
3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury


 , and
, and 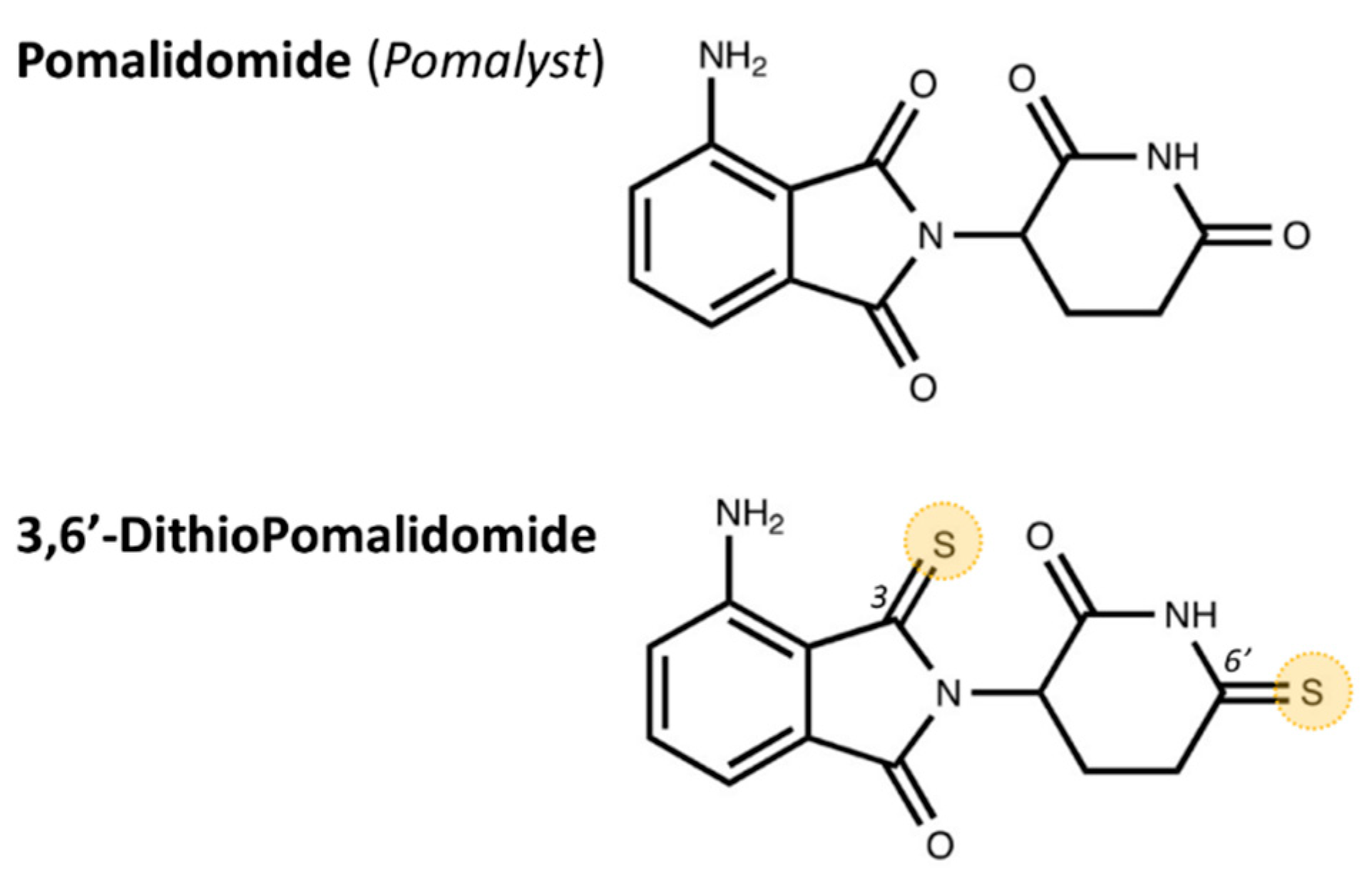{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
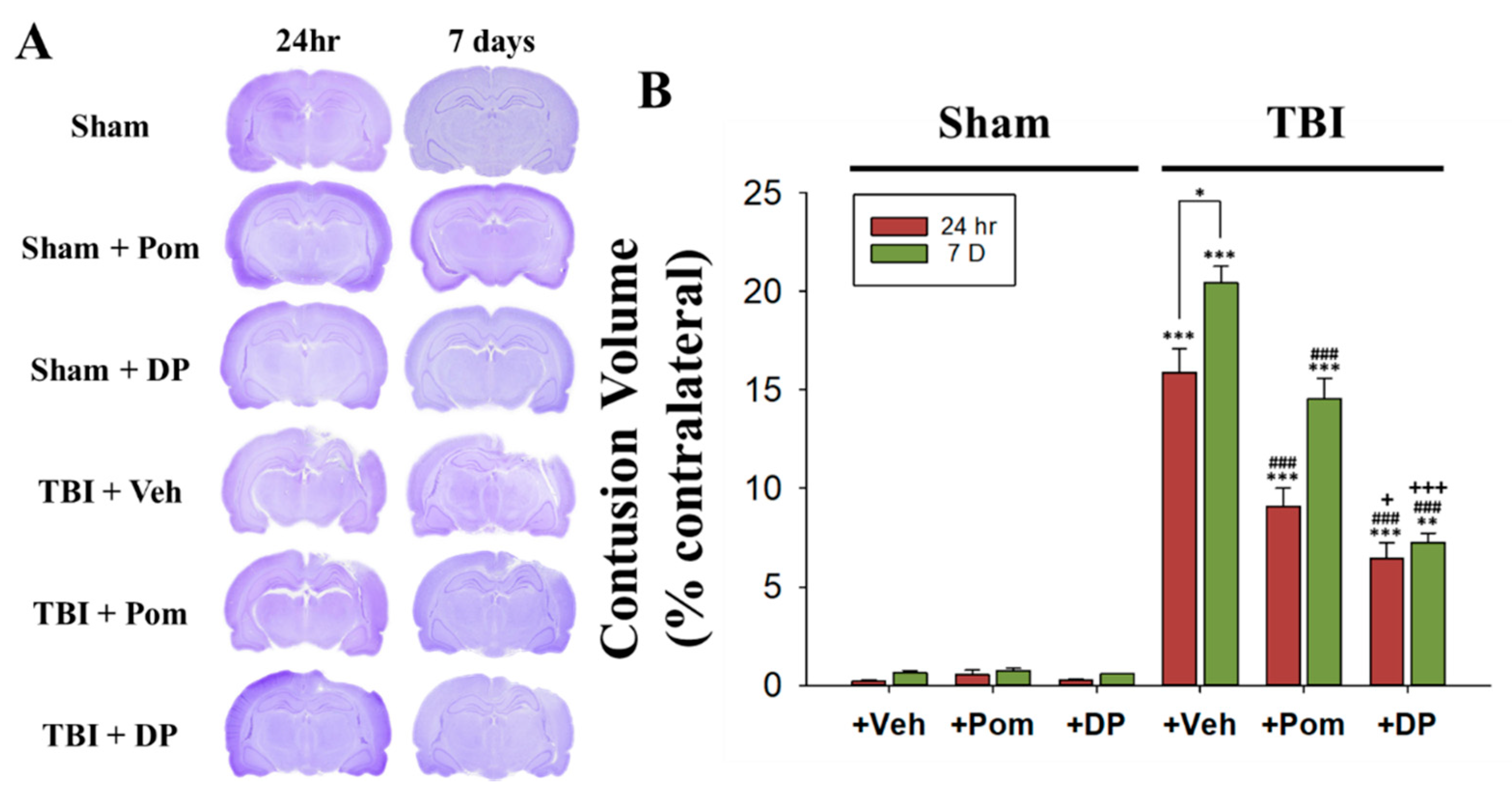
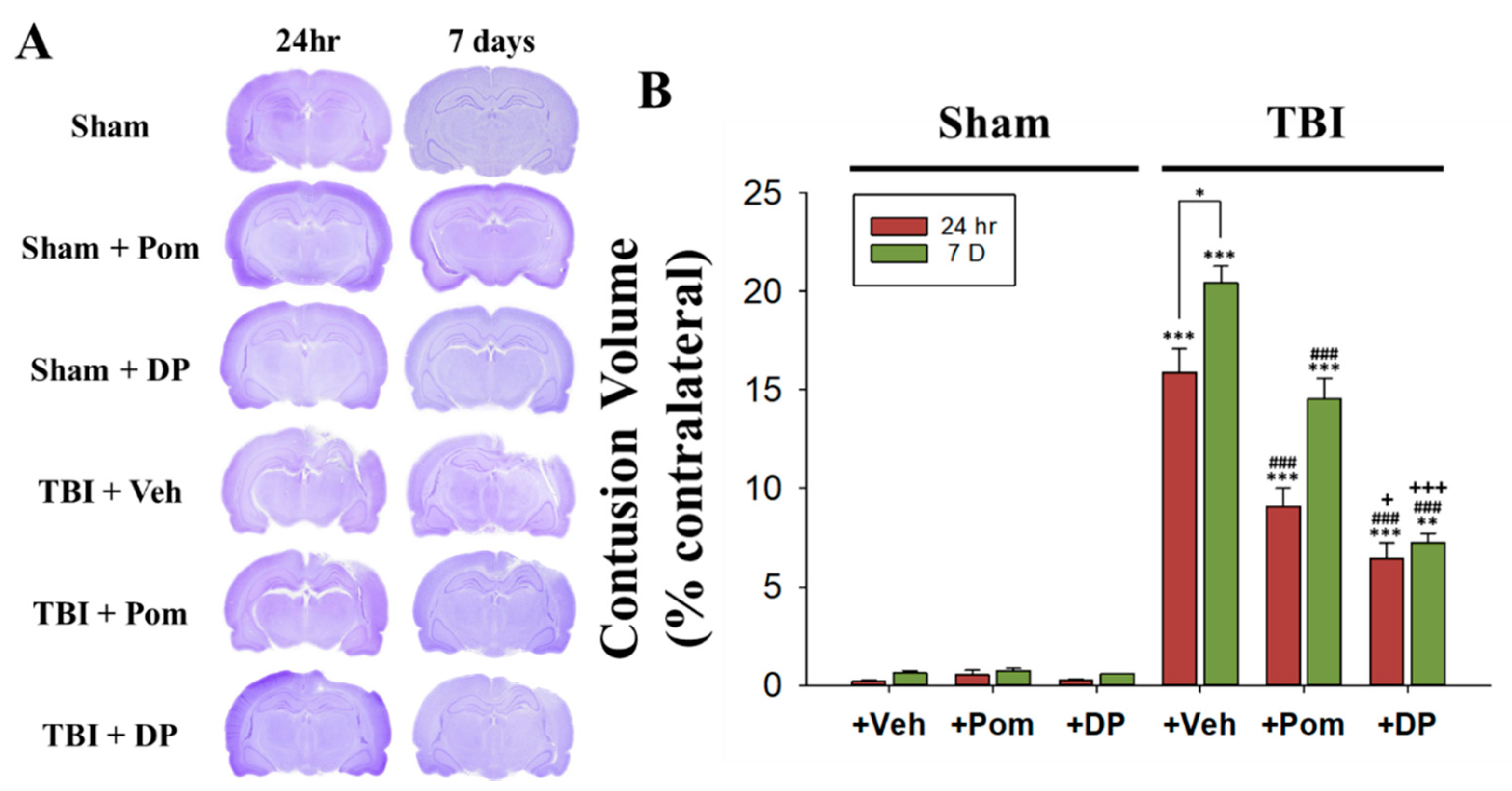
2.1. Post-Injury Treatment with Pom and DP Reduced Hippocampal Injury Caused by TBI with DP Proving More Efficacious in Reducing Injury Volume Than Pom at 24 h and 7 Days (7D) after TBI
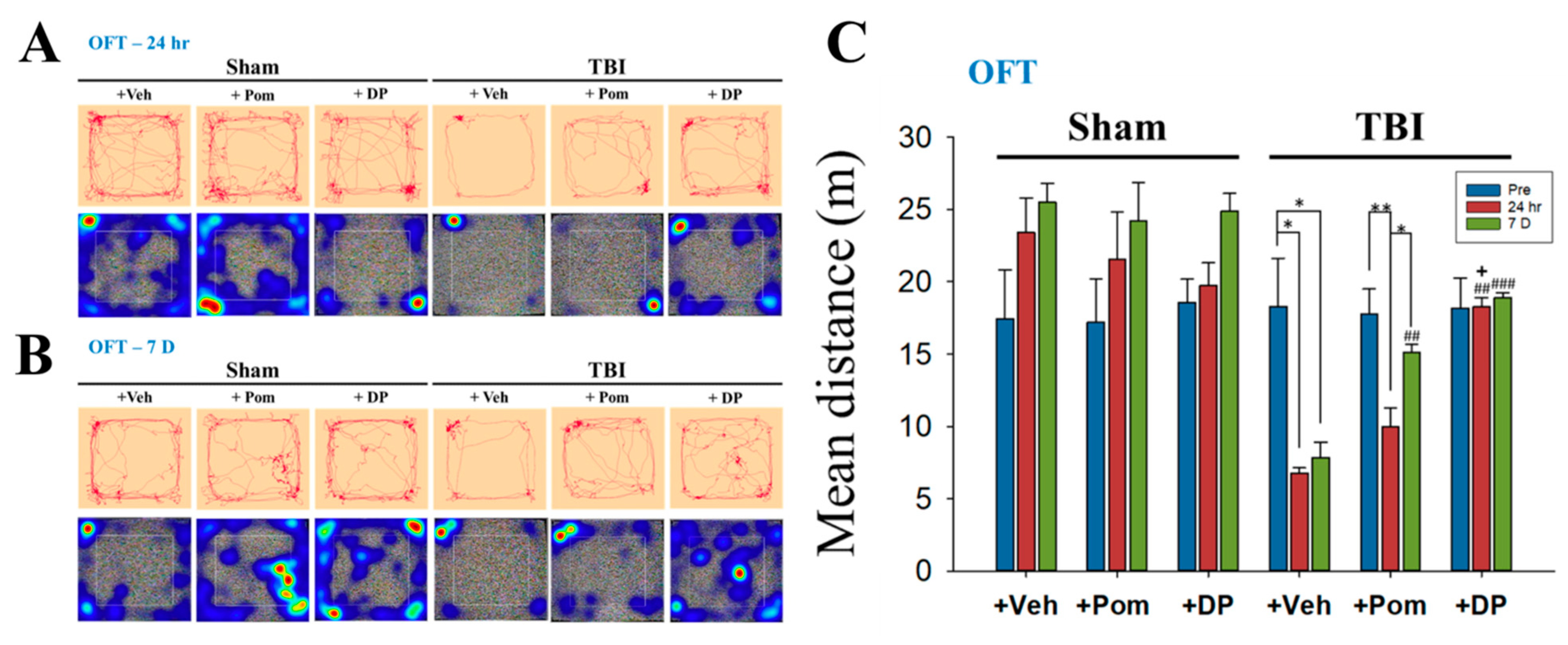
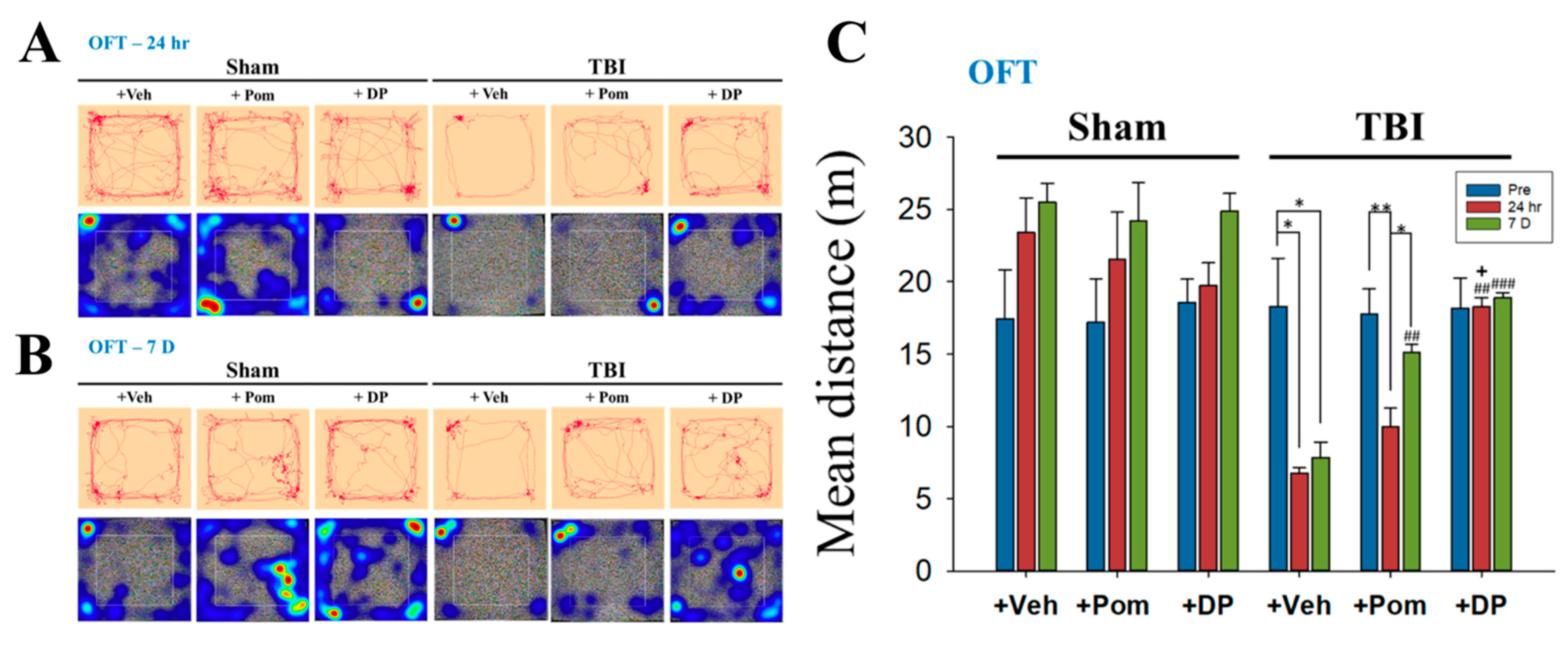
2.2. DP Exhibited an Earlier Onset of Improvement in TBI-Induced Anxiety-Like Behavior Than Pom within 24 h after TBI; the Effects of DP and Pom Were Comparable at 7D after TBI
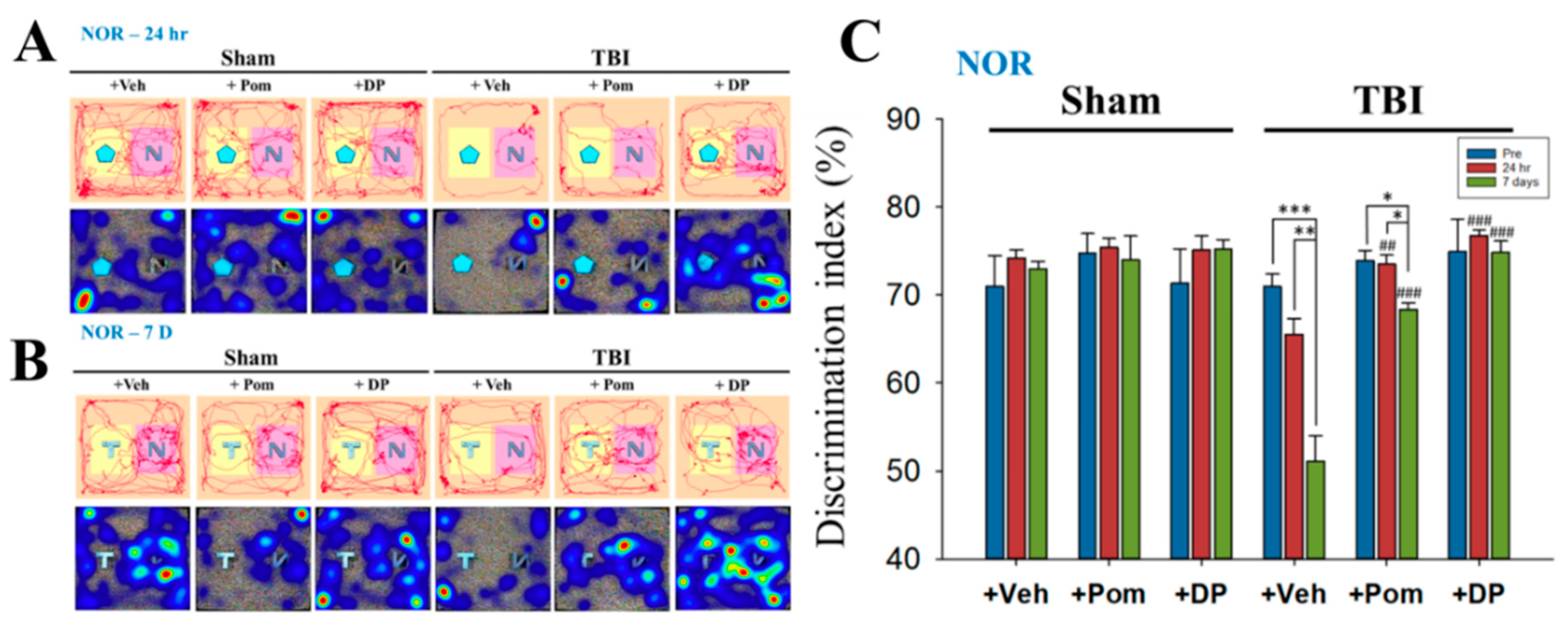
2.3. Short-Term Memory Impairment Was Significant at 7D after TBI and, although Both DP and Pom Improved Short-Term Memory, Only DP Fully Restored It at 7D after TBI to Pre-TBI Levels
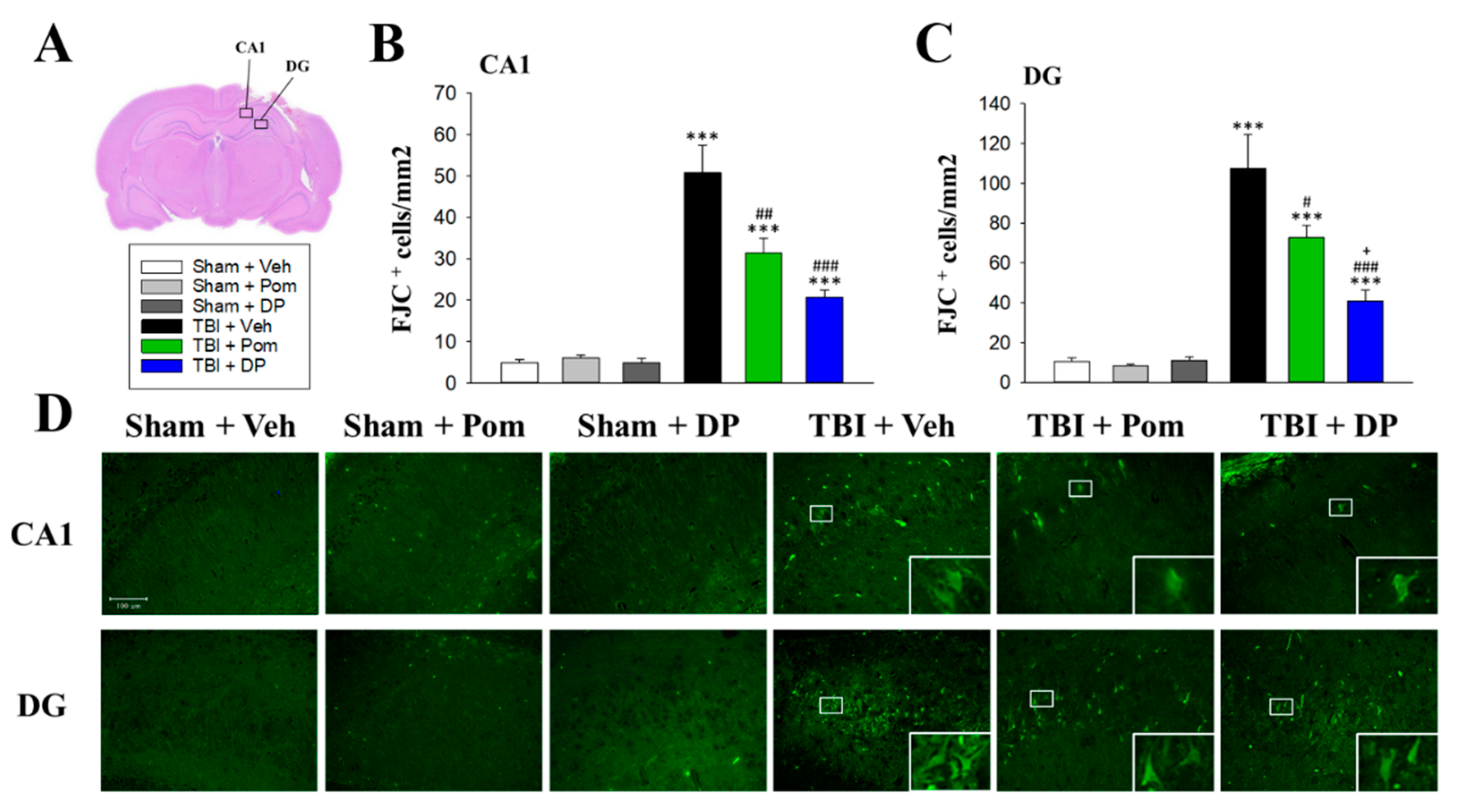
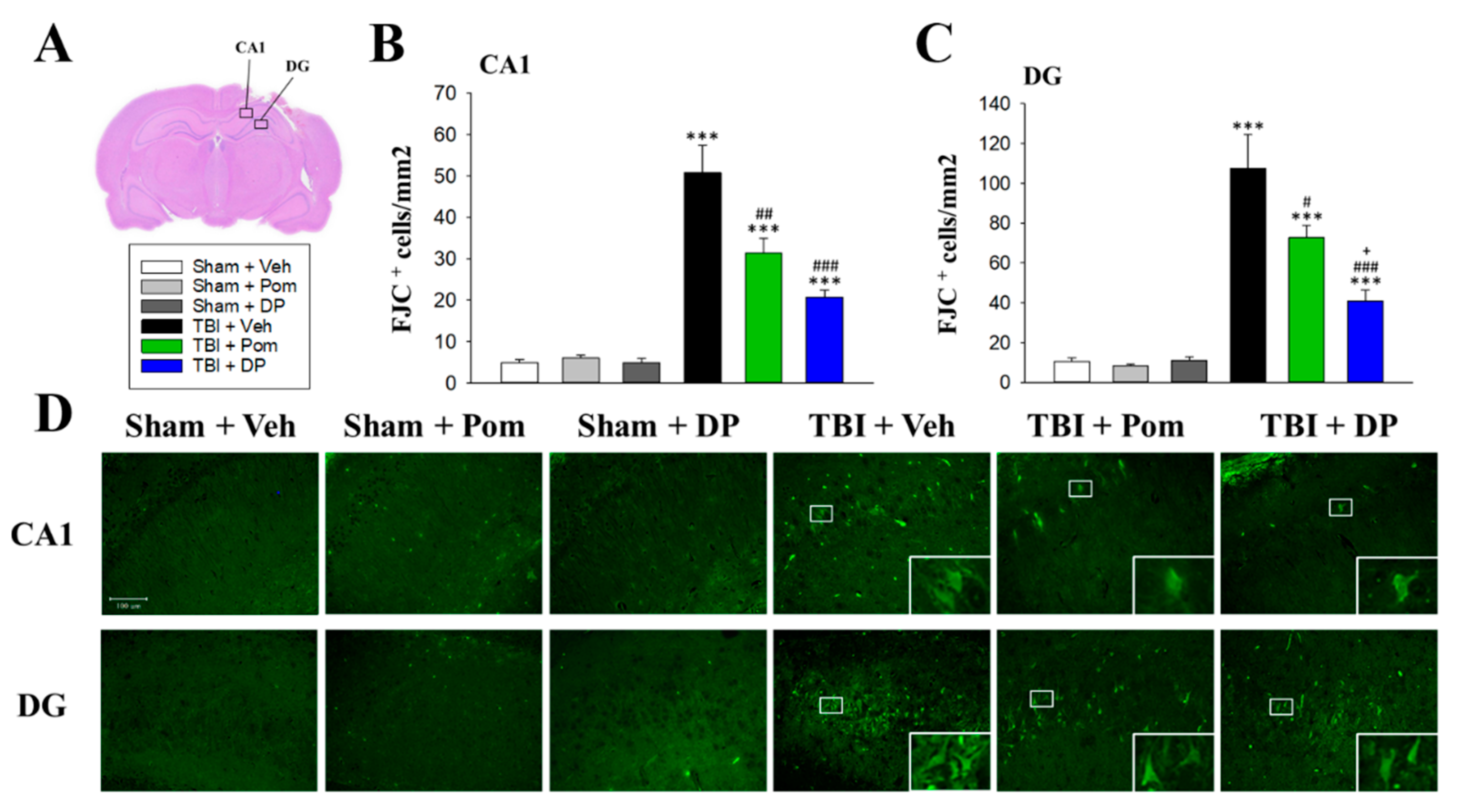
2.4. Pom and DP Reduced TBI-Induced Neurodegeneration in the Hippocampus at 7D after TBI but DP Reduced Numbers of Degenerating Neurons More Than Pom in the DG
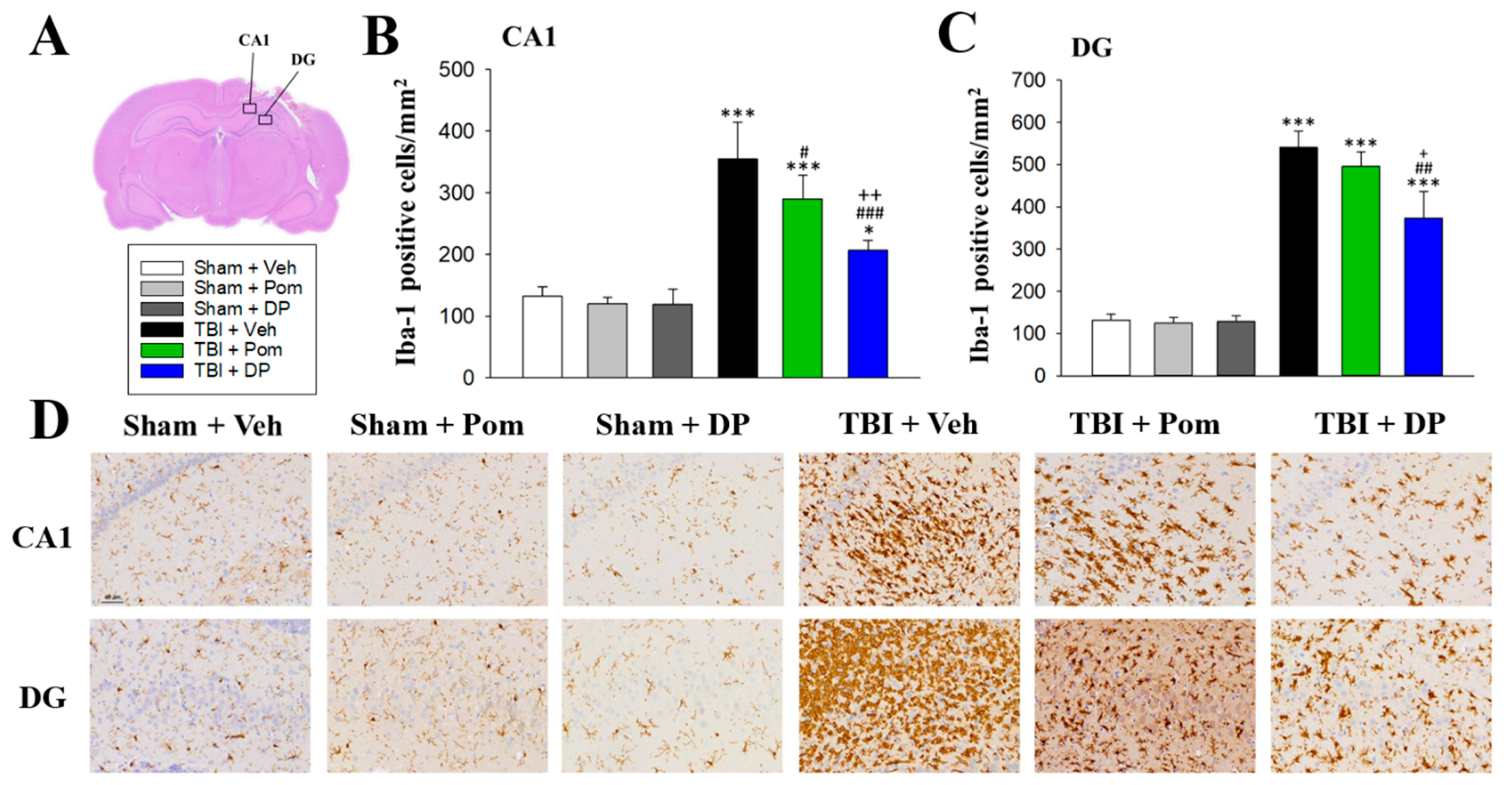
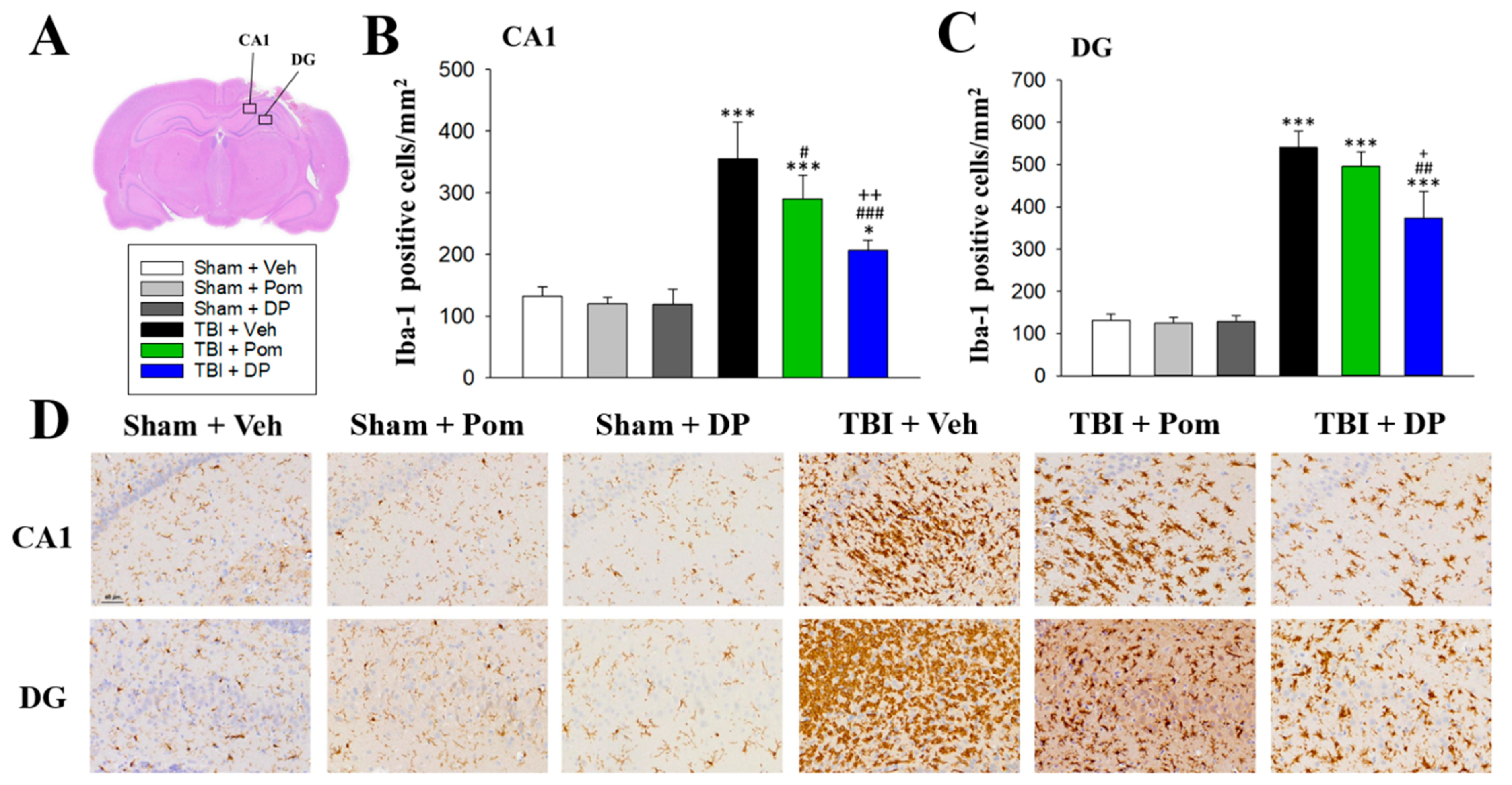
2.5. DP Reduced TBI-Induced Microgliosis in the Hippocampus at 7D after TBI
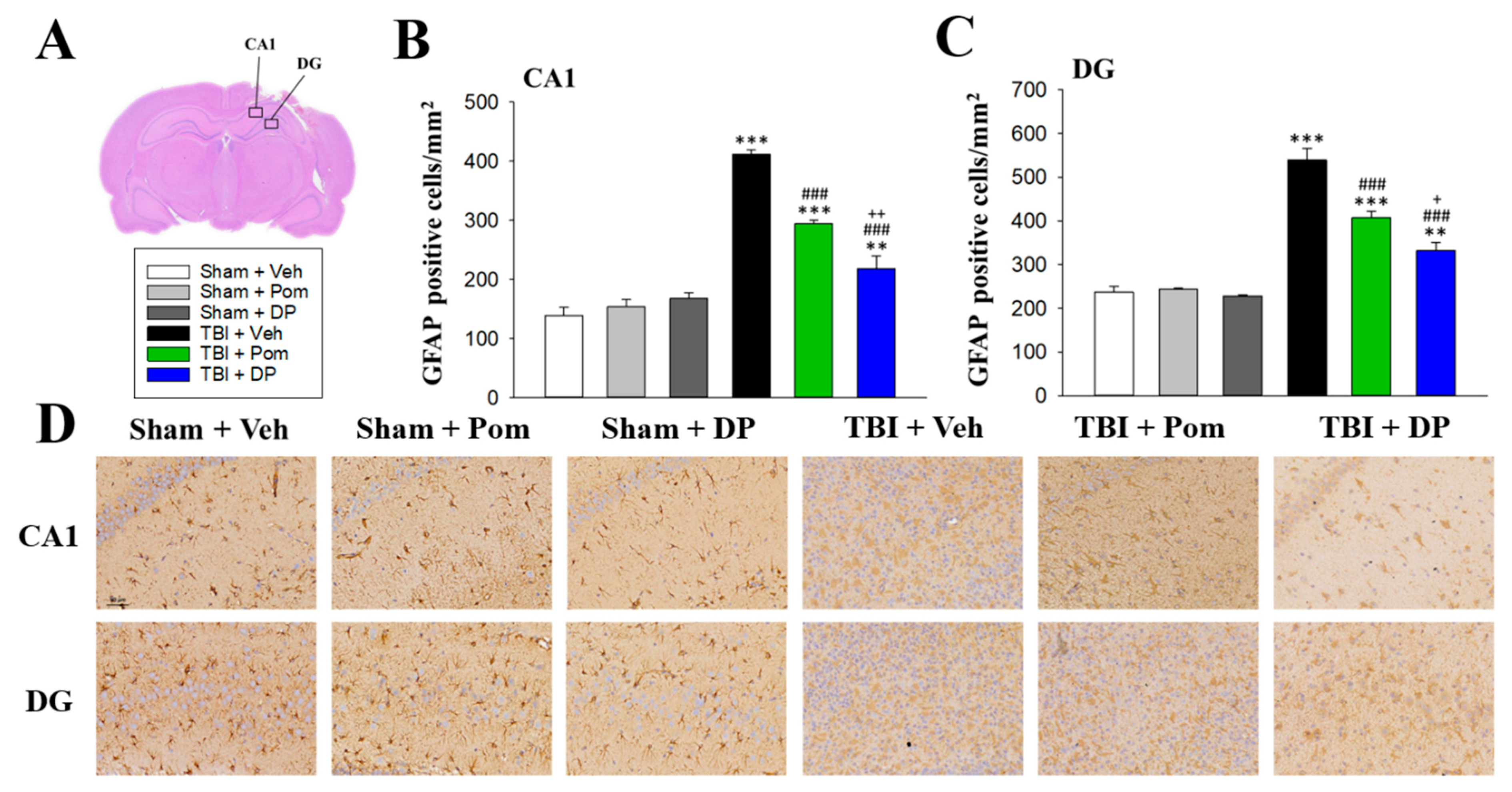
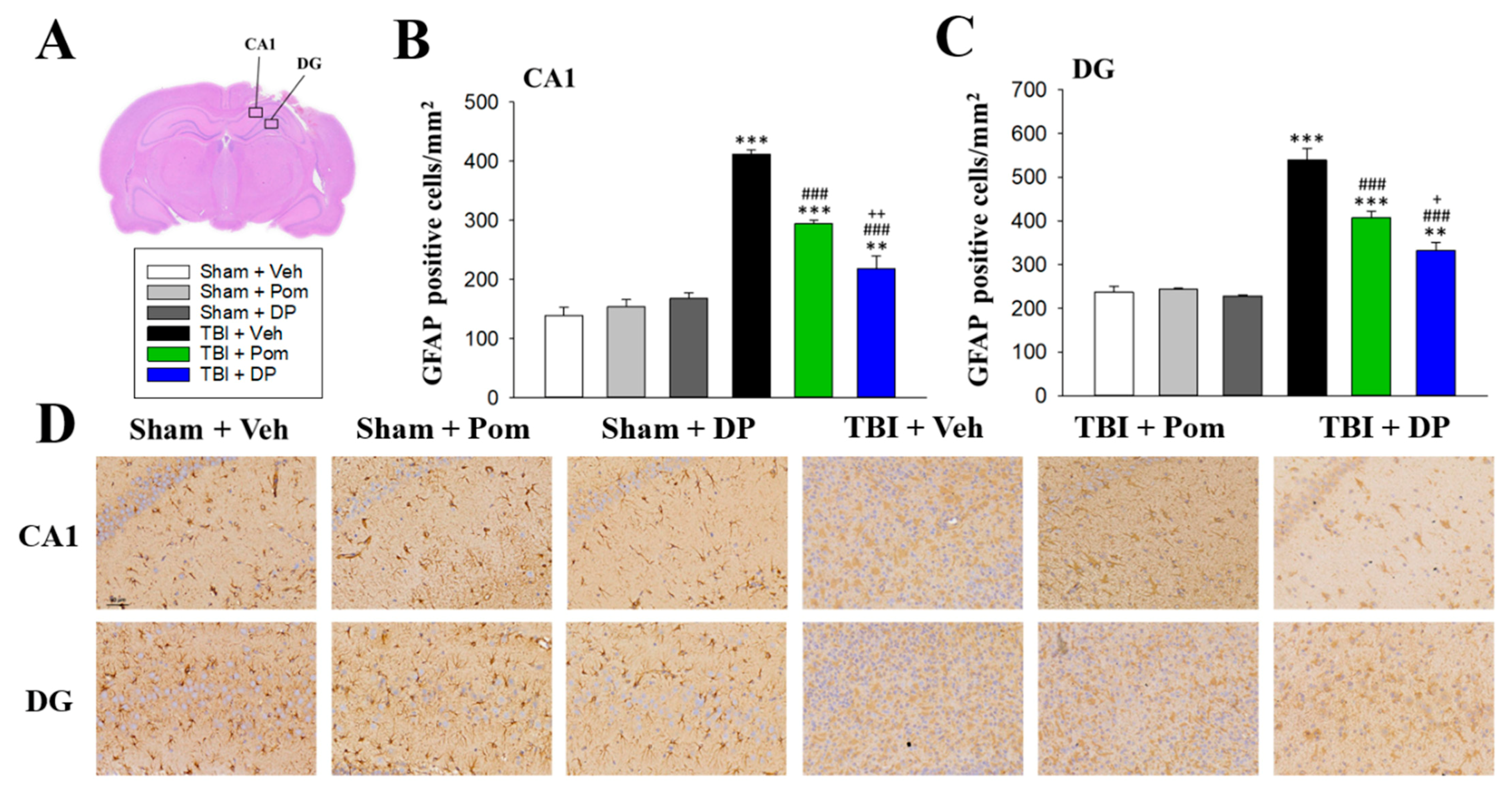
2.6. DP Was More Effective Than Pom at Attenuating TBI-Induced Astrogliosis in the Hippocampus
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animal Model of TBI
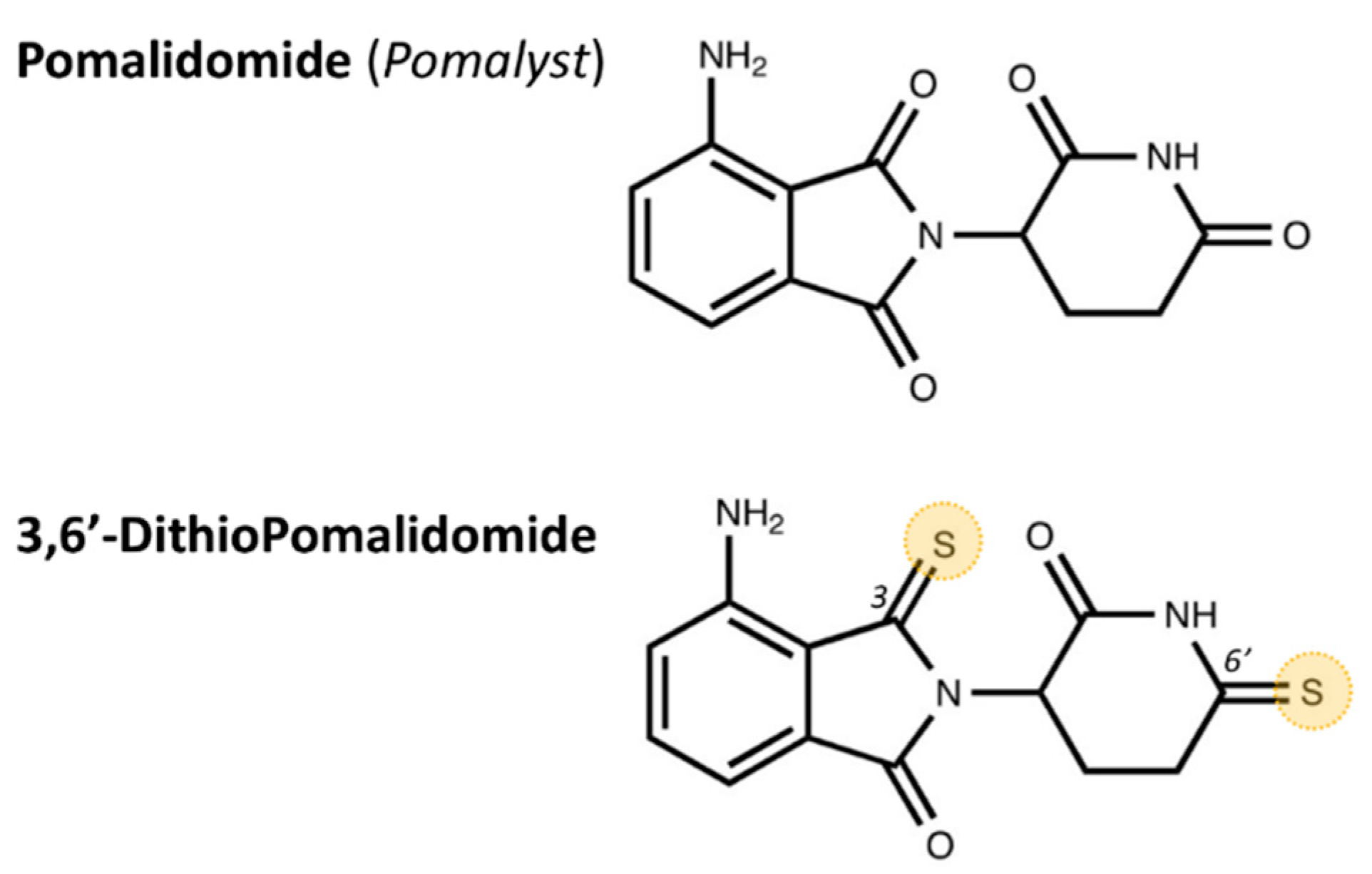
5.2. Drug Synthesis and Administration
5.3. Behavioral Evaluation of Neurological Outcomes
5.4. Open-Field Test (OFT)
5.5. Novel Object Recognition Test (NOR)
5.6. Contusion Volume
5.7. Fluoro-Jade C (FJC) Staining
5.8. Immunohistochemistry (IHC)
5.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [Green Version]
- LoBue, C.; Munro, C.; Schaffert, J.; Didehbani, N.; Hart, J.; Batjer, H.; Cullum, C.M. Traumatic Brain Injury and Risk of Long-Term Brain Changes, Accumulation of Pathological Markers, and Developing Dementia: A Review. J. Alzheimer Dis. 2019, 70, 629–654. [Google Scholar] [CrossRef]
