
Role of the Renin–Angiotensin–Aldosterone and Kinin–Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID

 , , and
, , and 
Abstract
:1. Introduction
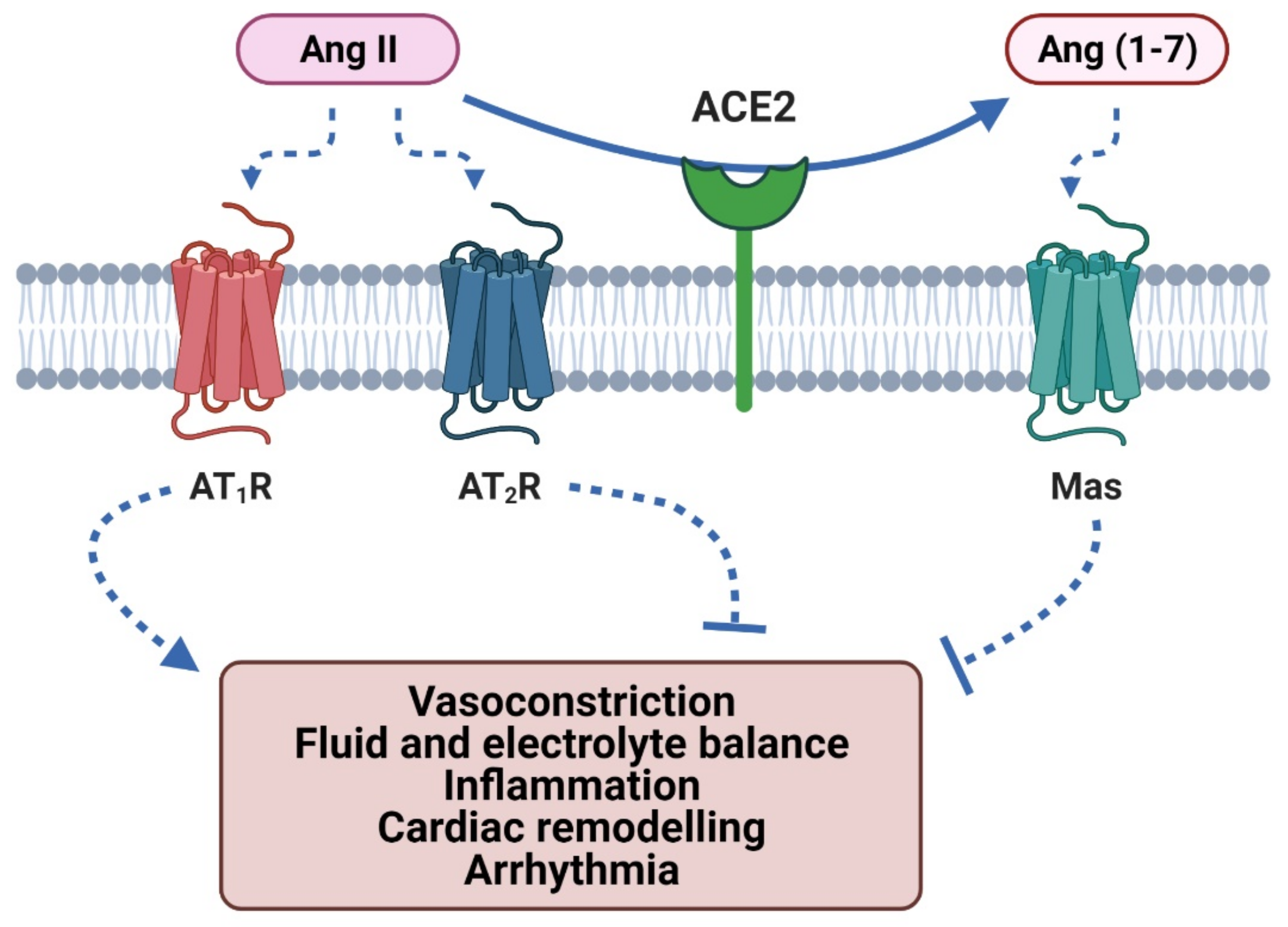
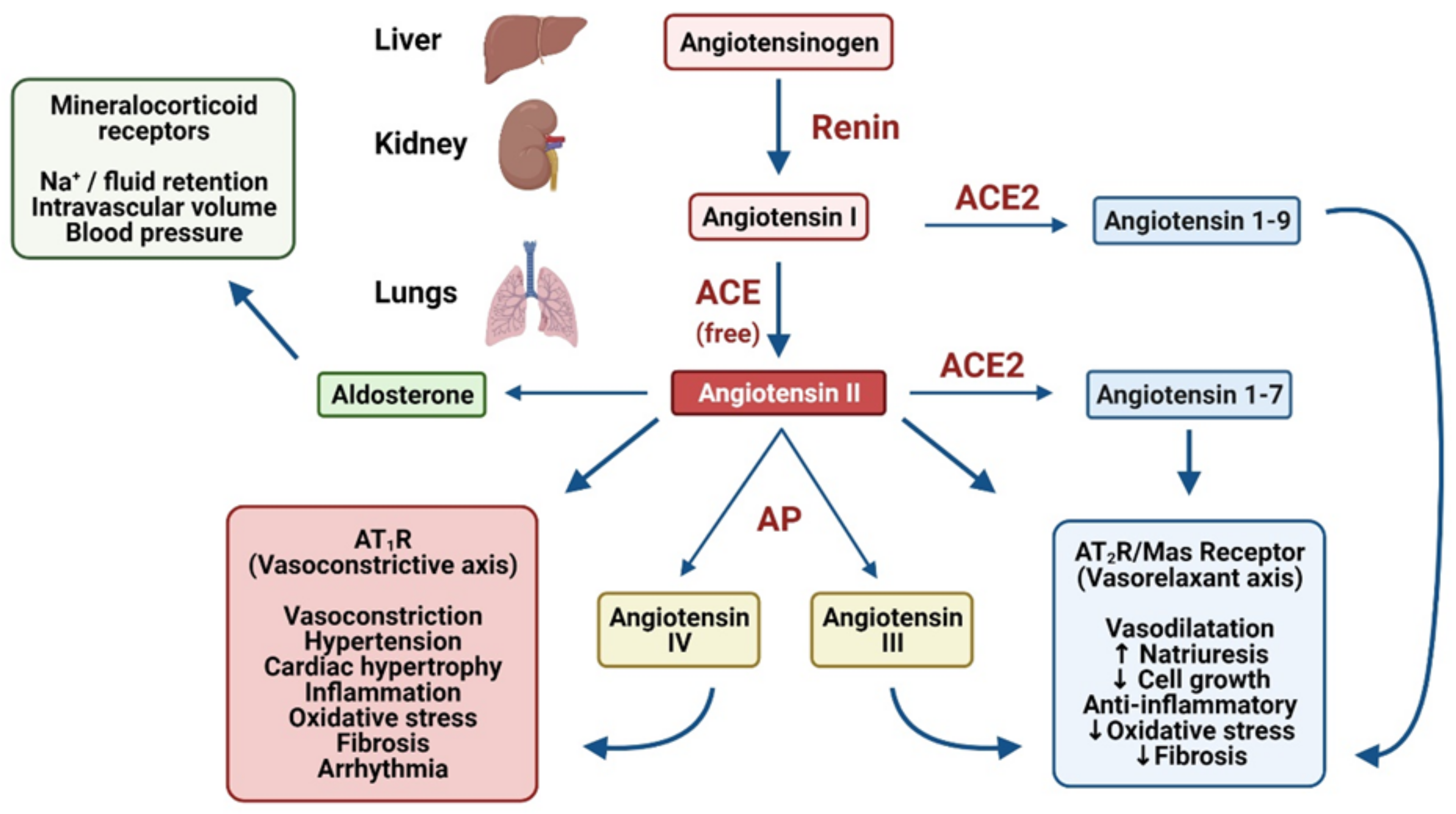
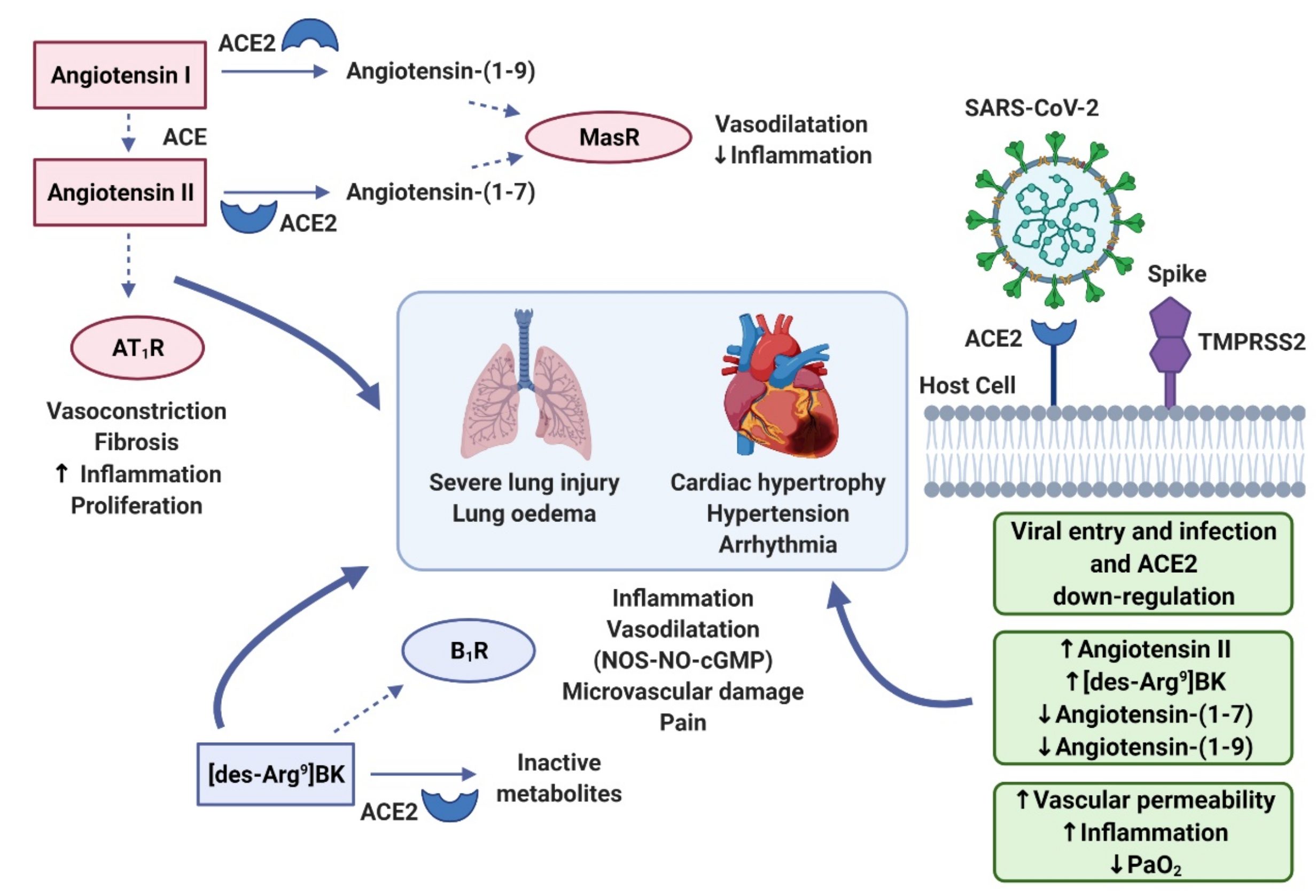
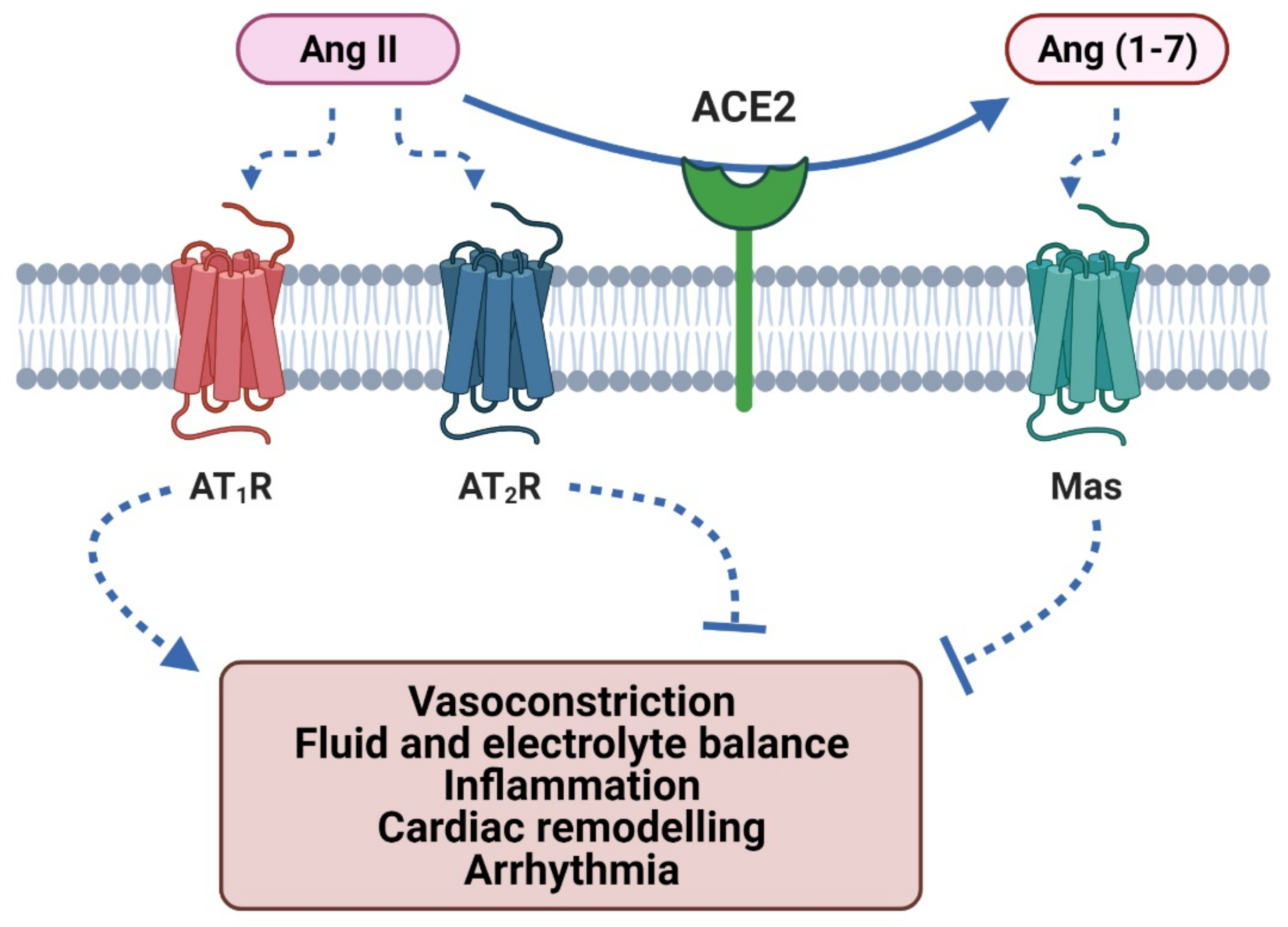
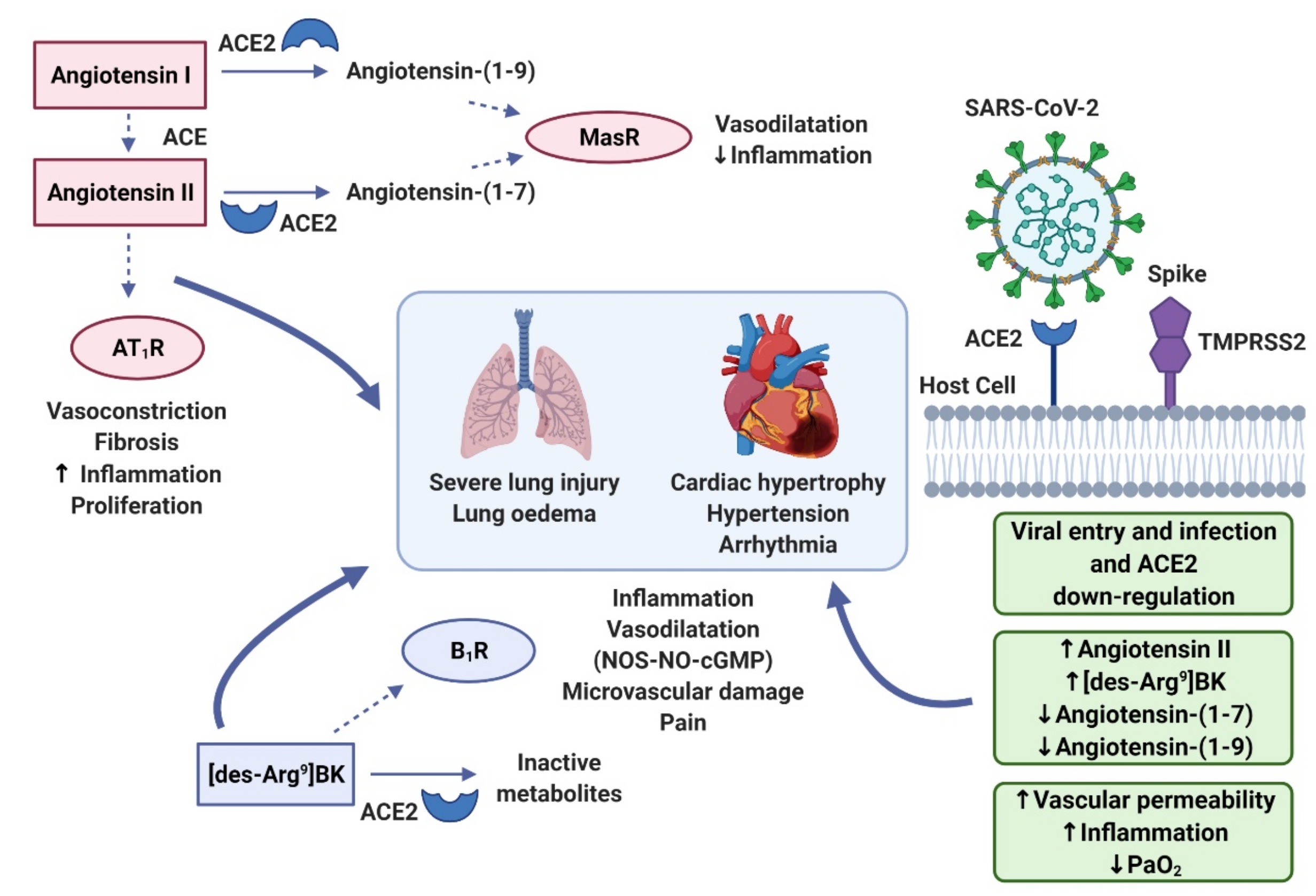
2. The Role of ACE2 in the RAAS and KKS
2.1. Angiotensin II Type-1 Receptor Activation
2.2. Angiotensin II Type-2 Receptor Activation
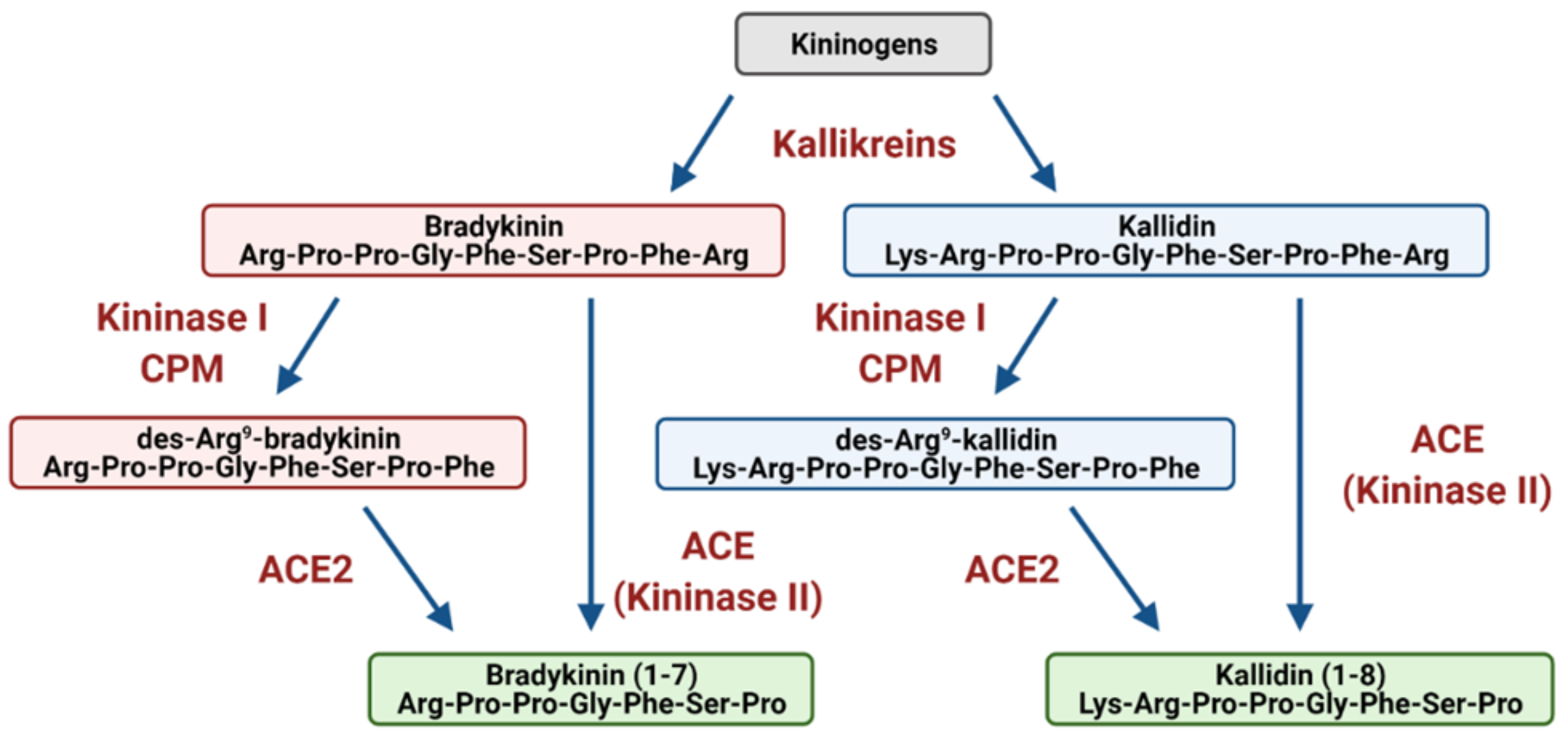
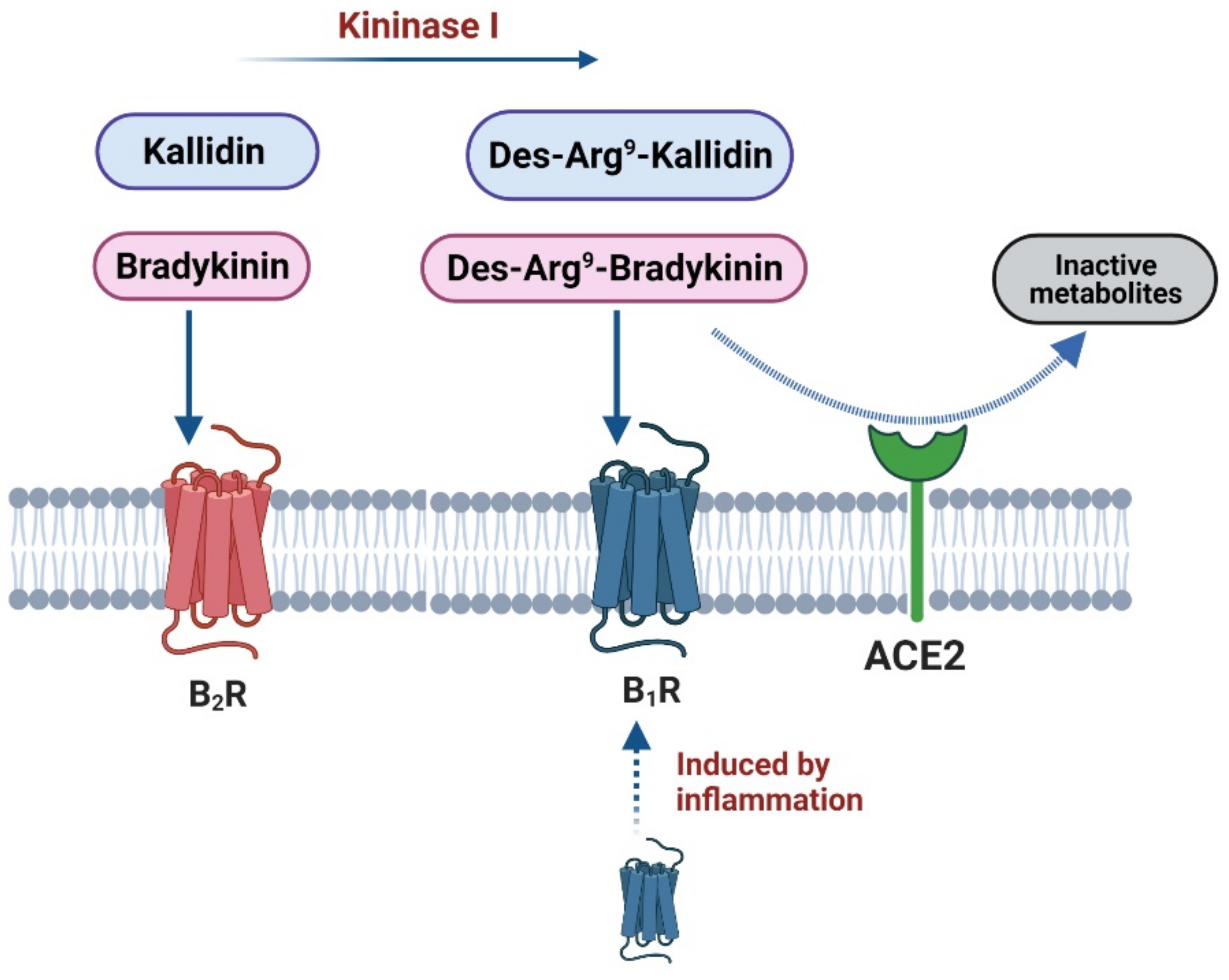
3. Bradykinin Signalling: The Kinin–Kallikrein System
4. Cytokine Storm
5. Bradykinin Storm
6. RAAS Involvement in Severe COVID-19 in Patients with Co-Morbidities
6.1. Thromboembolism
6.2. Hypertension
6.3. Cardiovascular Disease
6.4. Diabetes
7. Long COVID
8. Therapeutic Potential
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin converting enzyme 2 |
| ACEi | angiotensin converting enzyme inhibitor |
| Ang II | angiotensin II |
| AP | aminopeptidases |
| ARB | angiotensin receptor blockers |
| ARDS | acute respiratory distress syndrome |
| ASA | aldosterone synthase antagonist |
| AT1R/AT2R | angiotensin type 1/2 receptor |
| B1R/B2R | bradykinin type 1/2 receptor |
| BP | blood pressure |
| BK | bradykinin |
| COVID-19 | coronavirus disease 2019 |
| CVCs | cardiovascular complications |
| CPM | carboxypeptidase M |
| DABK | des-Arg9-bradykinin |
| DAKD | des-Arg10-kallidin |
| DPPD-4 | dipeptidyl peptidase 4 |
| GPCRs | G protein-coupled receptors |
| ICU | intensive care unit |
| IFN | interferon |
| IL-6 | interleukin-6 |
| KD | kallidin |
| KKS | kinin–kallikrein system |
| NETs | neutrophil extracellular traps |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| TMPRSS2 | transmembrane protease serine 2 |
| TNFα | tumour necrosis factor α |
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/ (accessed on 30 June 2021).
- Esakandari, H.; Nabi-Afjadi, M.; Fakkari-Afjadi, J.; Farahmandian, N.; Miresmaeili, S.M.; Bahreini, E. A comprehensive review of COVID-19 characteristics. Biol. Proced. Online 2020, 22, 19. [Google Scholar] [CrossRef]
- Vellas, C.; Delobel, P.; de Souto Barreto, P.; Izopet, J. COVID-19, Virology and Geroscience: A Perspective. J. Nutr. Health Aging 2020, 24, 685–691. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered from Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265–1273. [Google Scholar] [CrossRef]
- Venkatesan, P. NICE guideline on long COVID. Lancet Respir. Med. 2021, 9, 129. [Google Scholar] [CrossRef]
- Dennis, A.; Wamil, M.; Alberts, J.; Oben, J.; Cuthbertson, D.J.; Wootton, D.; Crooks, M.; Gabbay, M.; Brady, M.; Hishmeh, L.; et al. COVERSCAN study investigators. Multiorgan impairment in low-risk individuals with post-COVID-19 syndrome: A prospective, community-based study. BMJ Open 2021, 11, e048391. [Google Scholar]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrou, I.; Randeva, H.S.; Spandidos, D.A.; Karteris, E. Not only ACE2-the quest for additional host cell mediators of SARS-CoV-2 infection: Neuropilin-1 (NRP1) as a novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct. Target Ther. 2021, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.A.; Roche, R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. FASEB J. 2020, 34, 7265–7269. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Pawlowski, C.; Lenehan, P.; Puranik, A.; Agarwal, V.; Venkatakrishnan, A.J.; Niesen, M.J.M.; O’horo, J.C.; Badley, A.D.; Halamka, J.; Soundararajan, V. FDA-authorized COVID-19 vaccines are effective per real-world evidence synthesized across a multi-state health system. medRxiv 2021. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, L.; Huang, L.; Zhang, C.; Luo, R.; Zeng, L.; Liang, H.; Li, Q.; Lu, X.; Wang, X.; et al. Distinct disease severity between children and older adults with COVID-19: Impacts of ACE2 expression, distribution, and lung progenitor cells. Clin. Infect. Dis. 2021, 3, ciaa1911. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.R.; Ausiello, J. Functional ACE2 deficiency leading to angiotensin imbalance in the pathophysiology of COVID-19. Rev. Endocr. Metab. Disord. 2021, in press. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.A.; Valenzuela, R.; Garrido-Gil, P.; Labandeira, C.M.; Navarro, G.; Franco, R.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. Experimental data using candesartan and captopril indicate no double-edged sword effect in COVID-19. Clin. Sci. 2021, 135, 465–481. [Google Scholar] [CrossRef]
