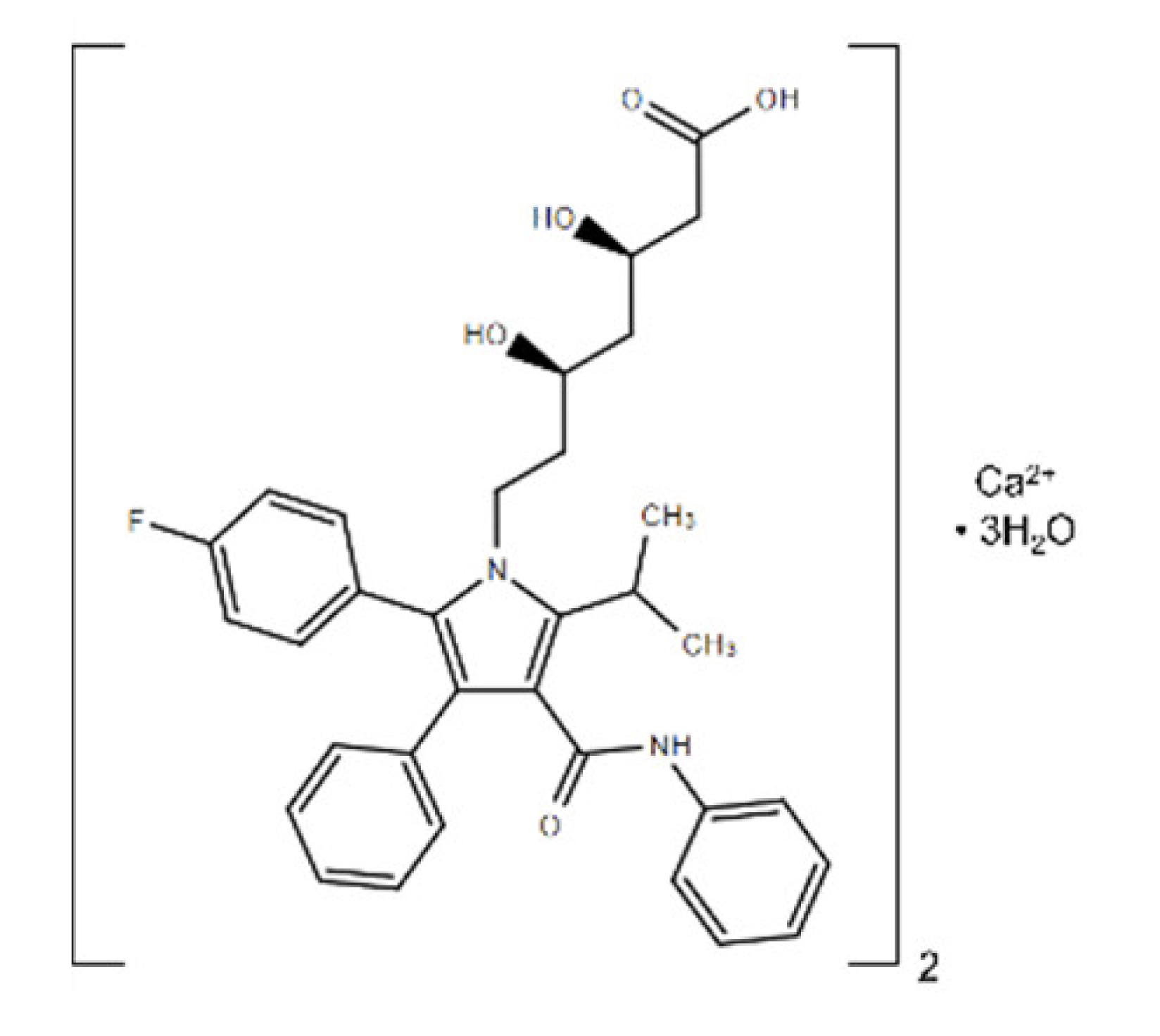 | HMG-CoA reductase |
| Avasimibe | 501.72 | 166518-60-1 | C29H43NO4S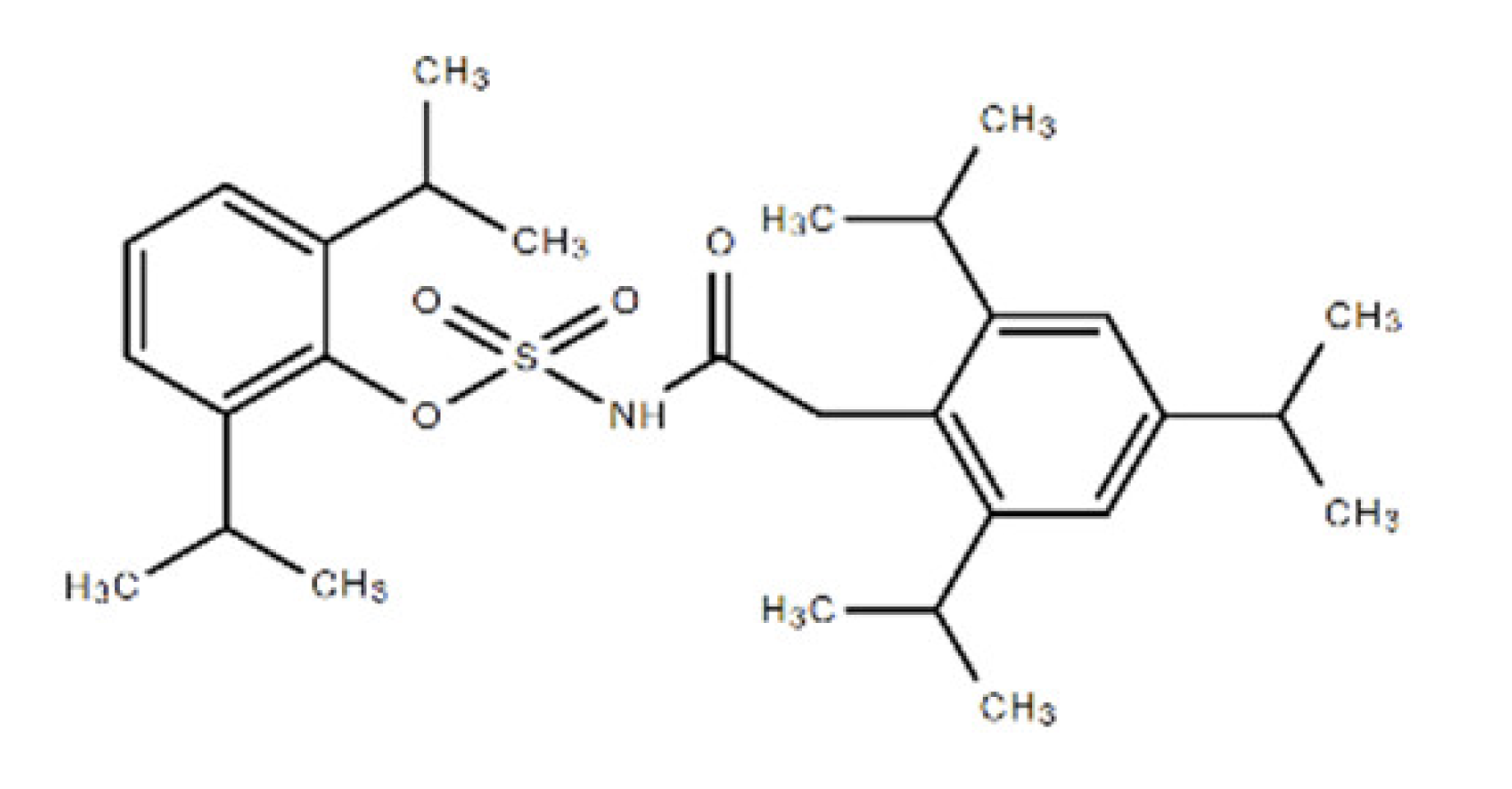 | Cholesterol acyltransferase (ACAT) and P450 isoenzymes CYP2C9, CYP1A2 and CYP2C19 |
| Baicalein | 270.24 | 491-67-8 | C15H10O5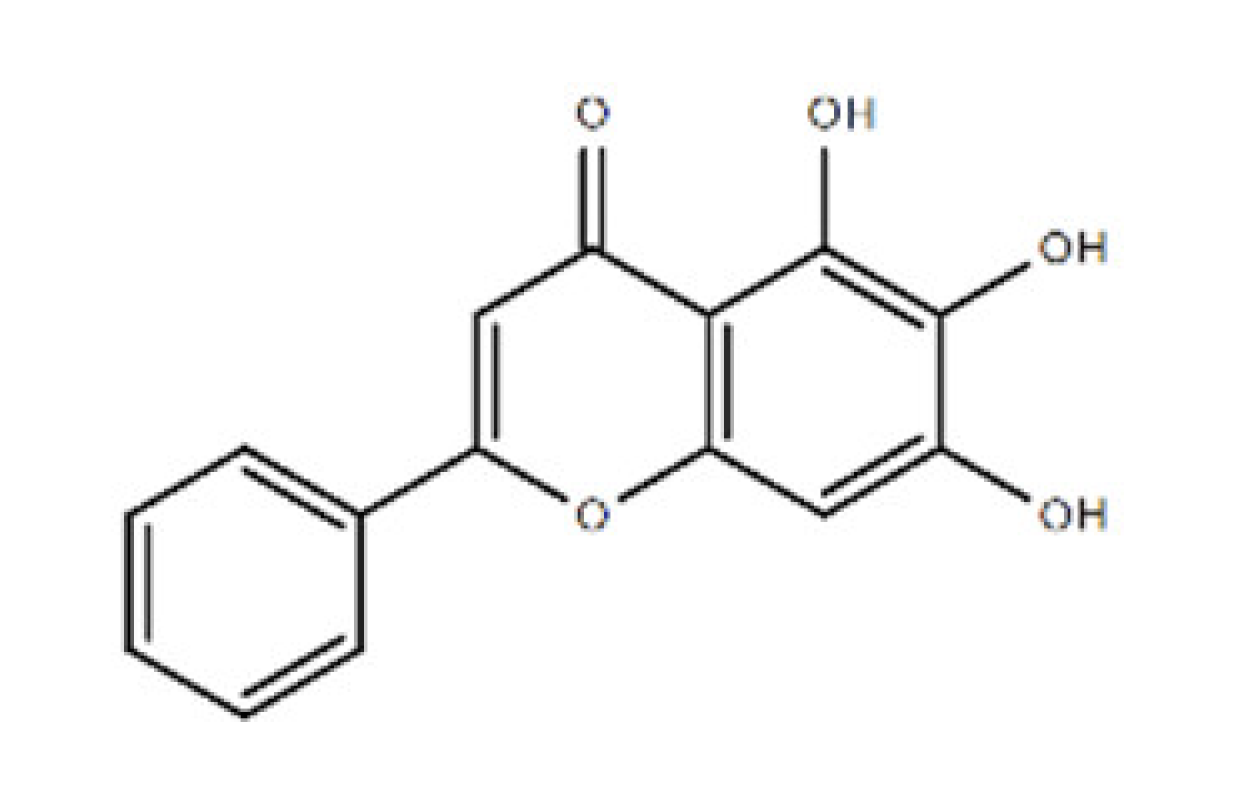 | P450 isozyme CYP2C9 and prolyl endopeptidase |
| Alizarin | 240.21 | 72-48-0 | C14H8O4 | P450 isoforms CYP1A1, CYP1A2 and CYP1B1 |