- Tagliaferri, F.; Compagnone, C.; Korsic, M.; Servadei, F.; Kraus, J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir. 2005, 148, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Popovic, V. Traumatic brain injury: Neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 2019, 22, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Kaup, A.; Kirby, K.A.; Byers, A.L.; Diaz-Arrastia, R.; Yaffe, K. Traumatic brain injury and risk of dementia in older veterans. Neurology 2014, 83, 312–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweedie, D.; Rachmany, L.; Rubovitch, V.; Lehrmann, E.; Zhang, Y.; Becker, K.G.; Perez, E.; Miller, J.; Hoffer, B.J.; Greig, N.H.; et al. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp. Neurol. 2013, 239, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweedie, D.; Rachmany, L.; Rubovitch, V.; Zhang, Y.; Becker, K.G.; Perez, E.; Hoffer, B.J.; Pick, C.G.; Greig, N.H. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol. Dis. 2013, 54, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hyder, A.A.; Wunderlich, C.A.; Puvanachandra, P.; Gururaj, G.; Kobusingye, O.C. The impact of traumatic brain injuries: A global perspective. NeuroRehabilitation 2007, 22, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Greig, N.H.; Tweedie, D.; Rachmany, L.; Li, Y.; Rubovitch, V.; Schreiber, S.; Chiang, Y.-H.; Hoffer, B.J.; Miller, J.; Lahiri, D.K.; et al. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimer Dement. 2014, 10, S62–S75. [Google Scholar] [CrossRef] [Green Version]
- Greve, M.W.; Zink, B.J. Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. 2009, 76, 97–104. [Google Scholar] [CrossRef]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury—An inflammatory disease? Brain Res. Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arrastia, R.; Kochanek, P.; Bergold, P.; Kenney, K.; Marx, C.E.; Grimes, C.J.B.; Loh, L.T.C.Y.; Adam, L.T.C.G.E.; Oskvig, D.; Curley, K.C.; et al. Pharmacotherapy of Traumatic Brain Injury: State of the Science and the Road Forward: Report of the Department of Defense Neurotrauma Pharmacology Workgroup. J. Neurotrauma 2014, 31, 135–158. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflammation 2008, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C.; Daneshvar, D.H. The neuropathology of traumatic brain injury. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 127, pp. 45–66. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.G.; Bruce-Keller, A.J.; Rabchevsky, A.G.; Christakos, S.; Clair, D.K.; Mattson, M.P.; Scheff, S.W. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci. 1999, 19, 6248–6256. [Google Scholar] [CrossRef] [PubMed]