- Sui, Y.; Li, J.; Venzon, D.J.; Berzofsky, J.A. SARS-CoV-2 Spike Protein Suppresses ACE2 and Type I Interferon Expression in Primary Cells from Macaque Lung Bronchoalveolar Lavage. Front. Immunol. 2021, 12, 658428. [Google Scholar] [CrossRef]
- Corey, K.E.; Shah, N.; Misdraji, J.; Abu Dayyeh, B.K.; Zheng, H.; Bhan, A.K.; Chung, R.T. The effect of angiotensin-blocking agents on liver fibrosis in patients with hepatitis C. Liver Int. 2009, 29, 748–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasa, S.; Burdo, T.H.; Williams, K.C.; Mitten, E.K.; Wong, K.; Fitch, K.V.; Stanley, T.; Adler, G.K.; Grinspoon, S.K. Effects of Sodium Restriction on Activation of the Renin-Angiotensin-Aldosterone System and Immune Indices During HIV Infection. J. Infect. Dis. 2016, 214, 1336–1340. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, S.; Zhou, G.; Jin, Y.; Zhang, R.; Yang, H.; Xi, Y.; Ren, J.; Duan, G. Involvement of the renin-angiotensin system in the progression of severe hand-foot-and-mouth disease. PLoS ONE 2018, 13, e0197861. [Google Scholar]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56, 2000912. [Google Scholar] [CrossRef] [PubMed]
- Crajoinas, R.O.; Polidoro, J.Z.; Carneiro de Morais, C.P.; Castelo-Branco, R.C.; Girardi, A.C. Angiotensin II counteracts the effects of cAMP/PKA on NHE3 activity and phosphorylation in proximal tubule cells. Am. J. Physiol. Cell Physiol. 2016, 311, C768–C776. [Google Scholar] [CrossRef] [Green Version]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A. Renin release. Physiology 2007, 22, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Kobori, H. Independent regulation of renin-angiotensin-aldosterone system in the kidney. Clin. Exp. Nephrol. 2018, 22, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Emdin, M.; Fatini, C.; Mirizzi, G.; Poletti, R.; Borrelli, C.; Prontera, C.; Latini, R.; Passino, C.; Clerico, A.; Vergaro, G. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: How useful, how feasible? Clin. Chim. Acta 2015, 443, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cure, E.; Ilcol, T.B.; Cumhur Cure, M. Angiotensin II, III, and IV may be important in the progression of COVID-19. J. Renin Angiotensin Aldosterone Syst. 2020, 21, 1470320320972019. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Gαq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Cong, H.; Li, X.; Ma, L.; Jiang, H.; Mao, Y.; Xu, M. Angiotensin II receptor type 1 is upregulated in atrial tissue of patients with rheumatic valvular disease with atrial fibrillation. J. Thorac. Cardiovasc. Surg. 2010, 140, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Coromilas, E.J.; Kochav, S.; Goldenthal, I.; Biviano, A.; Garan, H.; Goldbarg, S.; Kim, J.-H.; Yeo, I.; Tracy, C.; Ayanian, S.; et al. Worldwide Survey of COVID-19-Associated Arrhythmias. Circ. Arrhythmia Electrophysiol. 2021, 14, 285–295. [Google Scholar] [CrossRef]
- Giustino, G.; Croft, L.B.; Stefanini, G.G.; Bragato, R.; Silbiger, J.J.; Vicenzi, M.; Danilov, T.; Kukar, N.; Shaban, N.; Kini, A.; et al. Characterization of Myocardial Injury in Patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Cheng, W.H.; Hung, Y.; Lin, W.Y.; Chao, T.F.; Liao, J.N.; Lin, Y.J.; Lin, W.S.; Chen, Y.J.; Chen, S.A. Management of Atrial Fibrillation in COVID-19 Pandemic. Circ. J. 2020, 84, 1679–1685. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Namkung, Y.; LeGouill, C.; Kumar, S.; Cao, Y.; Teixeira, L.B.; Lukasheva, V.; Giubilaro, J.; Simões, S.C.; Longpré, J.M.; Devost, D.; et al. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 2018, 11, eaat1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porrello, E.R.; Delbridge, L.M.; Thomas, W.G. The angiotensin II type 2 (AT2) receptor: An enigmatic seven transmembrane receptor. Front. Biosci. 2009, 14, 958–972. [Google Scholar] [CrossRef] [Green Version]
- Asada, H.; Inoue, A.; Ngako Kadji, F.M.; Hirata, K.; Shiimura, Y.; Im, D.; Shimamura, T.; Nomura, N.; Iwanari, H.; Hamakubo, T.; et al. The Crystal Structure of Angiotensin II Type 2 Receptor with Endogenous Peptide Hormone. Structure 2020, 28, 418–425.e4. [Google Scholar] [CrossRef] [PubMed]
