
High-Density Lipoproteins in Kidney Disease

 ,
, {kind=link}
{kind=link}
Abstract
1. A Shift in Focus from HDL Cholesterol Levels to HDL Function
2. Kidney Regulation of HDL Metabolism
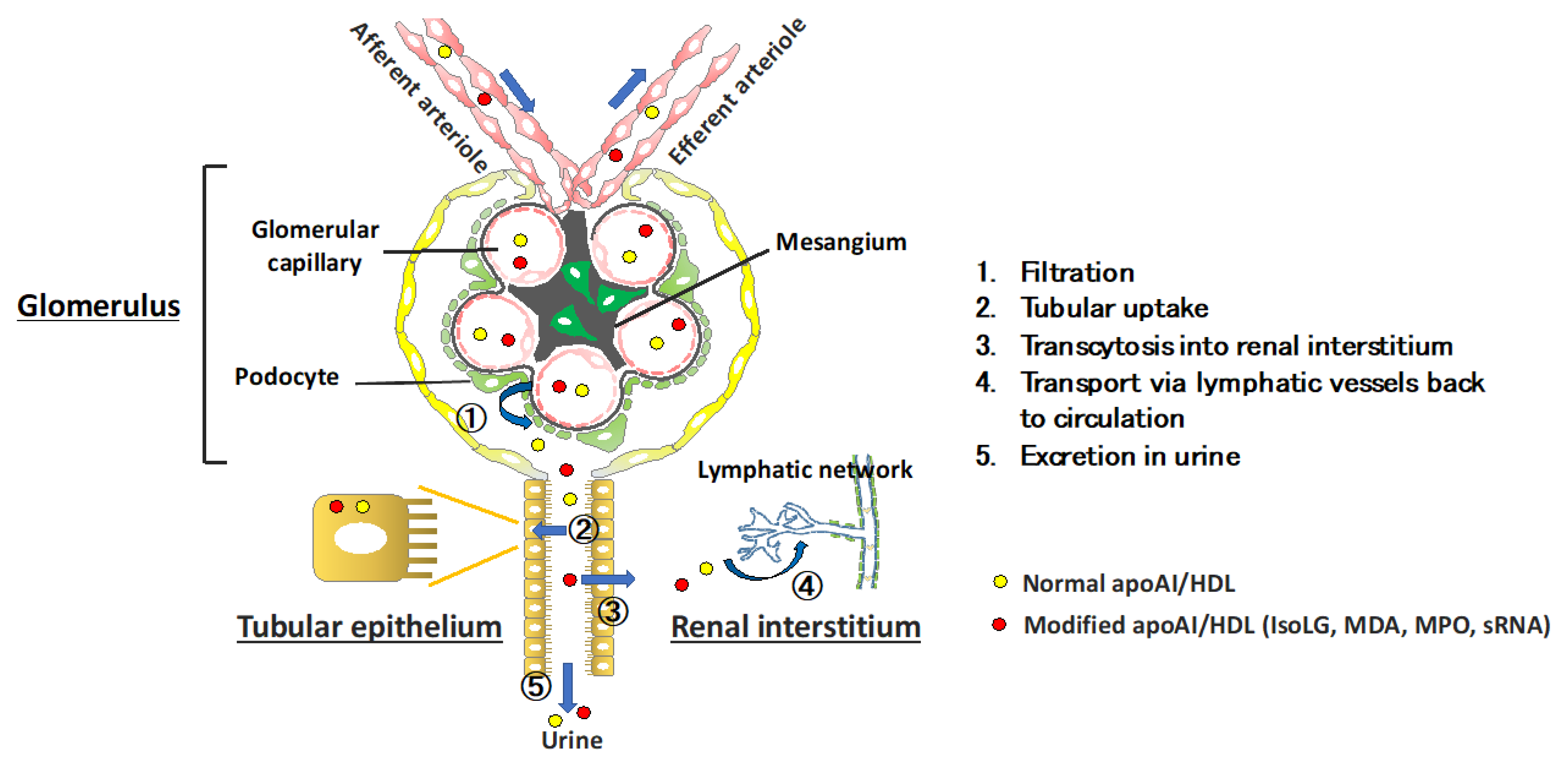
Renal Processes Regulating HDL Homeostasis
3. Renal Impact on Extra-Renal Metabolism of HDL
4. Kidney Disease Modulation of HDL
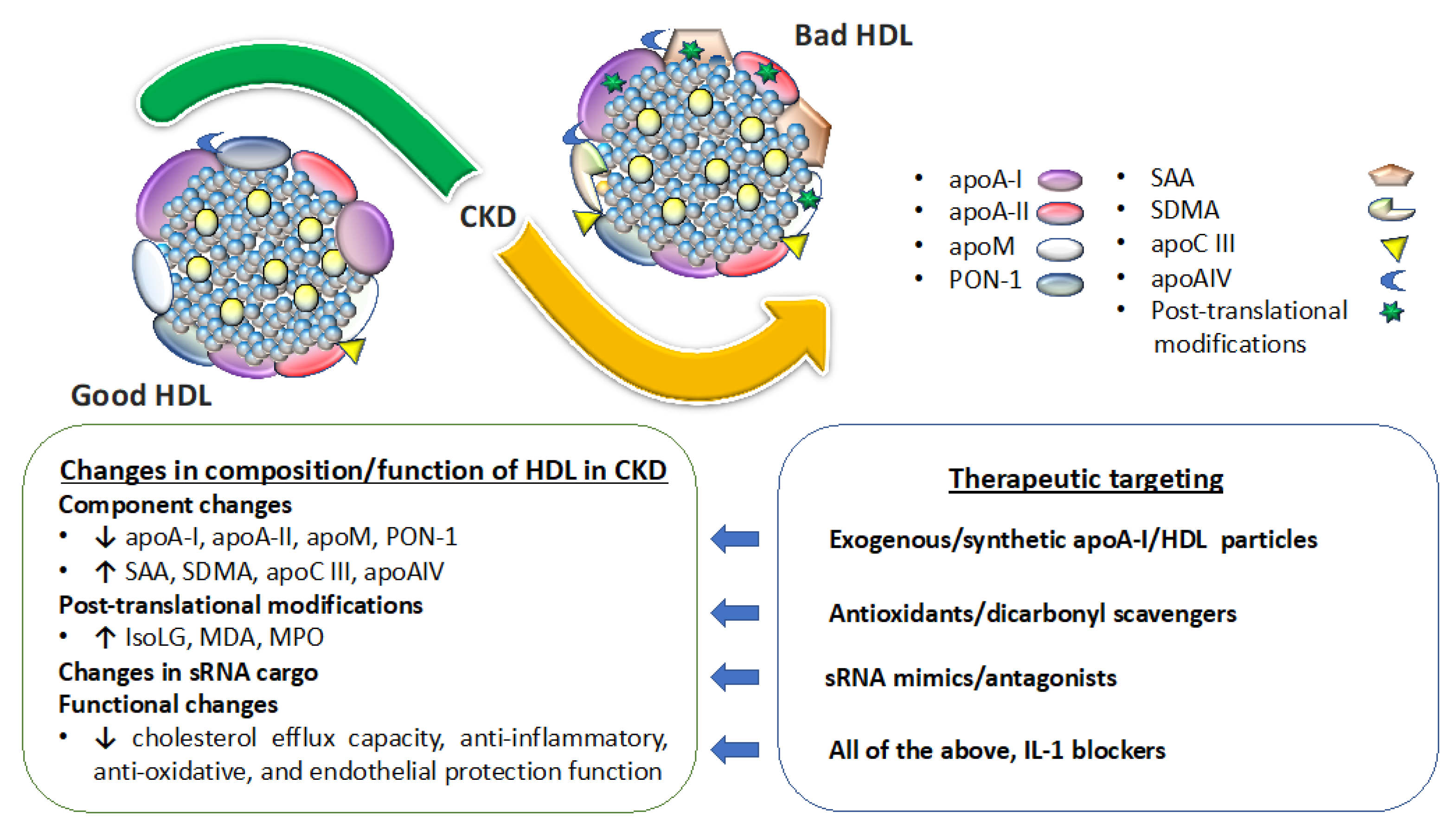
4.1. Kidney Disease Effect on Circulating Levels and Composition of HDL
4.2. Kidney Disease Effect on HDL Functionality
4.3. Kidney Disease and HDL-sRNAs
5. HDL Modulates Renal Parenchymal Cells and Function
6. Targeting Abnormal Levels and Functionality of HDL to Benefit Renal Disease
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Castelli, W.P. Epidemiology of coronary heart disease: The Framingham study. Am. J. Med. 1984, 76, 4–12. [Google Scholar] [CrossRef]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The role of lipids and lipoproteins in atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.C.; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Novel insights from human studies on the role of high-density lipoprotein in mortality and noncardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 128–140. [Google Scholar]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Ajala, O.N.; Demler, O.V.; Liu, Y.; Farukhi, Z.; Adelman, S.J.; Collins, H.L.; Ridker, P.M.; Rader, D.J.; Glynn, R.J.; Mora, S. Anti-Inflammatory HDL function, incident cardiovascular events, and mortality: A secondary analysis of the JUPITER randomized clinical trial. J. Am. Heart Assoc. 2020, 9, e016507. [Google Scholar] [CrossRef]
- Jia, C.; Anderson, J.L.C.; Gruppen, E.G.; Lei, Y.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. High-density lipoprotein anti-inflammatory capacity and incident cardiovascular events. Circulation 2021, 143, 1935–1945. [Google Scholar] [CrossRef]
- Ferro, C.J.; Mark, P.B.; Kanbay, M.; Sarafidis, P.; Heine, G.H.; Rossignol, P.; Massy, Z.A.; Mallamaci, F.; Valdivielso, J.M.; Malyszko, J.; et al. Lipid management in patients with chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 727–749. [Google Scholar] [CrossRef]
- Wanner, C.; Krane, V.; Marz, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E.; German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Fellstrom, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Gronhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Huang, J.; Yancey, P.G.; Yermalitsky, V.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Zagol-Ikapitte, I.; Ye, F.; Amarnath, V.; et al. Scavenging of reactive dicarbonyls with 2-hydroxybenzylamine reduces atherosclerosis in hypercholesterolemic Ldlr(−/−) mice. Nat. Commun. 2020, 11, 4084. [Google Scholar] [CrossRef]
- Vaziri, N.D. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 37–47. [Google Scholar] [CrossRef]
