
New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation

{kind=link}
{kind=link}
Abstract
1. Introduction
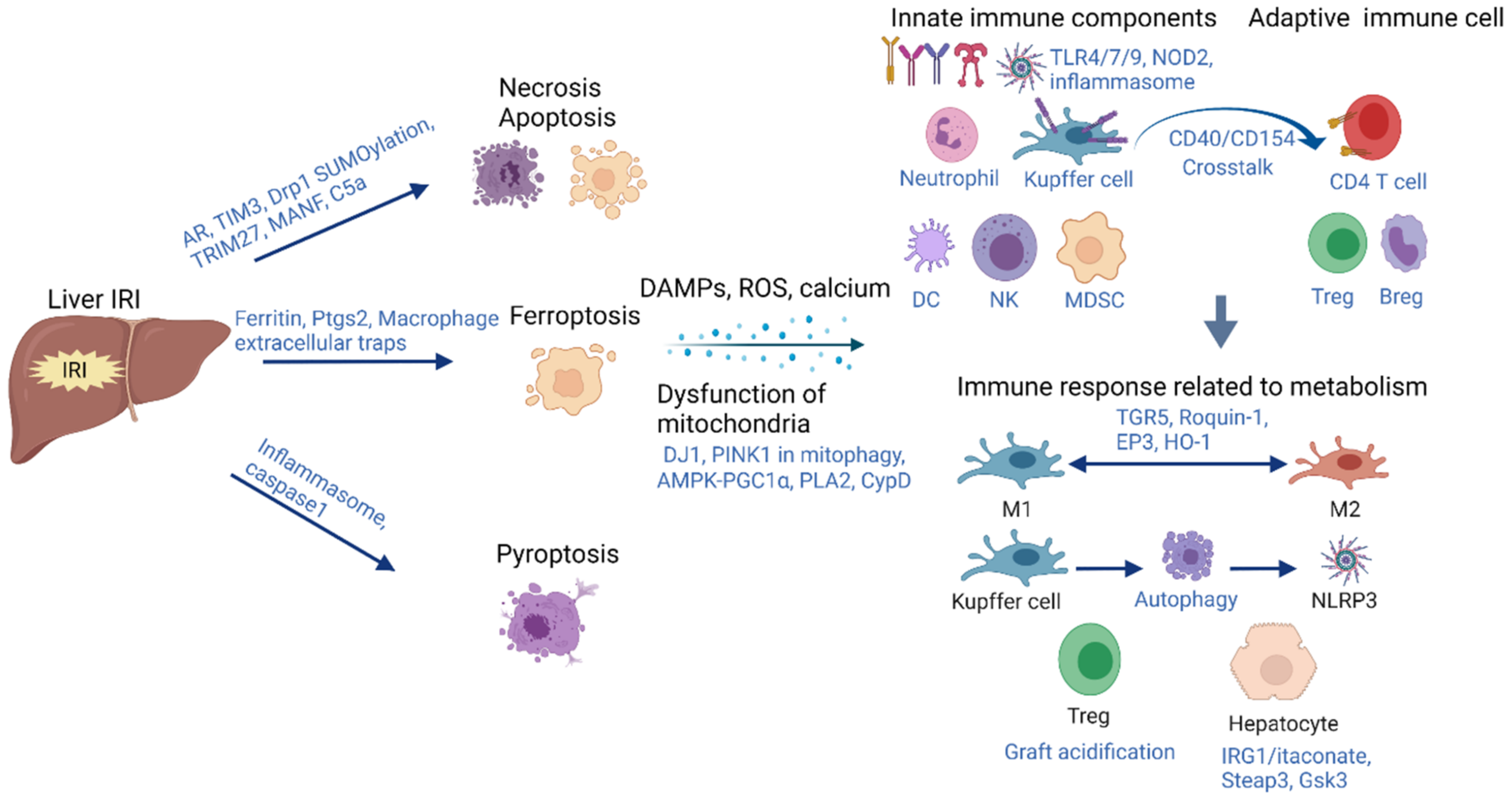
2. Acute-Phase Inflammation Initiated by Hepatic IRI
2.1. Cell Death
2.1.1. Apoptosis and Necrosis
2.1.2. Ferroptosis
2.1.3. Pytoptosis
2.2. Dysfunction of Mitochondria and Metabolism Alteration during Hepatic IRI
2.2.1. Mitophagy
2.2.2. Permeability Alteration of Mitochondria
2.2.3. Metabolic Alteration in IRI of Steatotic Liver
2.3. Immune Responses Associated with Metabolic Alteration during Hepatic IRI
2.3.1. Kupffer Cells and Tregs
2.3.2. Hepatocytes
3. Long-Term Impact of Inflammation Induced by Liver IRI
3.1. Cancer Recurrence
3.2. Fibrosis
4. Therapeutic Strategies to Reduce Both Short- and Long-Term Impacts of Inflammation in Hepatic IRI
4.1. Ischemic Preconditioning
4.2. Pharmacological Inhibitors
4.3. Machine Perfusion
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38, 2–6. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Czigany, Z.; Lurje, I.; Schmelzle, M.; Schöning, W.; Öllinger, R.; Raschzok, N.; Sauer, I.M.; Tacke, F.; Strnad, P.; Trautwein, C. Ischemia-reperfusion injury in marginal liver grafts and the role of hypothermic machine perfusion: Molecular mechanisms and clinical implications. J. Clin. Med. 2020, 9, 846. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4–dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Zhai, Y.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Liver Ischemia and Reperfusion Injury: New Insights into Mechanisms of Innate—Adaptive Immune-Mediated Tissue Inflammation. Am. J. Transplant. 2011, 11, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Piña, E.; Geller, D.A. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Bentley, R.C.; Madden, J.F.; Clavien, P.A. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology 1998, 27, 1652–1660. [Google Scholar] [CrossRef]
- Kohli, V.; Selzner, M.; Madden, J.F.; Bentley, R.C.; Clavien, P.-A. Endothelial Cell and Hepatocyte Deaths Occur by Apoptosis After Ischemia-reperfusion Injury In The Rat Liver1, 2. Transplantation 1999, 67, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.V. Necrapoptosis and the mitochondrial permeability transition: Shared pathways to necrosis and apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 1999, 276, G1–G6. [Google Scholar] [CrossRef]
- Li, C.X.; Ng, K.T.-P.; Shao, Y.; Liu, X.B.; Ling, C.C.; Ma, Y.Y.; Geng, W.; Qi, X.; Cheng, Q.; Chung, S.K. The inhibition of aldose reductase attenuates hepatic ischemia-reperfusion injury through reducing inflammatory response. Ann. Surg. 2014, 260, 317–328. [Google Scholar] [CrossRef]
- Fujii, T.; Duarte, S.; Lee, E.; Ke, B.; Busuttil, R.W.; Coito, A.J. Tissue Inhibitor of Metalloproteinase 3 Deficiency Disrupts the Hepatocyte E-Cadherin/β-Catenin Complex and Induces Cell Death in Liver Ischemia/Reperfusion Injury. Liver Transpl. 2020, 26, 113–126. [Google Scholar] [CrossRef]
- Huang, J.; Xie, P.; Dong, Y.; An, W. Inhibition of Drp1 SUMOylation by ALR protects the liver from ischemia-reperfusion injury. Cell Death Differ. 2021, 28, 1174–1192. [Google Scholar] [CrossRef]
- Chen, S.Y.; Zhang, H.P.; Li, J.; Shi, J.H.; Tang, H.W.; Zhang, Y.; Zhang, J.K.; Wen, P.H.; Wang, Z.H.; Shi, X.Y.; et al. Tripartite Motif-Containing 27 Attenuates Liver Ischemia/Reperfusion Injury by Suppressing Transforming Growth Factor beta-Activated Kinase 1 (TAK1) by TAK1 Binding Protein 2/3 Degradation. Hepatology 2021, 73, 738–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, P.; Zhang, C.; Huang, F.; Pang, G.; Wei, C.; Lv, C.; Chhetri, G.; Jiang, T.; Liu, J. Hepatocyte-derived MANF alleviates hepatic ischaemia-reperfusion injury via regulating endoplasmic reticulum stress-induced apoptosis in mice. Liver Int. 2021, 41, 623–639. [Google Scholar] [CrossRef]
- Kusakabe, J.; Hata, K.; Tamaki, I.; Tajima, T.; Miyauchi, H.; Wang, Y.; Nigmet, Y.; Okamura, Y.; Kubota, T.; Tanaka, H. Complement 5 inhibition ameliorates hepatic ischemia/reperfusion injury in mice, dominantly via the C5a-mediated cascade. Transplantation 2020, 104, 2065–2077. [Google Scholar] [CrossRef] [PubMed]
- Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’h, K. Ferroptosis in liver diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage Extracellular Traps Aggravate Iron Overload-Related Liver Ischemia/Reperfusion Injury. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Fagenson, A.M.; Xu, K.; Saaoud, F.; Nanayakkara, G.; Jhala, N.C.; Liu, L.; Drummer, C.; Sun, Y.; Lau, K.N.; Di Carlo, A. Liver Ischemia Reperfusion Injury, Enhanced by Trained Immunity, Is Attenuated in Caspase 1/Caspase 11 Double Gene Knockout Mice. Pathogens 2020, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.L.; Lopez, C.; Shen, M.; Shayakhmetov, D.; Gupta, N.A. Ischemia reperfusion injury induces pyroptosis and mediates injury in steatotic liver thorough Caspase 1 activation. Apoptosis 2021, 26, 361–370. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.; Jia, L.; Lyu, J.; Sun, Y.; Yu, H.; Li, H.; Liu, W.; Weng, Y.; Yu, W. Exosomes mediate hippocampal and cortical neuronal injury induced by hepatic ischemia-reperfusion injury through activating pyroptosis in rats. Oxid. Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef]
- Liu, H.; Lo, C.M.; Yeung, O.W.H.; Li, C.X.; Liu, X.B.; Qi, X.; Ng, K.T.P.; Liu, J.; Ma, Y.Y.; Lam, Y.F. NLRP3 inflammasome induced liver graft injury through activation of telomere-independent RAP1/KC axis. J. Pathol. 2017, 242, 284–296. [Google Scholar] [CrossRef]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [PubMed]
- Videla, L.A.; Fernández, V. Biochemical aspects of cellular oxidative stress. Arch. Biol. Med. Exp. 1988, 21, 85–92. [Google Scholar] [PubMed]
- Teoh, N.C.; Farrell, G.C. Hepatic ischemia reperfusion injury: Pathogenic mechanisms and basis for hepatoprotection. J. Gastroenterol. Hepatol. 2003, 18, 891–902. [Google Scholar] [CrossRef]
- Xu, M.; Hang, H.; Huang, M.; Li, J.; Xu, D.; Jiao, J.; Wang, F.; Wu, H.; Sun, X.; Gu, J. DJ-1 deficiency in hepatocytes improves liver ischemia-reperfusion injury by enhancing mitophagy. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, L.; Lu, T.; Zhang, Y.; Sui, X.; Li, Y.; Huang, X.; He, L.; Cai, J.; Zhou, C. MSCs ameliorate hepatocellular apoptosis mediated by PINK1-dependent mitophagy in liver ischemia/reperfusion injury through AMPKα activation. Cell Death Dis. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Liu, J.; Pang, L.; Ng, K.T.; Chiu, T.S.; Liu, H.; Liu, X.; Xu, A.; Lo, C.-M.; Man, K. Compromised AMPK-PGC1α Axis Exacerbated Steatotic Graft Injury by Dysregulating Mitochondrial Homeostasis in Living Donor Liver Transplantation. Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Halestrap, A. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.D.; Pierce, J.; Nicoud, I.; Belous, A.; Knox, C.D.; Chari, R.S. Modulation of mitochondrial calcium management attenuates hepatic warm ischemia-reperfusion injury. Liver Transpl. 2005, 11, 663–668. [Google Scholar] [CrossRef]
- Upadhya, G.A.; Topp, S.A.; Hotchkiss, R.S.; Anagli, J.; Strasberg, S.M. Effect of cold preservation on intracellular calcium concentration and calpain activity in rat sinusoidal endothelial cells. Hepatology 2003, 37, 313–323. [Google Scholar] [CrossRef]
- Oliva-Vilarnau, N.; Hankeova, S.; Vorrink, S.U.; Mkrtchian, S.; Andersson, E.R.; Lauschke, V.M. Calcium signaling in liver injury and regeneration. Front. Med. 2018, 5, 192. [Google Scholar] [CrossRef]
- Gu, J.; Zhang, T.; Guo, J.; Chen, K.; Wang, G.; Li, H.; Wang, J. Ursodeoxycholyl lysophosphatidylethanolamide protects against hepatic ischemia/reperfusion injury via phospholipid metabolism-mediated mitochondrial quality control. FASEB J. 2020, 34, 6198–6214. [Google Scholar] [CrossRef]
- Panel, M.; Ruiz, I.; Brillet, R.; Lafdil, F.; Teixeira-Clerc, F.; Nguyen, C.T.; Calderaro, J.; Gelin, M.; Allemand, F.; Guichou, J.-F. Small-molecule inhibitors of cyclophilins block opening of the mitochondrial permeability transition pore and protect mice from hepatic ischemia/reperfusion injury. Gastroenterology 2019, 157, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Kreuzer, J.; Kumsta, C.; Wu, L.; Kamer, K.J.; Cedillo, L.; Zhang, Y.; Li, S.; Kacergis, M.C.; Webster, C.M. Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell 2019, 177, 299–314.e16. [Google Scholar] [CrossRef] [PubMed]