- Frankola, K.A.; Greig, N.H.; Luo, W.; Tweedie, D. Targeting TNF-α to Elucidate and Ameliorate Neuroinflammation in Neurodegenerative Diseases. CNS Neurol. Disord. Drug Targets 2011, 10, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, J.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, T.; Wulff, P. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience 2015, 309, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tate, D.F.; Bigler, E.D. Fornix and hippocampal atrophy in traumatic brain injury. Learn. Mem. 2000, 7, 442–446. [Google Scholar] [CrossRef] [Green Version]
- White, E.R.; Pinar, C.; Bostrom, C.A.; Meconi, A.; Christie, B.R. Mild Traumatic Brain Injury Produces Long-Lasting Deficits in Synaptic Plasticity in the Female Juvenile Hippocampus. J. Neurotrauma 2017, 34, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Zhang, J.; Dong, J.-F.; Shi, F.-D. Dissemination of brain inflammation in traumatic brain injury. Cell. Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef]
- Redell, J.B.; Maynard, M.E.; Underwood, E.L.; Vita, S.M.; Dash, P.K.; Kobori, N. Traumatic brain injury and hippocampal neurogenesis: Functional implications. Exp. Neurol. 2020, 331, 113372. [Google Scholar] [CrossRef]
- Almeida-Suhett, C.; Li, Z.; Marini, A.M.; Braga, M.F.; Eiden, L.E. Temporal Course of Changes in Gene Expression Suggests a Cytokine-Related Mechanism for Long-Term Hippocampal Alteration after Controlled Cortical Impact. J. Neurotrauma 2014, 31, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-T.; Lecca, D.; Yang, L.-Y.; Luo, W.; Scerba, M.T.; Tweedie, D.; Huang, P.-S.; Jung, Y.-J.; Kim, D.S.; Yang, C.-H.; et al. 3,6’-Dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. Elife 2020, 9, 9. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Huang, Y.-N.; Chiu, C.-C.; Tweedie, D.; Luo, W.; Pick, C.G.; Chou, S.-Y.; Luo, Y.; Hoffer, B.J.; Greig, N.H.; et al. Pomalidomide mitigates neuronal loss, neuroinflammation, and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflammation 2016, 13, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borlongan, C.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Lozano, D.; Gonzales-Portillo, G.S. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, D.; Schimmel, S.J.; Acosta, S. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ. 2017, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Baratz, R.; Tweedie, D.; Wang, J.