- Turu, G.; Szidonya, L.; Gáborik, Z.; Buday, L.; Spät, A.; Clark, A.J.; Hunyady, L. Differential beta-arrestin binding of AT1 and AT2 angiotensin receptors. FEBS Lett. 2006, 580, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; Abrao, E.P.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Santos, R.A.; Bradford, C.N.; Mecca, A.P.; Sumners, C.; Katovich, M.J.; Raizada, M.K. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension 2010, 55, 207–213. [Google Scholar] [CrossRef]
- Calixto, J.B.; Medeiros, R.; Fernandes, E.S.; Ferreira, J.; Cabrini, D.A.; Campos, M.M. Kinin B1 receptors: Key G-protein-coupled receptors and their role in inflammatory and painful processes. Br. J. Pharmacol. 2004, 143, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Onohara, N.; Maruyama, Y.; Tanabe, S.; Kobayashi, H.; Fukutomi, M.; Nagamatsu, Y.; Nishihara, N.; Inoue, R.; Sumimoto, H.; et al. Galpha12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J. Biol. Chem. 2005, 280, 23041–23047. [Google Scholar] [CrossRef] [Green Version]
- Shatanawi, A.; Romero, M.J.; Iddings, J.A.; Chandra, S.; Umapathy, N.S.; Verin, A.D.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 2011, 300, C1181–C1192. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Kuen, D.S.; Chung, Y.; Kurose, H.; Kim, S.G. Gα12/13 signaling in metabolic diseases. Exp. Mol. Med. 2020, 52, 896–910. [Google Scholar] [CrossRef]
- Nanba, K.; Vaidya, A.; Rainey, W.E. Aging and Adrenal Aldosterone Production. Hypertension 2018, 71, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Maning, J.; Negussie, S.; Clark, M.A.; Lymperopoulos, A. Biased agonism/antagonism at the AngII-AT1 receptor: Implications for adrenal aldosterone production and cardiovascular therapy. Pharmacol. Res. 2017, 125, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Burrello, J.; Tizzani, D.; Bertello, C.; Viola, A.; Buffolo, F.; Gabetti, L.; Mengozzi, G.; Williams, T.A.; Rabbia, F.; et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J. Am. Coll. Cardiol. 2017, 69, 1811–1820. [Google Scholar] [CrossRef]
- Funder, J.W. Aldosterone and Mineralocorticoid Receptors-Physiology and Pathophysiology. Int. J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, X.H.; Yuan, H. Angiotensin II type-2 receptor-specific effects on the cardiovascular system. Cardiovasc. Diagn. Ther. 2012, 2, 56–62. [Google Scholar]
- Kaschina, E.; Namsolleck, P.; Unger, T. AT2 receptors in cardiovascular and renal diseases. Pharmacol. Res. 2017, 125, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Kemp, B.A.; Howell, N.L.; Keller, S.R.; Gildea, J.J.; Padia, S.H.; Carey, R.M. AT2 Receptor Activation Prevents Sodium Retention and Reduces Blood Pressure in Angiotensin II-Dependent Hypertension. Circ. Res. 2016, 119, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, L.; Novoa, U.; Moya, J.; Gabrielli, L.; Jalil, J.E.; García, L.; Chiong, M.; Lavandero, S.; Ocaranza, M.P. Angiotensin-(1-9) reduces cardiovascular and renal inflammation in experimental renin-independent hypertension. Biochem. Pharmacol. 2018, 156, 357–370. [Google Scholar] [CrossRef]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef]
- Mercure, C.; Yogi, A.; Callera, G.E.; Aranha, A.B.; Bader, M.; Ferreira, A.J.; Santos, R.A.; Walther, T.; Touyz, R.M.; Reudelhuber, T.L. Angiotensin (1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ. Res. 2008, 103, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.S.; Yeoh, S.F.; Long, C.M. COVID-19: Critical Role of Angiotensin 1-7 in ACE2 Modulation. Ann. Acad. Med. Singap. 2020, 49, 398–400. [Google Scholar] [CrossRef]
- Gaidarov, I.; Adams, J.; Frazer, J.; Anthony, T.; Chen, X.; Gatlin, J.; Semple, G.; Unett, D.J. Angiotensin (1-7) does not interact directly with MAS1, but can potently antagonize signaling from the AT1 receptor. Cell Signal. 2018, 50, 9–24. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.T.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177. [Google Scholar] [CrossRef] [PubMed]
- Ancion, A.; Tridetti, J.; Nguyen Trung, M.L.; Oury, C.; Lancellotti, P. A Review of the Role of Bradykinin and Nitric Oxide in the Cardioprotective Action of Angiotensin-Converting Enzyme Inhibitors: Focus on Perindopril. Cardiol. Ther. 2019, 8, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Abbasi, J. Researchers investigate what COVID-19 does to the heart. JAMA 2021, 325, 808–811. [Google Scholar] [CrossRef]