- Linares, I.; Hamar, M.; Selzner, N.; Selzner, M. Steatosis in liver transplantation: Current limitations and future strategies. Transplantation 2019, 103, 78–90. [Google Scholar] [CrossRef]
- D’Alessandro, A.M.; Kalayoglu, M.; Sollinger, H.W.; Hoffmann, R.M.; Reed, A.; Knechtle, S.J.; Pirsch, J.D.; Hafez, G.R.; Lorentzen, D.; Belzer, F.O. The predictive value of donor liver biopsies for the development of primary nonfunction after orthotopic liver transplantation. Transplantation 1991, 51, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Reynes, M.; Johann, M.; Morino, M.; Astarcioglu, I. The outcome of steatotic grafts in liver transplantation. Transplant Proc 1991, 1 Pt 2, 1538–1540. [Google Scholar]
- Nocito, A.; El-Badry, A.M.; Clavien, P.-A. When is steatosis too much for transplantation? J. Hepatol. 2006, 45, 494–499. [Google Scholar] [CrossRef]
- Chu, M.J.; Hickey, A.J.; Phillips, A.R.; Bartlett, A.S. The impact of hepatic steatosis on hepatic ischemia-reperfusion injury in experimental studies: A systematic review. BioMed Res. Int. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic fatty liver disease: A challenge from mechanisms to therapy. J. Clin. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Ng, K.T.; Fan, S.T.; Lim, Z.X.; Guo, D.Y.; Liu, X.B.; Liu, Y.; Poon, R.T.; Lo, C.M.; Man, K. Distinct mechanism of small-for-size fatty liver graft injury--Wnt4 signaling activates hepatic stellate cells. Am. J. Transpl. 2010, 10, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Ng, K.T.; Xu, A.; Li, C.X.; Liu, X.B.; Guo, D.Y.; Poon, R.T.; Fan, S.T.; Lo, C.M.; Man, K. The roles of lipocalin-2 in small-for-size fatty liver graft injury. Ann. Surg. 2014, 260, 1062–1072. [Google Scholar] [CrossRef]
- Li, C.X.; Lo, C.M.; Lian, Q.; Ng, K.T.-P.; Liu, X.B.; Ma, Y.Y.; Qi, X.; Yeung, O.W.H.; Tergaonkar, V.; Yang, X.X. Repressor and activator protein accelerates hepatic ischemia reperfusion injury by promoting neutrophil inflammatory response. Oncotarget 2016, 7, 27711. [Google Scholar] [CrossRef]
- Yang, X.; Li, C.; Ng, K.T.P.; Liu, J.; Liu, H.; Zhang, W.; Xiao, F.; Li, X.; Lo, C.M.; Lu, L. IL-17a exacerbates hepatic ischemia–reperfusion injury in fatty liver by promoting neutrophil infiltration and mitochondria-driven apoptosis. J. Leukoc. Biol. 2020, 108, 1603–1613. [Google Scholar] [CrossRef]
- Sosa, R.A.; Rossetti, M.; Naini, B.V.; Groysberg, V.M.; Kaldas, F.M.; Busuttil, R.W.; Chang, Y.-L.; Gjertson, D.W.; Kupiec-Weglinski, J.W.; Reed, E.F. Pattern recognition receptor-reactivity screening of liver transplant patients: Potential for personalized and precise organ matching to reduce risks of ischemia-reperfusion injury. Ann. Surg. 2020, 271, 922–931. [Google Scholar] [CrossRef]
- Han, S.J.; Kim, M.; Novitsky, E.; D’Agati, V.; Lee, H.T. Intestinal TLR9 deficiency exacerbates hepatic IR injury via altered intestinal inflammation and short-chain fatty acid synthesis. FASEB J. 2020, 34, 12083–12099. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, S.; Shi, Y.; Wang, Q.; Wei, S.; Wang, P.; Cheng, F.; Auwerx, J.; Schoonjans, K.; Lu, L. TGR5/Cathepsin E signaling regulates macrophage innate immune activation in liver ischemia and reperfusion injury. Am. J. Transplant. 2021, 21, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ling, W.; Zhu, D.; Li, Z.; Kong, L. Roquin-1 Regulates Macrophage Immune Response and Participates in Hepatic Ischemia–Reperfusion Injury. J. Immunol. 2020, 204, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, S.; Ito, Y.; Nishizawa, N.; Goto, T.; Kojo, K.; Kumamoto, Y.; Watanabe, M.; Narumiya, S.; Majima, M. EP3 signaling in dendritic cells promotes liver repair by inducing IL-13-mediated macrophage differentiation in mice. FASEB J. 2020, 34, 5610–5627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Nakamura, K.; Kageyama, S.; Lawal, A.O.; Gong, K.W.; Bhetraratana, M.; Fujii, T.; Sulaiman, D.; Hirao, H.; Bolisetty, S. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight 2018, 3, e120596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, S.; Chen, X.; Li, X.; Xia, N.; Pu, L. Eva1a inhibits NLRP3 activation to reduce liver ischemia-reperfusion injury via inducing autophagy in kupffer cells. Mol. Immunol. 2021, 132, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kageyama, S.; Kaldas, F.M.; Hirao, H.; Ito, T.; Kadono, K.; Dery, K.J.; Kojima, H.; Gjertson, D.W.; Sosa, R.A. Hepatic CEACAM1 expression indicates donor liver quality and prevents early transplantation injury. J. Clin. Investig. 2020, 130, 2689–2704. [Google Scholar] [CrossRef]