-Y.; Rubovitch, V.; Luo, W.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflammation 2015, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Roberts, I.; Yates, D.; Sandercock, P.; Farrell, B.; Wasserberg, J.; Lomas, G.; Cottingham, R.; Svoboda, P.; Brayley, N.; Mazairac, G.; et al. Effect of intravenous corticosteroids on death within 14 days in 10 008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial. Lancet 2004, 364, 1321–1328. [Google Scholar] [CrossRef]
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 2018, 141, 459–471. [Google Scholar] [CrossRef]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Kim, D.S.; Palmas, M.F.; Pisanu, A.; Carta, A.R.; Greig, N.H. Repurposing Immunomodulatory Imide Drugs (IMiDs) in Neuropsychiatric and Neurodegenerative Disorders. Front. Neurosci. 2021, 15, 656921. [Google Scholar] [CrossRef]
- Lu, J.; Goh, S.J.; Tng, P.Y.; Deng, Y.Y.; Ling, E.A.; Moochhala, S. Systemic inflammatory response following acute traumatic brain injury. Front. Biosci. 2009, 14, 3795–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohami, E.; Gallily, R.; Mechoulam, R.; Bass, R.; Ben-Hur, T. Cytokine production in the brain following closed head injury: Dexanabinol (HU-211) is a novel TNF-α inhibitor and an effective neuroprotectant. J. Neuroimmunol. 1997, 72, 169–177. [Google Scholar] [CrossRef]
- Chio, C.-C.; Lin, M.-T.; Chang, C.-P. Microglial activation as a compelling target for treating acute traumatic brain injury. Curr. Med. Chem. 2015, 22, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Chio, C.-C.; Chang, C.-H.; Wang, C.-C.; Cheong, C.-U.; Chao, C.-M.; Cheng, B.-C.; Yang, C.-Z.; Chang, C.-P. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-α. BMC Neurosci. 2013, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Tobinick, E.; Kim, N.M.; Reyzin, G.; Rodriguez-Romanacce, H.; DePuy, V. Selective TNF inhibition for chronic stroke and traumatic brain injury: An observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 2012, 26, 1051–1070. [Google Scholar] [CrossRef]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef]