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current understanding of the relationship of HDL composition, structure and function to their cardioprotective properties in chronic kidney disease. Biomolecules 2020, 10, 1348. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Amar, M.J.; Vaisman, B.; Bocharov, A.V.; Vishnyakova, T.G.; Freeman, L.A.; Kurlander, R.J.; Patterson, A.P.; Becker, L.C.; Remaley, A.T. Scavenger receptor-BI is a receptor for lipoprotein(a). J. Lipid Res. 2013, 54, 2450–2457. [Google Scholar] [CrossRef]
- Yang, X.; Sethi, A.; Yanek, L.R.; Knapper, C.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Becker, D.M.; Mathias, R.A.; Remaley, A.T.; Becker, L.C. SCARB1 gene variants are associated with the phenotype of combined high high-density lipoprotein cholesterol and high lipoprotein (a). Circ. Cardiovasc. Genet. 2016, 9, 408–418. [Google Scholar] [CrossRef]
- Graversen, J.H.; Castro, G.; Kandoussi, A.; Nielsen, H.; Christensen, E.I.; Norden, A.; Moestrup, S.K. A pivotal role of the human kidney in catabolism of HDL protein components apolipoprotein A-I and A-IV but not of A-II. Lipids 2008, 43, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Bulum, T.; Kolaric, B.; Duvnjak, L. Lower levels of total HDL and HDL3 cholesterol are associated with albuminuria in normoalbuminuric Type 1 diabetic patients. J. Endocrinol. Investig. 2013, 36, 574–578. [Google Scholar]
- Aseem, O.; Smith, B.T.; Cooley, M.A.; Wilkerson, B.A.; Argraves, K.M.; Remaley, A.T.; Argraves, W.S. Cubilin maintains blood levels of HDL and albumin. J. Am. Soc. Nephrol. JASN 2014, 25, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, N.; Lioudaki, E.; Lygerou, D.; Dermitzaki, E.K.; Papakitsou, I.; Kounali, V.; Holleboom, A.G.; Stratigis, S.; Belogianni, C.; Syngelaki, P.; et al. The P274S mutation of Lecithin-Cholesterol Acyltransferase (LCAT) and its clinical manifestations in a large kindred. Am. J. Kidney Dis. 2019, 74, 510–522. [Google Scholar] [CrossRef]
- Hirashio, S.; Ueno, T.; Naito, T.; Masaki, T. Characteristic kidney pathology, gene abnormality and treatments in LCAT deficiency. Clin. Exp. Nephrol. 2014, 18, 189–193. [Google Scholar] [CrossRef]
- Joles, J.A.; Willekes-Koolschijn, N.; Scheek, L.M.; Koomans, H.A.; Rabelink, T.J.; van Tol, A. Lipoprotein phospholipid composition and LCAT activity in nephrotic and analbuminemic rats. Kidney Int. 1994, 46, 97–104. [Google Scholar] [CrossRef][Green Version]
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern. Med. 2015, 277, 552–561. [Google Scholar] [CrossRef]
- Tsun, J.G.; Yung, S.; Chau, M.K.; Shiu, S.W.; Chan, T.M.; Tan, K.C. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS ONE 2014, 9, e105787. [Google Scholar]
- Lee, J.Y.; Timmins, J.M.; Mulya, A.; Smith, T.L.; Zhu, Y.; Rubin, E.M.; Chisholm, J.W.; Colvin, P.L.; Parks, J.S. HDLs in apoA-I transgenic Abca1 knockout mice are remodeled normally in plasma but are hypercatabolized by the kidney. J. Lipid Res. 2005, 46, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.; Wang, C.; Nyegaard, S.; Heit, B.; Fairn, G.D.; Lee, W.L. SR-BI mediated transcytosis of HDL in brain microvascular endothelial cells is independent of caveolin, clathrin, and PDZK1. Front. Physiol. 2017, 8, 841. [Google Scholar] [CrossRef]