- Gan, X.; Zhang, R.; Gu, J.; Ju, Z.; Wu, X.; Wang, Q.; Peng, H.; Qiu, J.; Zhou, J.; Cheng, F. Acidic Microenvironment Regulates the Severity of Hepatic Ischemia/Reperfusion Injury by Modulating the Generation and Function of Tregs via the PI3K-mTOR Pathway. Front. Immunol. 2020, 10, 2945. [Google Scholar] [CrossRef]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W. Immune-Responsive Gene 1/Itaconate Activates Nuclear Factor Erythroid 2–Related Factor 2 in Hepatocytes to Protect Against Liver Ischemia–Reperfusion Injury. Hepatology 2020, 72, 1394–1411. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.Z.; Fang, H.B.; Cao, S.L.; Chen, S.Y.; Li, J.; Shi, J.H.; Tang, H.W.; Zhang, Y.; Wen, P.H.; Zhang, J.K. Six-Transmembrane Epithelial Antigen of the Prostate 3 Deficiency in Hepatocytes Protects the Liver Against Ischemia-Reperfusion Injury by Suppressing Transforming Growth Factor-β-Activated Kinase 1. Hepatology 2020, 71, 1037–1054. [Google Scholar] [CrossRef]
- Ni, M.; Zhou, H.; Zhang, J.; Jin, D.; Lu, T.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Wang, X.; Zhai, Y. Isoform-and Cell Type–Specific Roles of Glycogen Synthase Kinase 3 N-Terminal Serine Phosphorylation in Liver Ischemia Reperfusion Injury. J. Immunol. 2020, 205, 1147–1156. [Google Scholar] [CrossRef]
- Clavien, P.A.; Lesurtel, M.; Bossuyt, P.M.; Gores, G.J.; Langer, B.; Perrier, A.; OLT for HCC Consensus Group. Recommendations for liver transplantation for hepatocellular carcinoma: An international consensus conference report. Lancet Oncol. 2012, 13, e11–e22. [Google Scholar] [CrossRef]
- Man, K.; Ng, K.T.; Lo, C.M.; Ho, J.W.; Sun, B.S.; Sun, C.K.; Lee, T.K.; Poon, R.T.; Fan, S.T. Ischemia-reperfusion of small liver remnant promotes liver tumor growth and metastases—Activation of cell invasion and migration pathways. Liver Transpl. 2007, 13, 1669–1677. [Google Scholar] [CrossRef]
- Nagai, S.; Yoshida, A.; Facciuto, M.; Moonka, D.; Abouljoud, M.S.; Schwartz, M.E.; Florman, S.S. Ischemia time impacts recurrence of hepatocellular carcinoma after liver transplantation. Hepatology 2015, 61, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, C.-H.; Moeckli, B.; Berney, T.; Toso, C.; Orci, L.A. Shorter survival after liver pedicle clamping in patients undergoing liver resection for hepatocellular carcinoma revealed by a systematic review and meta-analysis. Cancers 2021, 13, 637. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Lo, C.-M.; Ng, I.O.-L.; Wong, Y.-C.; Qin, L.-F.; Fan, S.-T.; Wong, J. Liver transplantation in rats using small-for-size grafts: A study of hemodynamic and morphological changes. Arch. Surg. 2001, 136, 280–285. [Google Scholar] [CrossRef]
- Liang, T.B.; Man, K.; Kin-Wah Lee, T.; Hong-Teng Tsui, S.; Lo, C.M.; Xu, X.; Zheng, S.S.; Fan, S.T.; Wong, J. Distinct intragraft response pattern in relation to graft size in liver transplantation. Transplantation 2003, 75, 673–678. [Google Scholar] [CrossRef]
- Man, K.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Fung, P.C.; Liang, T.B.; Lee, T.K.; Tsui, S.H.; Ng, I.O.; Zhang, Z.W.; et al. Graft injury in relation to graft size in right lobe live donor liver transplantation: A study of hepatic sinusoidal injury in correlation with portal hemodynamics and intragraft gene expression. Ann. Surg. 2003, 237, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Lacotte, S.; Oldani, G.; Slits, F.; De Vito, C.; Crowe, L.; Rubbia-Brandt, L.; Vallée, J.-P.; Morel, P.; Toso, C. Effect of ischaemic preconditioning on recurrence of hepatocellular carcinoma in an experimental model of liver steatosis. J. Br. Surg. 2016, 103, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Lo, C.M.; Xiao, J.W.; Ng, K.T.; Sun, B.S.; Ng, I.O.; Cheng, Q.; Sun, C.K.; Fan, S.T. The significance of acute phase small-for-size graft injury on tumor growth and invasiveness after liver transplantation. Ann. Surg. 2008, 247, 1049–1057. [Google Scholar] [CrossRef]