- Yoon, J.S.; Lee, J.-H.; Tweedie, D.; Mughal, M.R.; Chigurupati, S.; Greig, N.H.; Mattson, M.P. 3,6′-dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J. Neurosci. Res. 2013, 91, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, S.A.; Tajiri, N.; de la Pena, I.; Bastawrous, M.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J. Cell Physiol. 2015, 230, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Wilson, J.; Ritter, A.; Dardis, C.; DiFilippo, F.P.; Zhuang, X.; Cordes, D.; Lee, G.; Fulkerson, N.D.; Rose, T.S.; et al. MCLENA-1: A Phase II Clinical Trial for the Assessment of Safety, Tolerability, and Efficacy of Lenalidomide in Patients with Mild Cognitive Impairment Due to Alzheimer’s Disease. Open Access J. Clin. Trials 2020, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, L.; Lee, S.E.; Madiha, S.; Gaire, B.P.; Jin, M.; Yumnam, S.; Kim, S.Y. Phytochemicals against TNFα-Mediated Neuroinflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, S.C.; Luo, W.; Tweedie, D.; Kim, D.S.; Kim, Y.K.; Hwang, I.; Gil, J.-E.; Han, B.-S.; Chiang, Y.-H.; Selman, W.; et al. N-Adamantyl Phthalimidine: A New Thalidomide-like Drug That Lacks Cereblon Binding and Mitigates Neuronal and Synaptic Loss, Neuroinflammation, and Behavioral Deficits in Traumatic Brain Injury and LPS Challenge. ACS Pharmacol. Transl. Sci. 2021, 4, 980–1000. [Google Scholar] [CrossRef]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflammation 2012, 9, 106. [Google Scholar] [CrossRef]
- Terpos, E.; Kanellias, N.; Christoulas, D.; Kastritis, E.; Dimopoulos, M.A. Pomalidomide: A novel drug to treat relapsed and refractory multiple myeloma. OncoTargets Ther. 2013, 6, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-Y.; Greig, N.H.; Huang, Y.-N.; Hsieh, T.-H.; Tweedie, D.; Yu, Q.-S.; Hoffer, B.J.; Luo, Y.; Kao, Y.-C.; Wang, J.-Y. Post-traumatic administration of the p53 inactivator pifithrin-α oxygen analogue reduces hippocampal neuronal loss and improves cognitive deficits after experimental traumatic brain injury. Neurobiol. Dis. 2016, 96, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Schwarzbold, M.L.; Rial, D.; De Bem, T.; Machado, D.G.; Cunha, M.P.; Dos Santos, A.A.; Dos Santos, D.B.; Figueiredo, C.P.; Farina, M.; Goldfeder, E.M.; et al. Effects of Traumatic Brain Injury of Different Severities on Emotional, Cognitive, and Oxidative Stress-Related Parameters in Mice. J. Neurotrauma 2010, 27, 1883–1893. [Google Scholar] [CrossRef]
- Tucker, L.B.; Burke, J.; Fu, A.H.; McCabe, J.T. Neuropsychiatric Symptom Modeling in Male and Female C57BL/6J Mice after Experimental Traumatic Brain Injury. J. Neurotrauma 2017, 34, 890–905. [Google Scholar] [CrossRef] [Green Version]