- Yazdani, S.; Poosti, F.; Kramer, A.B.; Mirkovic, K.; Kwakernaak, A.J.; Hovingh, M.; Slagman, M.C.; Sjollema, K.A.; de Borst, M.H.; Navis, G.; et al. Proteinuria triggers renal lymphangiogenesis prior to the development of interstitial fibrosis. PLoS ONE 2012, 7, e50209. [Google Scholar] [CrossRef]
- Kerjaschki, D.; Huttary, N.; Raab, I.; Regele, H.; Bojarski-Nagy, K.; Bartel, G.; Krober, S.M.; Greinix, H.; Rosenmaier, A.; Karlhofer, F.; et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat. Med. 2006, 12, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Jabs, K.; Hunley, T.E.; Jones, D.P.; van de Voorde, R.G.; Anderson, C.; Du, L.; Zhong, J.; Fogo, A.B.; Yang, H.; et al. Urinary apolipoprotein AI in children with kidney disease. Pediatr. Nephrol. 2019, 34, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- May-Zhang, L.S.; Yermalitsky, V.; Huang, J.; Pleasent, T.; Borja, M.S.; Oda, M.N.; Jerome, W.G.; Yancey, P.G.; Linton, M.F.; Davies, S.S. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters HDL structure and function. J. Biol. Chem. 2018, 24, 9176–9187. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hellin, J.; Cantarell, C.; Jimeno, L.; Sanchez-Fructuoso, A.; Puig-Gay, N.; Guirado, L.; Vilarino, N.; Gonzalez-Roncero, F.M.; Mazuecos, A.; Lauzurica, R.; et al. A form of apolipoprotein a-I is found specifically in relapses of focal segmental glomerulosclerosis following transplantation. Am. J. Transplant. 2013, 13, 493–500. [Google Scholar] [CrossRef]
- Chindhy, S.; Joshi, P.; Khera, A.; Ayers, C.R.; Hedayati, S.S.; Rohatgi, A. Impaired renal function on cholesterol efflux capacity, HDL particle number, and cardiovascular events. J. Am. Coll. Cardiol. 2018, 72, 698–700. [Google Scholar] [CrossRef]
- Holzer, M.; Schilcher, G.; Curcic, S.; Trieb, M.; Ljubojevic, S.; Stojakovic, T.; Scharnagl, H.; Kopecky, C.M.; Rosenkranz, A.R.; Heinemann, A.; et al. Dialysis modalities and HDL composition and function. J. Am. Soc. Nephrol. JASN 2015, 26, 2267–2276. [Google Scholar] [CrossRef]
- Weichhart, T.; Kopecky, C.; Kubicek, M.; Haidinger, M.; Doller, D.; Katholnig, K.; Suarna, C.; Eller, P.; Tolle, M.; Gerner, C.; et al. Serum amyloid A in uremic HDL promotes inflammation. J. Am. Soc. Nephrol. JASN 2012, 23, 934–947. [Google Scholar] [CrossRef]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef]
- Rubinow, K.B.; Henderson, C.M.; Robinson-Cohen, C.; Himmelfarb, J.; de Boer, I.H.; Vaisar, T.; Kestenbaum, B.; Hoofnagle, A.N. Kidney function is associated with an altered protein composition of high-density lipoprotein. Kidney Int. 2017, 92, 1526–1535. [Google Scholar] [CrossRef]