- Man, K.; Shih, K.C.; Ng, K.T.; Xiao, J.W.; Guo, D.Y.; Sun, C.K.; Lim, Z.X.; Cheng, Q.; Liu, Y.; Fan, S.T.; et al. Molecular signature linked to acute phase injury and tumor invasiveness in small-for-size liver grafts. Ann. Surg. 2010, 251, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Govaert, K.M.; Emmink, B.L.; Nijkamp, M.W.; Cheung, Z.J.; Steller, E.J.; Fatrai, S.; de Bruijn, M.T.; Kranenburg, O.; Rinkes, I.H.B. Hypoxia after liver surgery imposes an aggressive cancer stem cell phenotype on residual tumor cells. Ann. Surg. 2014, 259, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Witt, U.; Kornberg, J.; Müller, K.; Friess, H.; Thrum, K. Postoperative peak serum C-reactive protein is a predictor of outcome following liver transplantation for hepatocellular carcinoma. Biomarkers 2016, 21, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Shen, X.D.; Gao, F.; Zhao, A.; Freitas, M.C.; Lassman, C.; Luster, A.D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology 2008, 47, 207–214. [Google Scholar] [CrossRef]
- Ling, C.C.; Ng, K.T.; Shao, Y.; Geng, W.; Xiao, J.W.; Liu, H.; Li, C.X.; Liu, X.B.; Ma, Y.Y.; Yeung, W.H.; et al. Post-transplant endothelial progenitor cell mobilization via CXCL10/CXCR3 signaling promotes liver tumor growth. J. Hepatol. 2014, 60, 103–109. [Google Scholar] [CrossRef]
- Li, C.X.; Ling, C.C.; Shao, Y.; Xu, A.; Li, X.C.; Ng, K.T.-P.; Liu, X.B.; Ma, Y.Y.; Qi, X.; Liu, H. CXCL10/CXCR3 signaling mobilized-regulatory T cells promote liver tumor recurrence after transplantation. J. Hepatol. 2016, 65, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ling, C.C.; Yeung, W.H.O.; Pang, L.; Liu, J.; Zhou, J.; Zhang, W.Y.; Liu, X.B.; Ng, T.P.K.; Yang, X.X. Monocytic MDSC mobilization promotes tumor recurrence after liver transplantation via CXCL10/TLR4/MMP14 signaling. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, Y.; Gao, F.; Ren, F.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology 2009, 50, 1537–1546. [Google Scholar] [CrossRef]
- Shao, Y.; Lo, C.M.; Ling, C.C.; Liu, X.B.; Ng, K.T.; Chu, A.C.; Ma, Y.Y.; Li, C.X.; Fan, S.T.; Man, K. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. 2014, 355, 264–272. [Google Scholar] [CrossRef]
- Yeung, O.W.; Lo, C.M.; Ling, C.C.; Qi, X.; Geng, W.; Li, C.X.; Ng, K.T.; Forbes, S.J.; Guan, X.Y.; Poon, R.T.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Orci, L.A.; Lacotte, S.; Delaune, V.; Slits, F.; Oldani, G.; Lazarevic, V.; Rossetti, C.; Rubbia-Brandt, L.; Morel, P.; Toso, C. Effects of the gut–liver axis on ischaemia-mediated hepatocellular carcinoma recurrence in the mouse liver. J. Hepatol. 2018, 68, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Venkatesan, A.M.; Mizuno, T.; Aloia, T.A.; Chun, Y.S.; Lee, J.E.; Vauthey, J.-N.; Conrad, C. Remnant liver ischemia as a prognostic factor for cancer-specific survival after resection of colorectal liver metastases. JAMA Surg. 2017, 152, e172986. [Google Scholar] [CrossRef] [PubMed]
- Van Der Bilt, J.D.; Kranenburg, O.; Nijkamp, M.W.; Smakman, N.; Veenendaal, L.M.; Te Velde, E.A.; Voest, E.E.; Van Diest, P.J.; Borel Rinkes, I.H. Ischemia/reperfusion accelerates the outgrowth of hepatic micrometastases in a highly standardized murine model. Hepatology 2005, 42, 165–175. [Google Scholar] [CrossRef]
- Nicoud, I.B.; Jones, C.M.; Pierce, J.M.; Earl, T.M.; Matrisian, L.M.; Chari, R.S.; Gorden, D.L. Warm hepatic ischemia-reperfusion promotes growth of colorectal carcinoma micrometastases in mouse liver via matrix metalloproteinase-9 induction. Cancer Res. 2007, 67, 2720–2728. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nijkamp, M.W.; Hoogwater, F.J.; Govaert, K.M.; Steller, E.J.; Verheem, A.; Kranenburg, O.; Borel Rinkes, I.H. A role for CD95 signaling in ischemia/reperfusion-induced invasion and outgrowth of colorectal micrometastases in mouse liver. J. Surg. Oncol. 2011, 104, 198–204. [Google Scholar] [CrossRef]