| Evacetrapib (LY2484595) | 638.65 | 1186486-62-3 | C31H36F6N6O2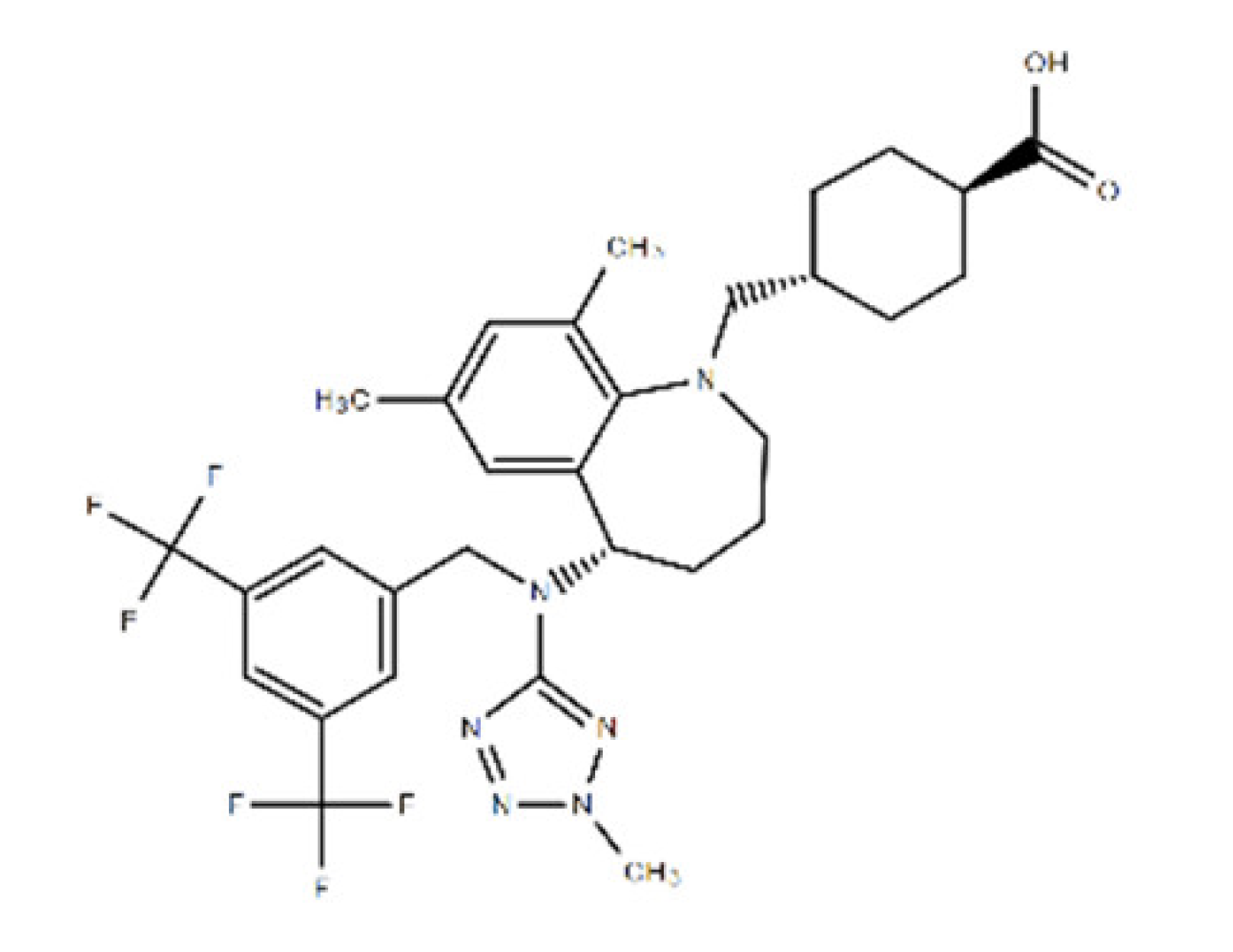 | cholesteryl ester transfer protein (CETP) |
| D4476 | 398.41 | 301836-43-1 | C23H18N4O3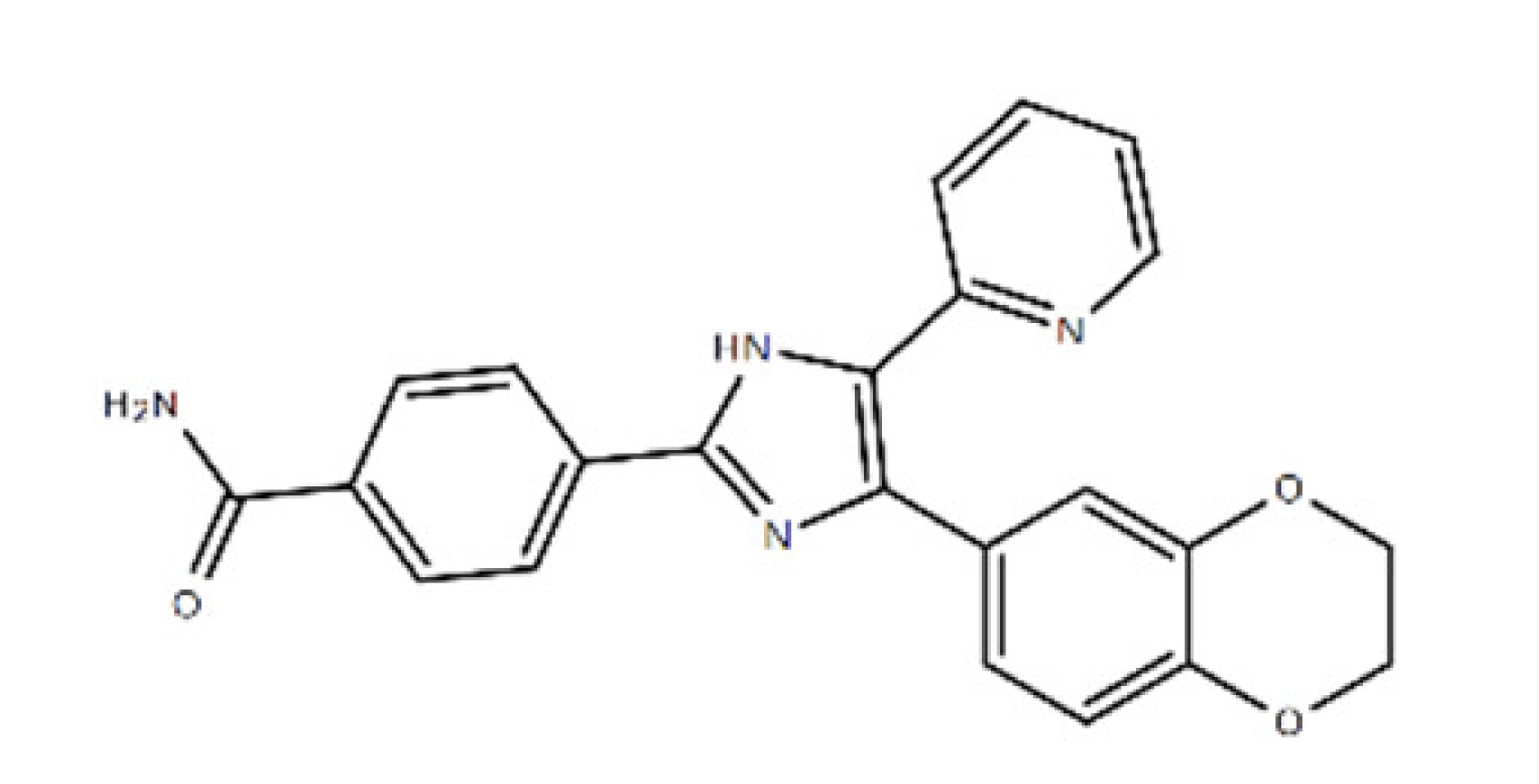 | Casein kinase 1 (CK1) and TGFβ type I receptor kinase (ALK5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-C.; Hung, C.-M.; Chen, C.