- Alhenc-Gelas, F.; Bouby, N.; Girolami, J.-P. Kallikrein/K1, Kinins, and ACE/Kininase II in Homeostasis and in disease insight from human and experimental genetic studies, therapeutic Implication. Front. Med. 2019, 6, 136. [Google Scholar] [CrossRef] [Green Version]
- Ceconi, C.; Francolini, G.; Olivares, A.; Comini, L.; Bachetti, T.; Ferrari, R. Angiotensin-converting enzyme (ACE) inhibitors have different selectivity for bradykinin binding sites of human somatic ACE. Eur. J. Pharmacol. 2007, 577, 1–6. [Google Scholar] [CrossRef]
- Pyne, S.; Pyne, N.J. Differential effects of B2 receptor antagonists upon bradykinin-stimulated phospholipase C and D in guinea-pig cultured tracheal smooth muscle. Br. J. Pharmacol. 1993, 110, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Radonjic-Hoesli, S.; Hofmeier, K.S.; Micaletto, S.; Micaletto, S.; Schmid-Grendelmeier, P.; Bircher, A.; Simon, D. Urticaria and Angioedema: An Update on Classification and Pathogenesis. Clin. Rev. Allerg. Immunol. 2018, 54, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.M.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4+ T Cells in COVID-19. Cell 2020, 183, 1340–1353. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Il-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The role of interleukin 6 during viral infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. NF-κB pathway as a potential target for treatment of critical stage COVID-19 patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.C.; Shao, S.C.; Cheng, C.W.; Chen, Y.C.; Hung, M.J. Incidence rate and clinical impacts of arrhythmia following COVID-19: A systematic review and meta-analysis of 17,435 patients. Crit. Care 2020, 24, 690. [Google Scholar] [CrossRef]
- Tse, G.; Yeo, J.M.; Chan, Y.W.; Lai, E.T.H.L.; Yan, B.P. What Is the Arrhythmic Substrate in Viral Myocarditis? Insights from Clinical and Animal Studies. Front. Physiol. 2016, 7, 308. [Google Scholar] [CrossRef] [Green Version]
- Alexander, L.K.; Keene, B.W.; Small, J.D.; Yount, B.; Baric, R.S. Electrocardiographic changes following rabbit coronavirus-induced myocarditis and dilated cardiomyopathy. Adv. Exp. Med. Biol. 1993, 342, 365–370. [Google Scholar] [PubMed] [Green Version]
- Hertanto, D.M.; Sutanto, H.; Wiratama, B.S.; Wungu, C.D.K. Modulating the host immune response to fight against COVID-19: Where are we in 2021? Virulence 2021, 12, 1732–1736. [Google Scholar] [CrossRef]
- Perrone, F.; Piccirillo, M.C.; Ascierto, P.A.; Salvarani, C.; Parrella, R.; Marata, A.M.; Popoli, P.; Ferraris, L.; Marrocco-Trischitta, M.M.; Ripamonti, D.; et al. TOCIVID-19 investigators, Italy. Tocilizumab for patients with COVID-19 pneumonia. The single-arm TOCIVID-19 prospective trial. J. Transl. Med. 2020, 18, 405. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Thakur, S.S. ACE2 as a potential therapeutic target for pandemic COVID-19. RSC Adv. 2020, 10, 39808–39813. [Google Scholar] [CrossRef]
- Campana, P.; Flocco, V.; Aruta, F.; Cacciatore, F.; Abete, P. Can aldosterone increase interleukin-6 levels in COVID-19 pneumonia? J. Med. Virol. 2021, 93, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, R.; Manetti, M.; Rosa, I.; Romano, E.; Galluccio, F.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Angiotensin II type 2 receptor (AT2R) as a novel modulator of inflammation in rheumatoid arthritis synovium. Sci. Rep. 2017, 7, 13293. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Cossa, L.G.; Vetrugno, C.; Marsigliante, S. Bradykinin stimulates prostaglandin E2 release in human skeletal muscular fibroblasts. Mol. Cell. Endocrinol. 2020, 507, 110771. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, C.; Terlizzi, M.; Pinto, A.; Sorrentino, R. A lesson from a saboteur: High-MW kininogen impact in coronavirus-induced disease 2019. Br. J. Pharmacol. 2020, 177, 4866–4872. [Google Scholar] [CrossRef]
- Riedl, M.A.; Maurer, M.; Bernstein, J.A.; Banerji, A.; Longhurst, H.J.; Li, H.H.; Lu, P.; Hao, J.; Juethner, S.; Lumry, W.R.; et al. Lanadelumab demonstrates rapid and sustained prevention of hereditary angioedema attacks. Allergy 2020, 75, 2879–2887. [Google Scholar] [CrossRef]