- Popovitz, J.; Mysore, S.; Adwanikar, H. Long-Term Effects of Traumatic Brain Injury on Anxiety-Like Behaviors in Mice: Behavioral and Neural Correlates. Front. Behav. Neurosci. 2019, 13, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, M.E.; Requena, D.F.; Casper, T.C.; Velhorst, A.K.; Lolofie, A.; McFarlane, K.E.; Otto, T.E.; Terry, C.; Gensel, J.C. Docosahexaenoic acid decreased neuroinflammation in rat pups after controlled cortical impact. Exp. Neurol. 2019, 320, 112971. [Google Scholar] [CrossRef] [PubMed]
- Tadepalli, S.A.; Bali, Z.K.; Bruszt, N.; Nagy, L.V.; Amrein, K.; Fazekas, B.; Büki, A.; Czeiter, E.; Hernadi, I. Long-term cognitive impairment without diffuse axonal injury following repetitive mild traumatic brain injury in rats. Behav. Brain Res. 2020, 378, 112268. [Google Scholar] [CrossRef] [PubMed]
- Leconte, C.; Benedetto, C.; Lentini, F.; Simon, K.; Ouaazizi, C.; Taib, T.; Cho, A.H.; Plotkine, M.; Mongeau, R.; Marchand-Leroux, C.; et al. Histological and Behavioral Evaluation after Traumatic Brain Injury in Mice: A Ten Months Follow-Up Study. J. Neurotrauma 2020, 37, 1342–1357. [Google Scholar] [CrossRef]
- Lorente, L.; Martín, M.M.; González-Rivero, A.F.; Argueso, M.; Ramos, L.; Sole-Violán, J.; Cáceres, J.J.; Jiménez, A.; Borreguero-León, J.M. Serum Levels of Caspase-Cleaved Cytokeratin-18 in Patients with Severe Traumatic Brain Injury Are Associated with Mortality: A Pilot Study. PLoS ONE 2015, 10, e0121739. [Google Scholar] [CrossRef]
- Saatman, K.E.; Feeko, K.J.; Pape, R.L.; Raghupathi, R. Differential Behavioral and Histopathological Responses to Graded Cortical Impact Injury in Mice. J. Neurotrauma 2006, 23, 1241–1253. [Google Scholar] [CrossRef]
- Tong, W.; Igarashi, T.; Ferriero, D.M.; Noble, L.J. Traumatic brain injury in the immature mouse brain: Characterization of regional vulnerability. Exp. Neurol. 2002, 176, 105–116. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, L.; Gao, X.; Luo, B.; Chen, J. Moderate Traumatic Brain Injury Triggers Rapid Necrotic Death of Immature Neurons in the Hippocampus. J. Neuropathol. Exp. Neurol. 2012, 71, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Deng-Bryant, Y.; Cho, W.; Carrico, K.M.; Hall, E.D.; Chen, J. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J. Neurosci. Res. 2008, 86, 2258–2270. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Madathil, S.K.; Wilfred, B.S.; Urankar, S.E.; Yang, W.; Leung, L.Y.; Gilsdorf, J.S.; Shear, D.A. Early Microglial Activation Following Closed-Head Concussive Injury Is Dominated by Pro-Inflammatory M-1 Type. Front. Neurol. 