- Van Der Bilt, J.D.; Soeters, M.E.; Duyverman, A.M.; Nijkamp, M.W.; Witteveen, P.O.; Van Diest, P.J.; Kranenburg, O.; Rinkes, I.H.B. Perinecrotic hypoxia contributes to ischemia/reperfusion-accelerated outgrowth of colorectal micrometastases. Am. J. Pathol. 2007, 170, 1379–1388. [Google Scholar] [CrossRef][Green Version]
- Konishi, T.; Schuster, R.M.; Lentsch, A.B. Liver repair and regeneration after ischemia-reperfusion injury is associated with prolonged fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G323–G331. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Liu, H.; Liu, J.; Cheung, A.K.L.; Zheng, M.Z.; Cheng, J.L.; Liu, Q.S.; Lo, C.M.; Chen, Z.W.; Man, K. Cytomegalovirus Latency Exacerbated Small-for-Size Liver Graft Injury Through Activation of CCL19/CCR7 in Hepatic Stellate Cells. Transplantation 2021. [Google Scholar] [CrossRef]
- Xiao-Bing, L.; Chung-Mau, L.; Qiao CHENG, K.T.-P.N.; Yan, S.; Chang-Xian, L.; Sookja, K.C.; Irene Oi Lin, N.; Jun, Y.; Kwan, M. Oval cells contribute to fibrogenesis of marginal liver grafts under stepwise regulation of aldose reductase and notch signaling. Theranostics 2017, 7, 4879. [Google Scholar]
- Liu, Y.; Lu, T.; Zhang, C.; Xu, J.; Xue, Z.; Busuttil, R.W.; Xu, N.; Xia, Q.; Kupiec-Weglinski, J.W.; Ji, H. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J. Hepatol. 2019, 71, 719–730. [Google Scholar] [CrossRef]
- Clavien, P.-A.; Yadav, S.; Sindram, D.; Bentley, R.C. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann. Surg. 2000, 232, 155. [Google Scholar] [CrossRef]
- Makuuchi, M.; Mori, T.; Gunven, P.; Yamazaki, S.; Hasegawa, H. Safety of hemihepatic vascular occlusion during resection of the liver. Surg. Gynecol. Obstet. 1987, 164, 155–158. [Google Scholar]
- Selzner, N.; Rudiger, H.; Graf, R.; Clavien, P.-A. Protective strategies against ischemic injury of the liver. Gastroenterology 2003, 125, 917–936. [Google Scholar] [CrossRef]
- Peralta, C.; Hotter, G.; Closa, D.; Prats, N.; Xaus, C.; Gelpí, E.; Roselló-Catafau, J. The protective role of adenosine in inducing nitric oxide synthesis in rat liver ischemia preconditioning is mediated by activation of adenosine A2 receptors. Hepatology 1999, 29, 126–132. [Google Scholar] [CrossRef]
- Cursio, R.; Gugenheim, J.; Ricci, J.; Crenesse, D.; Rostagno, P.; Maulon, L.; Saint-Paul, M.-C.; Ferrua, B.; Mouiel, J.; Auberger, P. Caspase inhibition protects from liver injury following ischemia and reperfusion in rats. Transpl. Int. 2000, 13, S568–S572. [Google Scholar] [CrossRef]
- Araki, H.; Lefer, A.M. Cytoprotective actions of prostacyclin during hypoxia in the isolated perfused cat liver. Am. J. Physiol. Heart Circ. Physiol. 1980, 238, H176–H181. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, G.A.; Strasberg, S.M. Glutathione, lactobionate, and histidine: Cryptic inhibitors of matrix metalloproteinases contained in University of Wisconsin and histidine/tryptophan/ketoglutarate liver preservation solutions. Hepatology 2000, 31, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Strubelt, O.; Younes, M.; Li, Y. Protection by albumin against ischaemia-and hypoxia-induced hepatic injury. Pharmacol. Toxicol. 1994, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Man, K.; Lo, C.M.; Ng, K.T.; Li, X.L.; Sun, C.K.; Lee, T.K.; Dai, X.W.; Fan, S.T. Attenuation of small-for-size liver graft injury by FTY720: Significance of cell-survival Akt signaling pathway. Am. J. Transplant. 2004, 4, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Egawa, H.; Inomata, Y.; Uemoto, S.; Asonuma, K.; Kiuchi, T.; Yamaoka, Y.; Tanaka, K. Efficiency of pentoxifylline in donor pretreatment in rat liver transplantation. J. Surg. Res. 1997, 72, 170–176. [Google Scholar] [CrossRef][Green Version]
- Klein, M.; Geoghegan, J.; Wangemann, R.; Böckler, D.; Schmidt, K.; Scheele, J. Preconditioning of donor livers with prostaglandin I2 before retrieval decreases hepatocellular ischemia-reperfusion injury. Transplantation 1999, 67, 1128–1132. [Google Scholar] [CrossRef]