- Byrd, J.B.; Touzin, K.; Sile, S.; Gainer, J.V.; Yu, C.; Nadeau, J.; Adam, A.; Brown, N.J. Dipeptidyl peptidase IV in angiotensin-converting enzyme inhibitor-associated angioedema. Hypertension 2008, 51, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Ulbricht, D.; Tindall, C.A.; Oertwig, K.; Hanke, S.; Sträter, N.; Heiker, J.T. Kallikrein-related peptidase 14 is the second KLK protease targeted by the serpin vaspin. Biol. Chem. 2018, 399, 1079–1084. [Google Scholar] [CrossRef]
- Van de Veerdonk, F.; Janssen, N.; Grondman, I.; de Nooijer, A.; Koeken, V.; Matzaraki, V.; Boahen, C.; Kumar, V.; Kox, M.; Koenen, H.; et al. A systems approach to inflammation identifies therapeutic targets in SARS-CoV-2 infection. medRxiv 2020, 2, 100166. [Google Scholar]
- Dawson, P.; Rabold, E.M.; Laws, R.L.; Conners, E.E.; Gharpure, R.; Yin, S.; Buono, S.A.; Dasu, T.; Bhattacharyya, S.; Westergaard, R.P.; et al. Loss of Taste and Smell as Distinguishing Symptoms of Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 72, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients with COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the Coronavirus Disease 2019 (COVID-19) outbreak in China: Summary of a report of 72,314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef]
- Verdery, A.M.; Newmyer, L.; Wagner, B.; Margolis, R. National Profiles of Coronavirus Disease 2019 Mortality Risks by Age Structure and Pre-existing Health Conditions. Gerontologist 2021, 61, 71–77. [Google Scholar] [CrossRef]
- Giannis, D.; Barish, M.A.; Goldin, M.; Cohen, S.L.; Kohn, N.; Gianos, E.; Chatterjee, S.; Lesser, M.; Coppa, K.; Hirsch, J.S.; et al. Incidence of Venous Thromboembolism and Mortality in Patients with Initial Presentation of COVID-19. J. Thromb. Thrombolysis 2021, 51, 897–901. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Der Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Gandet, F.F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Jenner, W.J.; Kanji, R.; Mirsadraee, S.; Gue, Y.X.; Price, S.; Prasad, S.; Gorog, D.A. Thrombotic complications in 2928 patients with COVID-19 treated in intensive care: A systematic review. J. Thromb. Thrombolysis 2021, 51, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Ortega-Paz, L.; Capodanno, D.; Montalescot, G.; Angiolillo, D. COVID-19 Associated Thrombosis and Coagulopathy: Review of the Pathophysiology and Implications for Antithrombotic Management. J. Am. Heart Assoc. 2021, 10, e019650. [Google Scholar]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine storm associated coagulation complications in COVID-19 patients: Pathogenesis and Management. Expert Rev. Anti-Infect. Ther. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Page, E.M.; Ariëns, R.A.S. Mechanisms of thrombosis and cardiovascular complications in COVID-19. Thromb. Res. 2021, 200, 1–8. [Google Scholar] [CrossRef]
- Dmitrieva, N.I.; Burg, M.B. Elevated Sodium and Dehydration Stimulate Inflammatory Signaling in Endothelial Cells and Promote Atherosclerosis. PLoS ONE 2015, 10, e0128870. [Google Scholar] [CrossRef] [Green Version]
- McGonagle, D.; De Marco, G.; Bridgewood, C. Mechanisms of Immunothrombosis in Vaccine-Induced Thrombotic Thrombocytopenia (VITT) Compared to Natural SARS-CoV-2 Infection. J. Autoimmun. 2021, 121, 102662. [Google Scholar] [CrossRef]
- Billy, E.; Clarot, F.; Depagne, C.; Korsia-Meffre, S.; Rochoy, M.; Zores, F. Thrombotic events after AstraZeneca vaccine: What if it was related to dysfunctional immune response? Therapie 2021, in press. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. COVID-19 Lombardy ICU Network. Baseline Characteristics and Outcomes of 1591 Patients Infected with SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Grant, M.B.; Richards, E.M.; Raizada, M.K. ACE2 as therapeutic agent. Clin. Sci. 2020, 134, 2581–2595. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of COVID-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef]
- Bosso, M.; Thanaraj, T.A.; Abu-Farha, M.; Alanbaei, M.; Abubaker, J.; Al-Mulla, F. The Two Faces of ACE2: The Role of ACE2 Receptor and Its Polymorphisms in Hypertension and COVID-19. Mol. Ther. Methods Clin. Dev. 2020, 18, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Rolain, J.-M.; Raoult, D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. Immunol. Infect. 2020, 53, 425–435. [Google Scholar] [CrossRef]