-H.; Hsu, Y.-C.; Huang, Y.-P.; Huang, T.-B.; Lee, M.-J. Novel Aptamer-Based Small-Molecule Drug Screening Assay to Identify Potential Sclerostin Inhibitors against Osteoporosis. Int. J. Mol. Sci. 2021, 22, 8320. https://doi.org/10.3390/ijms22158320
Lee C-C, Hung C-M, Chen C-H, Hsu Y-C, Huang Y-P, Huang T-B, Lee M-J. Novel Aptamer-Based Small-Molecule Drug Screening Assay to Identify Potential Sclerostin Inhibitors against Osteoporosis. International Journal of Molecular Sciences. 2021; 22(15):8320. https://doi.org/10.3390/ijms22158320
Chicago/Turabian StyleLee, Chien-Ching, Chao-Ming Hung, Chung-Hwan Chen, Yi-Chiang Hsu, Yuan-Pin Huang, Tsung-Bin Huang, and Mon-Juan Lee. 2021. "Novel Aptamer-Based Small-Molecule Drug Screening Assay to Identify Potential Sclerostin Inhibitors against Osteoporosis" International Journal of Molecular Sciences 22, no. 15: 8320. https://doi.org/10.3390/ijms22158320
APA StyleLee, C.-C., Hung, C.-M., Chen, C.-H., Hsu, Y.-C., Huang, Y.-P., Huang, T.-B., & Lee, M.-J. (2021). Novel Aptamer-Based Small-Molecule Drug Screening Assay to Identify Potential Sclerostin Inhibitors against Osteoporosis. International Journal of Molecular Sciences, 22(15), 8320. https://doi.org/10.3390/ijms22158320