- Bogetti, D.; Sankary, H.N.; Jarzembowski, T.M.; Manzelli, A.; Knight, P.S.; Thielke, J.; Chejfec, G.; Cotler, S.; Oberholzer, J.; Testa, G. Thymoglobulin induction protects liver allografts from ischemia/reperfusion injury. Clin. Transplant. 2005, 19, 507–511. [Google Scholar] [CrossRef]
- Luntz, S.P.; Unnebrink, K.; Seibert-Grafe, M.; Bunzendahl, H.; Kraus, T.W.; Büchler, M.W.; Klar, E.; Schemmer, P. HEGPOL: Randomized, placebo controlled, multicenter, double-blind clinical trial to investigate hepatoprotective effects of glycine in the postoperative phase of liver transplantation [ISRCTN69350312]. BMC Surg. 2005, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.; Lipshutz, G.; Kupiec-Weglinski, J.; Ponthieux, S.; Gjertson, D.; Cheadle, C.; Watkins, T.; Ehrlich, E.; Katz, E.; Squiers, E. rPSGL-Ig for improvement of early liver allograft function: A double-blind, placebo-controlled, single-center phase II study. Am. J. Transplant. 2011, 11, 786–797. [Google Scholar] [CrossRef]
- Hong, J.C.; Koroleff, D.; Xia, V.; Chang, C.M.; Duarte, S.M.; Xu, J.; Lassman, C.; Kupiec-Weglinski, J.; Coito, A.J.; Busuttil, R.W. Regulated hepatic reperfusion mitigates ischemia-reperfusion injury and improves survival after prolonged liver warm ischemia: A pilot study on a novel concept of organ resuscitation in a large animal model. J. Am. Coll. Surg. 2012, 214, 505–515. [Google Scholar] [CrossRef]
- Nasralla, D.; Coussios, C.C.; Mergental, H.; Akhtar, M.Z.; Butler, A.J.; Ceresa, C.D.; Chiocchia, V.; Dutton, S.J.; García-Valdecasas, J.C.; Heaton, N. A randomized trial of normothermic preservation in liver transplantation. Nature 2018, 557, 50. [Google Scholar] [CrossRef]
- Arai, M.; Thurman, R.G.; Lemasters, J.J. Contribution of adenosine A2 receptors and cyclic adenosine monophosphate to protective ischemic preconditioning of sinusoidal endothelial cells against storage/reperfusion injury in rat livers. Hepatology 2000, 32, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Yamamoto, Y.; Kume, M.; Yamagami, K.; Yamamoto, H.; Kimoto, S.; Ishikawa, Y.; Ozaki, N.; Shimahara, Y.; Yamaoka, Y. Pharmacologic stimulation of adenosine A2 receptor supplants ischemic preconditioning in providing ischemic tolerance in rat livers. Surgery 1999, 126, 945–954. [Google Scholar] [CrossRef]
- Peralta, C.; Hotter, G.; Closa, D.; Gelpí, E.; Bulbena, O.; Rosello-Catafau, J. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: Role of nitric oxide and adenosine. Hepatology 1997, 25, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Carini, R.; De Cesaris, M.G.; Splendore, R.; Vay, D.; Domenicotti, C.; Nitti, M.P.; Paola, D.; Pronzato, M.A.; Albano, E. Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology 2001, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Carini, R.; De Cesaris, M.G.; Splendore, R.; Albano, E. Stimulation of p38 MAP kinase reduces acidosis and Na+ overload in preconditioned hepatocytes. FEBS Lett. 2001, 491, 180–183. [Google Scholar] [CrossRef]
- Peralta, C.; Serafin, A.; Bl, C.; Guzm, M.; Prats, N.; Xaus, C.; Cutillas, B.; Gelp, E.; Rosell, J. Adenosine monophosphate [ndash] activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology 2001, 34, 1164–1173. [Google Scholar] [CrossRef]
- Teoh, N.; Pena, A.D.; Farrell, G. Hepatic ischemic preconditioning in mice is associated with activation of NF-κB, p38 kinase, and cell cycle entry. Hepatology 2002, 36, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Oldani, G.; Crowe, L.A.; Orci, L.A.; Slits, F.; Rubbia-Brandt, L.; De Vito, C.; Morel, P.; Mentha, G.; Berney, T.; Vallée, J.-P. Pre-retrieval reperfusion decreases cancer recurrence after rat ischemic liver graft transplantation. J. Hepatol. 2014, 61, 278–285. [Google Scholar] [CrossRef]