- Shao, B.; de Boer, I.; Tang, C.; Mayer, P.S.; Zelnick, L.; Afkarian, M.; Heinecke, J.W.; Himmelfarb, J. A Cluster of Proteins Implicated in Kidney Disease Is Increased in High-Density Lipoprotein Isolated from Hemodialysis Subjects. J. Proteome Res. 2015, 14, 2792–2806. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Krankel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. JASN 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Heinecke, J.W. Impact of HDL oxidation by the myeloperoxidase system on sterol efflux by the ABCA1 pathway. J. Proteom. 2011, 74, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Kalantar-Zadeh, K.; Wang, Z.; Fu, X.; Tang, W.H.; Hazen, S.L. Protein carbamylation predicts mortality in ESRD. J. Am. Soc. Nephrol. JASN 2013, 24, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Morrow, J.D.; Roberts, L.J.; Evanson, J.A.; Becker, B.; Hakim, R.M.; Shyr, Y.; Himmelfarb, J. Plasma F2-isoprostane levels are elevated in chronic hemodialysis patients. Clin. Nephrol. 2002, 58, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Bacchetti, T.; Masciangelo, S.; Armeni, T.; Bicchiega, V.; Ferretti, G. Glycation of human high density lipoprotein by methylglyoxal: Effect on HDL-paraoxonase activity. Metab. Clin. Exp. 2014, 63, 307–311. [Google Scholar] [CrossRef]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. JASN 2011, 22, 1631–1641. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef]
- Luo, M.; Liu, A.; Wang, S.; Wang, T.; Hu, D.; Wu, S.; Peng, D. ApoCIII enrichment in HDL impairs HDL-mediated cholesterol efflux capacity. Sci. Rep. 2017, 7, 2312. [Google Scholar] [CrossRef] [PubMed]
- Kaseda, R.; Jabs, K.; Hunley, T.E.; Jones, D.; Bian, A.; Allen, R.M.; Vickers, K.C.; Yancey, P.G.; Linton, M.F.; Fazio, S.; et al. Dysfunctional high-density lipoproteins in children with chronic kidney disease. Metab. Clin. Exp. 2015, 64, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Kopecky, C.; Haidinger, M.; Birner-Grunberger, R.; Darnhofer, B.; Kaltenecker, C.C.; Marsche, G.; Holzer, M.; Weichhart, T.; Antlanger, M.; Kovarik, J.J.; et al. Restoration of renal function does not correct impairment of uremic HDL properties. J. Am. Soc. Nephrol. JASN 2015, 26, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Annema, W.; Dikkers, A.; de Boer, J.F.; Dullaart, R.P.; Sanders, J.S.; Bakker, S.J.; Tietge, U.J. HDL cholesterol efflux predicts graft failure in renal transplant recipients. J. Am. Soc. Nephrol. JASN 2016, 27, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Allen, R.M.; Zhao, S.; Ramirez Solano, M.A.; Zhu, W.; Michell, D.L.; Wang, Y.; Shyr, Y.; Sethupathy, P.; Linton, M.F.; Graf, G.A.; et al. Bioinformatic analysis of endogenous and exogenous small RNAs on lipoproteins. J. Extracell Vesicles 2018, 7, 1506198. [Google Scholar] [CrossRef]
- Canfran-Duque, A.; Ramirez, C.M.; Goedeke, L.; Lin, C.S.; Fernandez-Hernando, C. microRNAs and HDL life cycle. Cardiovasc. Res. 2014, 103, 414–422. [Google Scholar] [CrossRef]