2018, 9, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Aquila, M.; Maiese, A.; De Matteis, A.; Viola, R.V.; Arcangeli, M.; La Russa, R.; Fineschi, V. Traumatic brain injury: Estimate of the age of the injury based on neuroinflammation, endothelial activation markers and adhesion molecules. Histol. Histopathol. 2021, 18319. [Google Scholar]
- Caplan, H.W.; Cardenas, F.; Gudenkauf, F.; Zelnick, P.; Xue, H.; Cox, C.S.; Bedi, S.S. Spatiotemporal Distribution of Microglia After Traumatic Brain Injury in Male Mice. ASN Neuro 2020, 12, 1759091420911770. [Google Scholar] [CrossRef] [PubMed]
- Luoto, T.M.; Raj, R.; Posti, J.P.; Gardner, A.J.; Panenka, W.J.; Iverson, G.L. A Systematic Review of the Usefulness of Glial Fibrillary Acidic Protein for Predicting Acute Intracranial Lesions following Head Trauma. Front. Neurol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheff, S.W.; Ansari, M.A.; Roberts, K.N. Neuroprotective effect of Pycnogenol(R) following traumatic brain injury. Exp. Neurol. 2013, 239, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Wakade, C.; Sukumari-Ramesh, S.; Laird, M.D.; Dhandapani, K.M.; Vender, J.R. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J. Neurosurg. 2010, 113, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Jamjoom, A.A.B.; Rhodes, J.; Andrews, P.J.D.; Grant, S.G.N. The synapse in traumatic brain injury. Brain 2021, 144, 18–31. [Google Scholar] [CrossRef]
- Castejòn, O.J.; Valero, C.; Dìaz, M. Synaptic degenerative changes in human traumatic brain edema. An electron microscopic study of cerebral cortical biopsies. J. Neurosurg. Sci. 1995, 39, 47–65. [Google Scholar] [PubMed]
- Wen, Z.; Li, D.; Shen, M.; Chen, G. Therapeutic Potentials of Synapses after Traumatic Brain Injury: A Comprehensive Review. Neural Plast. 2017, 2017, 4296075. [Google Scholar] [CrossRef]
- Krukowski, K.; Chou, A.; Feng, X.; Tiret, B.; Paladini, M.S.; Riparip, L.-K.; Chaumeil, M.M.; Lemere, C.; Rosi, S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int. J. Mol. Sci. 2018, 19, 3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-F.; Hsu, C.-W.; Huang, W.-H.; Wang, J.-Y. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-F.; Hung, T.-H.; Chen, C.-C.; Lin, K.-H.; Huang, Y.-N.; Tsai, H.-C.; Wang, J.-Y. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 2007, 81, 288–298. [Google Scholar] [CrossRef]