- Nishizawa, N.; Ito, Y.; Eshima, K.; Ohkubo, H.; Kojo, K.; Inoue, T.; Raouf, J.; Jakobsson, P.-J.; Uematsu, S.; Akira, S. Inhibition of microsomal prostaglandin E synthase-1 facilitates liver repair after hepatic injury in mice. J. Hepatol. 2018, 69, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Witt, U.; Kornberg, J.; Friess, H.; Thrum, K. Treating ischaemia-reperfusion injury with prostaglandin E1 reduces the risk of early hepatocellular carcinoma recurrence following liver transplantation. Aliment. Pharmacol. Ther. 2015, 42, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kageyama, S.; Ito, T.; Hirao, H.; Kadono, K.; Aziz, A.; Dery, K.J.; Everly, M.J.; Taura, K.; Uemoto, S. Antibiotic pretreatment alleviates liver transplant damage in mice and humans. J. Clin. Investig. 2019, 129, 3420–3434. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Greten, T.F. Gut microbiome in HCC–Mechanisms, diagnosis and therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; O’Connell, R.; Gao, F.; Lassman, C.; Busuttil, R.W.; Cheng, G.; Kupiec-Weglinski, J.W. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J. Immunol. 2004, 173, 7115–7119. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Lin, Y.; Xu, D.; Tian, Y.; Zhan, Y.; Li, C.; Farmer, D.G.; Kupiec-Weglinski, J.W.; Ke, B. CD47-Mediated Hedgehog/SMO/GLI1 Signaling Promotes Mesenchymal Stem Cell Immunomodulation in Mouse Liver Inflammation. Hepatology 2021. [Google Scholar] [CrossRef]
- Yao, J.; Zheng, J.; Cai, J.; Zeng, K.; Zhou, C.; Zhang, J.; Li, S.; Li, H.; Chen, L.; He, L. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate rat hepatic ischemia-reperfusion injury by suppressing oxidative stress and neutrophil inflammatory response. FASEB J. 2019, 33, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yan, Y.; Wang, B.; Qian, H.; Zhang, X.; Shen, L.; Wang, M.; Zhou, Y.; Zhu, W.; Li, W. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013, 22, 845–854. [Google Scholar] [CrossRef]
- Qi, X.; Ng, K.T.; Lian, Q.Z.; Liu, X.B.; Li, C.X.; Geng, W.; Ling, C.C.; Ma, Y.Y.; Yeung, W.H.; Tu, W.W.; et al. Clinical significance and therapeutic value of glutathione peroxidase 3 (GPx3) in hepatocellular carcinoma. Oncotarget 2014, 5, 11103–11120. [Google Scholar] [CrossRef] [PubMed]
- Dutkowski, P.; Guarrera, J.V.; De Jonge, J.; Martins, P.N.; Porte, R.J.; Clavien, P.-A. Evolving trends in machine perfusion for liver transplantation. Gastroenterology 2019, 156, 1542–1547. [Google Scholar] [CrossRef]
- Schlegel, A.; Graf, R.; Clavien, P.-A.; Dutkowski, P. Hypothermic oxygenated perfusion (HOPE) protects from biliary injury in a rodent model of DCD liver transplantation. J. Hepatol. 2013, 59, 984–991. [Google Scholar] [CrossRef]
- Schlegel, A.; Kron, P.; Graf, R.; Clavien, P.-A.; Dutkowski, P. Hypothermic Oxygenated Perfusion (HOPE) downregulates the immune response in a rat model of liver transplantation. Ann. Surg. 2014, 260, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Kron, P.; Schlegel, A.; Mancina, L.; Clavien, P.-A.; Dutkowski, P. Hypothermic oxygenated perfusion (HOPE) for fatty liver grafts in rats and humans. J. Hepatol. 2018, 68, 82–91. [Google Scholar] [CrossRef]
- Matsushima, H.; Acevedo-Moreno, L.A.; Sasaki, K.; Fujiki, M.; Kwon, C.H.D.; Uso, T.D.; D’Amico, G.; Aucejo, F.; Eghtesad, B.; Miller, C. Does graft hemodynamics affect the risk of hepatocellular carcinoma recurrence after liver transplantation? Clin. Transplant. 2020, 34, e14004. [Google Scholar] [CrossRef]
- Mueller, M.; Kalisvaart, M.; Joanne, O.R.; Shetty, S.; Parente, A.; Muller, X.; Isaac, J.; Muellhaupt, B.; Muiesan, P.; Shah, T. Hypothermic oxygenated liver perfusion (HOPE) prevents tumor recurrence in liver transplantation from donation after circulatory death. Ann. Surg. 2020, 272, 759–765. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Man, K. New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation. Int. J. Mol. Sci. 2021, 22, 8210. https://doi.org/10.3390/ijms22158210
Liu H, Man K. New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation. International Journal of Molecular Sciences. 2021; 22(15):8210. https://doi.org/10.3390/ijms22158210
Chicago/Turabian StyleLiu, Hui, and Kwan Man. 2021. "New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation" International Journal of Molecular Sciences 22, no. 15: 8210. https://doi.org/10.3390/ijms22158210
APA StyleLiu, H., & Man, K. (2021). New Insights in Mechanisms and Therapeutics for Short- and Long-Term Impacts of Hepatic Ischemia Reperfusion Injury Post Liver Transplantation. International Journal of Molecular Sciences, 22(15), 8210. https://doi.org/10.3390/ijms22158210