- Scicali, R.; di Pino, A.; Pavanello, C.; Ossoli, A.; Strazzella, A.; Alberti, A.; di Mauro, S.; Scamporrino, A.; Urbano, F.; Filippello, A.; et al. Analysis of HDL-microRNA panel in heterozygous familial hypercholesterolemia subjects with LDL receptor null or defective mutation. Sci. Rep. 2019, 9, 20354. [Google Scholar] [CrossRef]
- Simionescu, N.; Niculescu, L.S.; Carnuta, M.G.; Sanda, G.M.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. Hyperglycemia determines increased specific MicroRNAs levels in sera and HDL of acute coronary syndrome patients and stimulates MicroRNAs production in human macrophages. PLoS ONE 2016, 11, e0161201. [Google Scholar] [CrossRef]
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and miR-92a identified in circulating HDL discriminate between stable and vulnerable coronary artery disease patients. PLoS ONE 2015, 10, e0140958. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, H.; Huang, Z.; Zheng, H.; Zhou, J. Emerging role of miRNAs in renal fibrosis. RNA Biol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Axmann, M.; Meier, S.M.; Karner, A.; Strobl, W.; Stangl, H.; Plochberger, B. Serum and lipoprotein particle miRNA profile in uremia patients. Genes 2018, 9, 533. [Google Scholar] [CrossRef]
- Umanath, K.; Lewis, J.B. Update on diabetic nephropathy: Core curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef]
- Florijn, B.W.; Duijs, J.; Levels, J.H.; Dallinga-Thie, G.M.; Wang, Y.; Boing, A.N.; Yuana, Y.; Stam, W.; Limpens, R.; Au, Y.W.; et al. Diabetic nephropathy alters the distribution of circulating angiogenic MicroRNAs among extracellular vesicles, HDL, and Ago-2. Diabetes 2019, 68, 2287–2300. [Google Scholar] [CrossRef]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef]
- Anand, S.; Majeti, B.K.; Acevedo, L.M.; Murphy, E.A.; Mukthavaram, R.; Scheppke, L.; Huang, M.; Shields, D.J.; Lindquist, J.N.; Lapinski, P.E.; et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat. Med. 2010, 16, 909–914. [Google Scholar] [CrossRef]
- Tsuchida, Y.; Zhong, J.; Otsuka, T.; Dikalova, A.; Pastan, I.; Anantharamaiah, G.M.; Linton, M.F.; Yancey, P.G.; Ikizler, T.A.; Fogo, A.B.; et al. Lipoprotein modulation of proteinuric renal injury. Lab. Investig. 2019, 99, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; Gupta, N.; De Maziere, A.M.; Posthuma, G.; Chiu, C.P.; Pierce, A.A.; Hotzel, K.; Tao, J.; Foreman, O.; Koukos, G.; et al. Apolipoprotein L1-specific antibodies detect endogenous APOL1 inside the endoplasmic reticulum and on the plasma membrane of podocytes. J. Am. Soc. Nephrol. JASN 2020, 31, 2044–2064. [Google Scholar] [CrossRef] [PubMed]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlback, B.; Kono, M.; Proia, R.L.; Smith, J.D.; et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.S.; Yang, D.; Swendeman, S.L.; Christoffersen, C.; Nielsen, L.B.; Friedman, S.L.; Powell, C.A.; Hla, T.; Cao, Z. Aging suppresses sphingosine-1-phosphate chaperone ApoM in circulation resulting in