- Huang, Y.-N.; Yang, L.-Y.; Greig, N.H.; Wang, Y.-C.; Lai, C.-C.; Wang, J.-Y. Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018, 8, 2368. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-Y.; Chu, Y.-H.; Tweedie, D.; Yu, Q.-S.; Pick, C.G.; Hoffer, B.J.; Greig, N.H.; Wang, J.-Y. Post-trauma administration of the pifithrin-α oxygen analog improves histological and functional outcomes after experimental traumatic brain injury. Exp. Neurol. 2015, 269, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prut, L.; Belzung, C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol. 2003, 463, 3–33. [Google Scholar] [CrossRef]
- Zhang, C.; Raghupathi, R.; Saatman, K.E.; Smith, D.H.; Stutzmann, J.-M.; Wahl, F.; Mcintosh, T.K. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J. Neurosci. Res. 1998, 52, 342–349. [Google Scholar] [CrossRef]
- Yang, L.Y.; Greig, N.H.; Tweedie, D.; Jung, Y.J.; Chiang, Y.H.; Hoffer, B.J.; Miller, J.P.; Chang, K.H.; Wang, J.Y. The p53 inactivators pifithrin-mu and pifithrin-alpha mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy. Exp. Neurol. 2020, 324, 113135. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, P.-S.; Tsai, P.-Y.; Yang, L.-Y.; Lecca, D.; Luo, W.; Kim, D.S.; Hoffer, B.J.; Chiang, Y.-H.; Greig, N.H.; Wang, J.-Y. 3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 8276. https://doi.org/10.3390/ijms22158276
Huang P-S, Tsai P-Y, Yang L-Y, Lecca D, Luo W, Kim DS, Hoffer BJ, Chiang Y-H, Greig NH, Wang J-Y. 3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury. International Journal of Molecular Sciences. 2021; 22(15):8276. https://doi.org/10.3390/ijms22158276
Chicago/Turabian StyleHuang, Pen-Sen, Ping-Yen Tsai, Ling-Yu Yang, Daniela Lecca, Weiming Luo, Dong Seok Kim, Barry J. Hoffer, Yung-Hsiao Chiang, Nigel H. Greig, and Jia-Yi Wang. 2021. "3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury" International Journal of Molecular Sciences 22, no. 15: 8276. https://doi.org/10.3390/ijms22158276
APA StyleHuang, P.-S., Tsai, P.-Y., Yang, L.-Y., Lecca, D., Luo, W., Kim, D. S., Hoffer, B. J., Chiang, Y.-H., Greig, N. H., & Wang, J.-Y. (2021). 3,6′-Dithiopomalidomide Ameliorates Hippocampal Neurodegeneration, Microgliosis and Astrogliosis and Improves Cognitive Behaviors in Rats with a Moderate Traumatic Brain Injury. International Journal of Molecular Sciences, 22(15), 8276. https://doi.org/10.3390/ijms22158276