- Hosseinzadeh, R.; Goharrizi, M.A.S.B.; Bahardoust, M.; Alvanegh, A.G.; Ataee, M.R.; Bagheri, M.; Navidiyan, E.S.; Zijoud, S.R.H.; Heiat, M. Should all patients with hypertension be worried about developing severe coronavirus disease 2019 (COVID-19)? Clin. Hypertens. 2021, 27, 3. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, S.E. Hypertension and cardiovascular risk: General aspects. Pharmacol. Res. 2018, 129, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Weiss, P.; Murdoch, D.R. Clinical course and mortality risk of severe COVID-19. Lancet 2020, 395, 1014–1015. [Google Scholar] [CrossRef]
- Fang, H.; Liu, Q.; Xi, M.; Xiong, D.; He, J.; Luo, P.; Li, Z.; Third, W.; Hospital, W.T. Impact of comorbidities on clinical prognosis in 1280 patients with different types of COVID-19. J. Investig. Med. 2021, 69, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Volpe, M.; Kreutz, R. Hypertension, a moving target in COVID-19: Current views and perspectives. Circ. Res. 2021, 128, 1062–1079. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, L.F.; Cheng, J.W.M.; Desai, A. Cardiovascular Pharmacology in the Time of COVID-19: A Focus on Angiotensin-Converting Enzyme 2. J. Cardiovasc. Pharmacol. 2020, 75, 526–529. [Google Scholar] [CrossRef]
- Kai, H.; Kai, M.; Niiyama, H.; Okina, N.; Sasaki, M.; Maeda, T.; Katoh, A. Overexpression of angiotensin-converting enzyme 2 by renin-angiotensin system inhibitors. Truth or myth? A systematic review of animal studies. Hypertens. Res. 2021, in press. [Google Scholar] [CrossRef]
- Kow, C.S.; Ming, L.C.; Hasan, S.S. Renin-angiotensin system inhibitor use and the risk of mortality in hospitalized patients with COVID-19: A meta-analysis of randomized controlled trials. Hypertens. Res. 2021, in press. [Google Scholar] [CrossRef]
- Bae, S.A.; Kim, S.R.; Kim, M.N.; Shim, W.J.; Park, S.M. Impact of cardiovascular disease and risk factors on fatal outcomes in patients with COVID-19 according to age: A systematic review and meta-analysis. Heart 2021, 107, 373–380. [Google Scholar] [CrossRef]
- Pranata, R.; Huang, I.; Lim, M.A.; Wahjoepramono, E.J.; July, J. Impact of cerebrovascular and cardiovascular diseases on mortality and severity of COVID-19–systematic review, meta-analysis, and meta-regression. J. Stroke Cerebrovasc. Dis. 2020, 29, 104949. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Benter, I.F.; Danjuma, M.I.; Doi, S.A.R.; Hasan, S.S.; Habib, A.M. Pharmacotherapy in COVID-19 patients: A review of ACE2-raising drugs and their clinical safety. J. Drug Target. 2020, 28, 683–699. [Google Scholar] [CrossRef]
- Dostal, D.E.; Hunt, R.A.; Kule, C.E.; Bhat, G.J.; Karoor, V.; McWhinney, C.D.; Baker, K.M. Molecular mechanisms of angiotensin II in modulating cardiac function: Intracardiac effects and signal transduction pathways. J. Mol. Cell Cardiol. 1997, 29, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Urbanek, K.; De Angelis, A.; Sessa, M.; Scavone, C.; Berrino, L.; Rosano, G.M.C.; Capuano, A.; Rossi, F. Angiotensin II and angiotensin 1-7: Which is their role in atrial fibrillation? Heart Fail. Rev. 2020, 25, 367–380. [Google Scholar] [CrossRef]
- Laurino, A.; Spinelli, V.; Gencarelli, M.; Balducci, V.; Dini, L.; Diolaiuti, L.; Ghionzoli, M.; Messineo, A.; Mugelli, A.; Cerbai, E.; et al. Angiotensin-II Drives Human Satellite Cells Toward Hypertrophy and Myofibroblast Trans-Differentiation by Two Independent Pathways. Int. J. Mol. Sci. 2019, 20, 4912. [Google Scholar] [CrossRef] [Green Version]
- Ceravolo, G.S.; Montezano, A.C.; Jordão, M.T.; Akamine, E.H.; Costa, T.J.; Takano, A.P.; Fernandes, D.C.; Barreto-Chaves, M.L.; Laurindo, F.R.; Tostes, R.C.; et al. An interaction of renin-angiotensin and kallikrein-kinin systems contributes to vascular hypertrophy in angiotensin II-induced hypertension: In vivo and in vitro studies. PLoS ONE 2014, 9, e111117110. [Google Scholar] [CrossRef]
- Skogestad, J.; Aronsen, J.M. Hypokalemia-induced arrhythmias and heart failure: New insights and implications for therapy. Front. Physiol. 2018, 9, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, G.; Pizzocaro, A.; Vena, W.; Rastrelli, G.; Semeraro, F.; Isidori, A.M.; Pivonello, R.; Salonia, A.; Sforza, A.; Maggi, M. Diabetes is most important cause for mortality in COVID-19 hospitalized patients: Systematic review and meta-analysis. Rev. Endocr. Metab. Disord. 2021, 22, 275–2968. [Google Scholar] [CrossRef]