maladaptive organ repair. Dev. Cell 2020, 53, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, I.M.; Bertelsen, M.; Freese, E.; Lindhard, K.; Ullum, H.; Feldt-Rasmussen, B.; Nielsen, L.B.; Christoffersen, C.; Bro, S. Apolipoprotein M in patients with chronic kidney disease. Atherosclerosis 2018, 275, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Tsuneyama, K.; Morimoto, Y.; Nishikawa, M.; Yatomi, Y. Apolipoprotein M suppresses the phenotypes of IgA nephropathy in hyper-IgA mice. FASEB J. 2019, 33, 5181–5195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, X.; Wu, J.; Liu, D.; Cai, H.; Fu, L.; Mei, C. Oxidized high-density lipoprotein enhances inflammatory activity in rat mesangial cells. Diabetes Metab. Res. Rev. 2010, 26, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wu, J.; Qian, Y.; Fu, L.; Wu, G.; Xu, C.; Mei, C. Oxidized high-density lipoprotein impairs the function of human renal proximal tubule epithelial cells through CD36. Int. J. Mol. Med. 2014, 34, 564–572. [Google Scholar] [CrossRef]
- Milasan, A.; Dallaire, F.; Mayer, G.; Martel, C. Effects of LDL receptor modulation on lymphatic function. Sci. Rep. 2016, 6, 27862. [Google Scholar] [CrossRef]
- Bisoendial, R.; Tabet, F.; Tak, P.P.; Petrides, F.; Cuesta Torres, L.F.; Hou, L.; Cook, A.; Barter, P.J.; Weninger, W.; Rye, K.A. Apolipoprotein A-I limits the negative effect of tumor necrosis factor on lymphangiogenesis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2443–2450. [Google Scholar] [CrossRef]
- Engelbrecht, E.; Levesque, M.V.; He, L.; Vanlandewijck, M.; Nitzsche, A.; Niazi, H.; Kuo, A.; Singh, S.A.; Aikawa, M.; Holton, K.; et al. Sphingosine 1-phosphate-regulated transcriptomes in heterogenous arterial and lymphatic endothelium of the aorta. eLife 2020, 9, e52690. [Google Scholar] [CrossRef]
- Yazdani, S.; Navis, G.; Hillebrands, J.L.; van Goor, H.; van den Born, J. Lymphangiogenesis in renal diseases: Passive bystander or active participant? Expert Rev. Mol. Med. 2014, 16, e15. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xian, H.; Balasubramanian, S.; Al-Aly, Z. Low levels of high-density lipoprotein cholesterol increase the risk of incident kidney disease and its progression. Kidney Int. 2016, 89, 886–896. [Google Scholar] [CrossRef]
- Fox, C.S.; Larson, M.G.; Leip, E.P.; Culleton, B.; Wilson, P.W.; Levy, D. Predictors of new-onset kidney disease in a community-based population. JAMA 2004, 291, 844–850. [Google Scholar] [CrossRef]
- Muntner, P.; Coresh, J.; Smith, J.C.; Eckfeldt, J.; Klag, M.J. Plasma lipids and risk of developing renal dysfunction: The atherosclerosis risk in communities study. Kidney Int. 2000, 58, 293–301. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Theriault, S.; Walsh, M.; Pare, G. HDL cholesterol, LDL cholesterol, and triglycerides as risk factors for CKD: A Mendelian randomization study. Am. J. Kidney Dis. 2018, 71, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Hu, Q.; Zhang, Q.; Su, G.Y.; Xiao, H.M.; Li, B.Y.; Shen, W.D.; Qiu, X.; Lv, W.Q.; Deng, H.W. Causal effects of genetically predicted cardiovascular risk factors on chronic kidney disease: A two-sample mendelian randomization study. Front. Genet. 2019, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, M.; Walley, K.R.; Russell, J.A.; Brunham, L.R.; Genga, K.R.; Boyd, J.H. Decreased high-density lipoprotein cholesterol level is an early prognostic marker for organ dysfunction and death in patients with suspected sepsis. J. Crit. Care 2017, 38, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.F.; Chen, C.Y.; Hsu, C.L.; Chen, K.Y.; Yu, C.J. Decreased serum level of lipoprotein cholesterol is a poor prognostic factor for patients with severe community-acquired pneumonia that required intensive care unit admission. J. Crit. Care 2015, 30, 506–510. [Google Scholar] [CrossRef]
- Roveran Genga, K.; Lo, C.; Cirstea, M.; Zhou, G.; Walley, K.R.; Russell, J.A.; Levin, A.; Boyd, J.H. Two-year follow-up of patients with septic shock presenting with low HDL: The effect upon acute kidney injury, death and estimated glomerular filtration rate. J. Intern. Med. 2017, 281, 518–529. [Google Scholar] [CrossRef]
- Genga, K.R.; Trinder, M.; Kong, H.J.; Li, X.; Leung, A.K.K.; Shimada, T.; Walley, K.R.; Russell, J.A.; Francis, G.A.; Brunham, L.R.; et al. CETP genetic variant rs1800777 (allele A) is associated with abnormally low HDL-C levels and increased risk of AKI during sepsis. Sci. Rep. 2018, 8, 16764. [Google Scholar] [CrossRef]
- Smith, L.E.; Smith, D.K.; Blume, J.D.; Linton, M.F.; Billings, F.T.T. High-density lipoprotein cholesterol concentration and acute kidney injury after cardiac surgery. J. Am. Heart Assoc. 2017, 6, 006975. [Google Scholar] [CrossRef]
- Smith, L.E.; Smith, D.K.; Yancey, P.G.; Kon, V.; Remaley, A.T.; Billings, F.T.T.; Linton, M.F. Perioperative high-density lipoproteins, oxidative stress, and kidney injury after cardiac surgery. J. Lipid Res. 2021, 62, 100024. [Google Scholar] [CrossRef]
- Kronenberg, F. HDL in CKD—The devil is in the detail. J. Am. Soc. Nephrol. JASN 2018, 29, 1356–1371. [Google Scholar] [CrossRef]
- Lamprea-Montealegre, J.A.; Sharrett, A.R.; Matsushita, K.; Selvin, E.; Szklo, M.; Astor, B.C. Chronic kidney disease, lipids and apolipoproteins, and coronary heart disease: The ARIC study. Atherosclerosis 2014, 234, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Cases, A.; Coll, E. Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int. Suppl. 2005, 68, S87–S93. [Google Scholar] [CrossRef] [PubMed]
- Schaeffner, E.S.; Kurth, T.; Curhan, G.C.; Glynn, R.J.; Rexrode, K.M.; Baigent, C.; Buring, J.E.; Gaziano, J.M. Cholesterol and the risk of renal dysfunction in apparently healthy men. J. Am. Soc. Nephrol. JASN 2003, 14, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Yang, W.; Akkina, S.; Alper, A.; Anderson, A.H.; Appel, L.J.; He, J.; Raj, D.S.; Schelling, J.; Strauss, L.; et al. Relation of serum lipids and lipoproteins with progression of CKD: The CRIC study. Clin. J. Am. Soc. Nephrol. CJASN 2014, 9, 1190–1198. [Google Scholar] [CrossRef]