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Azar, W.S.; Njeim, R.; Fares, A.H.; Azar, N.S.; Azar, S.T.; El Sayed, M.; Eid, A.A. COVID-19 and diabetes mellitus: How one pandemic worsens the other. Rev. Endocr. Metab. Disord. 2020, 21, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Campopiano, M.C.; Mantuano, M.; Mazoni, L.; Coppelli, A.; Del Prato, S. COVID-19 in people with diabetes: Understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol. 2020, 8, 782–792. [Google Scholar] [CrossRef]
- Alsadhan, I.; Alruwashid, S.; Alhamad, M.; Alajmi, S.; Alshehri, S.; Alfadhli, E.; Ekhzaimy, A. Diabetic ketoacidosis precipitated by Coronavirus disease 2019 infection: Case series. Curr. Ther. Res. Clin. Exp. 2020, 93, 100609. [Google Scholar] [CrossRef]
- Tang, X.; Uhl, S.; Zhang, T.; Xue, D.; Li, B.; Vandana, J.J.; Acklin, J.A.; Bonnycastle, L.L.; Narisu, N.; Erdos, M.R.; et al. SARS-CoV-2 infection induces beta cell transdifferentiation. Cell Metab. 2021, in press. [Google Scholar] [CrossRef]
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 Expression in Pancreas May Cause Pancreatic Damage after SARS-CoV-2 Infection. Clin. Gastroenterol. Hepatol. 2020, 18, 2128–2130. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Salvayre, R.; Augé, N.; Pamplona, R.; Portero-Otín, M. Hyperglycemia and Glycation in Diabetic Complications. Antioxid. Redox. Signal. 2009, 11, 3071–3109. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Egbuche, O.; Igwe, J.; Jegede, O.; Wagle, B.; Olanipekun, T.; Onwuanyi, A. Cardiovascular complications in COVID-19 patients with or without diabetes mellitus. Endocrinol. Diabetes Metab. 2021, 4, e00218. [Google Scholar] [CrossRef]
- Dennis, J.M.; McGovern, A.P.; Vollmer, S.J.; Mateen, B.A. Improving Survival of Critical Care Patients with Coronavirus Disease 2019 in England: A National Cohort Study, March to June 2020. Crit. Care Med. 2021, 49, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Sivan, M.; Rayner, C.; Delaney, B. Fresh evidence of the scale and scope of long COVID. BMJ 2021, 373, n853. [Google Scholar] [CrossRef]
- Halpin, S.J.; McIvor, C.; Whyatt, G.; Adams, A.; Harvey, O.; McLean, L.; Walshaw, C.; Kemp, S.; Corrado, J.; Singh, R.; et al. Post-discharge symptoms and rehabilitation needs in survivors of COVID-19 infection: A cross-sectional evaluation. J. Med. Virol. 2021, 93, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Patel, K.; Greenwood, D.C.; Halpin, S.; Lewthwaite, P.; Salawu, A.; Eyre, L.; Breen, A.; Jones, A.; Sivan, M. Long-term clinical outcomes in survivors of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) coronavirus outbreaks after hospitalisation or ICU admission: A systematic review and meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Ayoubkhani, D.; Khunti, K.; Nafilyan, V.; Maddox, T.; Humberstone, B.; Diamond, I.; Banerjee, A. Epidemiology of post-COVID syndrome following hospitalisation with coronavirus: A retrospective cohort study. BMJ 2021, 372, n693. [Google Scholar] [CrossRef]
- Dennis, A.; Wamil, M.; Kapur, S.; Alberts, J.; Badley, A.D.; Decker, G.A.; Rizza, S.A.; Banerjee, R.; Banerjee, A. Multi-organ impairment in low-risk individuals with long COVID. medRxiv 2020. [Google Scholar] [CrossRef]
- Ludvigsson, J.F. Case report and systematic review suggest that children may experience similar long-term effects to adults after clinical COVID-19. Acta Paediatr. Int. J. Paediatr. 2021, 110, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscha Steckelings, U.; Sumners, C. Correcting the imbalanced protective RAS in COVID-19 with angiotensin AT2-receptor agonists. Clin. Sci. 2020, 134, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Rathinasabapathy, A.; Horowitz, A.; Horton, K.; Kumar, A.; Gladson, S.; Unger, T.; Martinez, D.; Bedse, G.; West, J.; Raizada, M.K.; et al. The selective angiotensin II type 2 receptor agonist, compound 21, attenuates the progression of lung fibrosis and pulmonary hypertension in an experimental model of bleomycin-induced lung injury. Front. Physiol. 2018, 9, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namsolleck, P.; Moll, G.N. Does activation of the protective Renin-Angiotensin System have therapeutic potential in COVID-19? Mol. Med. 2020, 26, 80. [Google Scholar] [CrossRef]
- Hernández Prada, J.A.; Ferreira, A.J.; Katovich, M.J.; Shenoy, V.; Qi, Y.; Santos, R.A.S.; Castellano, R.K.; Lampkins, A.J.; Gubala, V.; Ostrov, D.A.; et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension 2008, 51, 1312–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prata, L.O.; Rodrigues, C.R.; Martins, J.M.; Vasconcelos, P.C.; Oliveira, F.M.S.; Ferreira, A.J.; Rodrigues-Machado, M.d.G.; Caliari, M.V. Original Research: ACE2 activator associated with physical exercise potentiates the reduction of pulmonary fibrosis. Exp. Biol. Med. 2017, 242, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Joshi, S.; Guo, L.; Baker, M.B.; Li, Y.; Castellano, R.K.; Raizada, M.K.; Jarajapu Yagna, Y.P.R. ACE2/Ang-(1-7)/Mas axis stimulates vascular repair-relevant functions of CD34+ cells. Am. J. Physiol. Hear. Circ. Physiol. 2015, 309, H1697–H1707. [Google Scholar] [CrossRef] [Green Version]
- Savergnini, S.Q.; Beiman, M.; Lautner, R.Q.; De Paula-Carvalho, V.; Allahdadi, K.; Caires Pessoa, D.; Pereira Costa-Fraga, F.; Araújo Fraga-Silva, R.; Cojocaru, G.; Cohen, Y.; et al. Vascular Relaxation, Antihypertensive Effect, and Cardioprotection of a Novel Peptide Agonist of the Mas Receptor. Hypertension 2010, 56, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Mansour, E.; Palma, A.C.; Ulaf, R.G.; Ribeiro, L.C.; Bernardes, A.F.; Nunes, T.A.; Agrela, M.V.; Bombassaro, B.; Monfort-Pires, M.; Camargo, R.L.; et al. Safety and outcomes associated with the pharmacological inhibition of the kinin–kallikrein system in severe COVID-19. Viruses 2021, 13, 309. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | G Protein Signalling | Cellular Actions | Physiological Response |
|---|---|---|---|
| AT1R | Gαq | DAG, PKC, ↑NO, ↑Ca2+, NHE3 activation | Vascular constriction, renal sodium retention (↑H+ secretion, ↑Na+ absorption), ↑ROS |
| Gαi2 Gαi3 | ↓cAMP, activates GIRKs | ↑parasympathetic pathways, ↓HR, ↓BP | |
| Gα12 βarrestin2 | Rho GTPase, TKs, NADPH oxidases Receptor desensitisation, internalisation | ↑actin stress fibres, ↑focal adhesions, ↑cell growth, ↑fibrosis, ↑hypertrophy Dampens AT1 physiological effects | |
| AT2R | Gαs Gai/0 Gαi2 Gαi3 Non-canonical | ↑cAMP ↑eNOS ↓TKs ↑BK/cGMP/NO | Muscle repair, vasorelaxation, ↑paracrine signalling |
| ↓IP3, ↑NOS, ↓Na+ATPase, ↓PLD ↓Rho | Inhibition of AT1 responses | ||
| MasR | Constitutively activates G proteins Gαq Gαi βarrestin2 | Exact role of ligand mediated G protein coupling not yet known. | NO-dependant vasorelaxation, protects endothelial function, ↓thrombosis ↓inflammation No measurable effect of Ca2+ |
| Receptor Internalisation, ↑ERK1/2, AKT, PLA2 ↓cAMP | Attenuates Ang((1-7)-)mediated activity at | ||
| B1 receptor | Gαq Gαi | PLC, AKT, iNOS, ↑NO, ↑Ca2+ ↓cAMP, activates GIRKs, PLA | Vasorelaxation Release of arachidonic acid and prostaglandins Sustained activation-long term inflammation |
| B2 receptor | Gαq Gαi βarrestin | PLC, AKT, iNOS, ↑NO, ↑Ca2+ ↓cAMP, activates GIRKs, PLA Internalisation/receptor recycling | Vasorelaxation Release of arachidonic acid and prostaglandins Desensitisation- short term effects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, S.L.; Boyle, E.; Jefferson, S.R.; Heslop, C.R.A.; Mohan, P.; Mohanraj, G.G.J.; Sidow, H.A.; Tan, R.C.P.; Hill, S.J.; Woolard, J. Role of the Renin–Angiotensin–Aldosterone and Kinin–Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. Int. J. Mol. Sci. 2021, 22, 8255. https://doi.org/10.3390/ijms22158255
Cooper SL, Boyle E, Jefferson SR, Heslop CRA, Mohan P, Mohanraj GGJ, Sidow HA, Tan RCP, Hill SJ, Woolard J. Role of the Renin–Angiotensin–Aldosterone and Kinin–Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. International Journal of Molecular Sciences. 2021; 22(15):8255. https://doi.org/10.3390/ijms22158255
Chicago/Turabian StyleCooper, Samantha L., Eleanor Boyle, Sophie R. Jefferson, Calum R. A. Heslop, Pirathini Mohan, Gearry G. J. Mohanraj, Hamza A. Sidow, Rory C. P. Tan, Stephen J. Hill, and Jeanette Woolard. 2021. "Role of the Renin–Angiotensin–Aldosterone and Kinin–Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID" International Journal of Molecular Sciences 22, no. 15: 8255. https://doi.org/10.3390/ijms22158255
APA StyleCooper, S. L., Boyle, E., Jefferson, S. R., Heslop, C. R. A., Mohan, P., Mohanraj, G. G. J., Sidow, H. A., Tan, R. C. P., Hill, S. J., & Woolard, J. (2021). Role of the Renin–Angiotensin–Aldosterone and Kinin–Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. International Journal of Molecular Sciences, 22(15), 8255. https://doi.org/10.3390/ijms22158255