- Kang, H.T.; Kim, J.K.; Kim, J.Y.; Linton, J.A.; Yoon, J.H.; Koh, S.B. Independent association of TG/HDL-C with urinary albumin excretion in normotensive subjects in a rural Korean population. Clin. Chim. Acta 2012, 413, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, K.; Yoshida, H.; Nagata, M.; Kitazono, T.; Iseki, K.; Iseki, C.; Fujimoto, S.; Konta, T.; Moriyama, T.; Yamagata, K.; et al. Impact of the triglycerides to high-density lipoprotein cholesterol ratio on the incidence and progression of CKD: A longitudinal study in a large Japanese population. Am. J. Kidney Dis. 2015, 66, 972–983. [Google Scholar] [CrossRef]
- Haynes, R.; Staplin, N.; Emberson, J.; Herrington, W.G.; Tomson, C.; Agodoa, L.; Tesar, V.; Levin, A.; Lewis, D.; Reith, C.; et al. Evaluating the contribution of the cause of kidney disease to prognosis in CKD: Results from the Study of Heart and Renal Protection (SHARP). Am. J. Kidney Dis. 2014, 64, 40–48. [Google Scholar] [CrossRef]
- Ong, K.L.; Waters, D.D.; Fayyad, R.; Vogt, L.; Melamed, S.; DeMicco, D.A.; Rye, K.A.; Barter, P.J. Relationship of high-density lipoprotein cholesterol with renal function in patients treated with atorvastatin. J. Am. Heart Assoc. 2018, 7, e007387. [Google Scholar] [CrossRef]
- Moradi, H.; Streja, E.; Kashyap, M.L.; Vaziri, N.D.; Fonarow, G.C.; Kalantar-Zadeh, K. Elevated high-density lipoprotein cholesterol and cardiovascular mortality in maintenance hemodialysis patients. Nephrol. Dial. Transplant. 2014, 29, 1554–1562. [Google Scholar] [CrossRef]
- Li, Y.; Dong, J.B.; Wu, M.P. Human ApoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur. J. Pharmacol. 2008, 590, 417–422. [Google Scholar] [CrossRef]
- Moreira, R.S.; Irigoyen, M.; Sanches, T.R.; Volpini, R.A.; Camara, N.O.; Malheiros, D.M.; Shimizu, M.H.; Seguro, A.C.; Andrade, L. Apolipoprotein A-I mimetic peptide 4F attenuates kidney injury, heart injury, and endothelial dysfunction in sepsis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R514–R524. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Ahmad, G.; Hossain, F.; Liu, Y.; Wang, X.; Dennis, J.; Freedman, B.; Witting, P.K. High-Density Lipoprotein (HDL) inhibits serum amyloid A (SAA)-induced vascular and renal dysfunctions in apolipoprotein e-deficient mice. Int. J. Mol. Sci. 2020, 21, 1316. [Google Scholar] [CrossRef]
- Smith, L.E. High-density lipoproteins and acute kidney injury. Semin. Nephrol. 2020, 40, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.M.; Barden, A.E.; Loke, W.M.; Croft, K.D.; Puddey, I.B.; Mori, T.A. HDL is the major lipoprotein carrier of plasma F2-isoprostanes. J. Lipid Res. 2009, 50, 716–722. [Google Scholar] [CrossRef]
- Takahashi, K.; Nammour, T.M.; Fukunaga, M.; Ebert, J.; Morrow, J.D.; Roberts, L.J., 2nd; Hoover, R.L.; Badr, K.F. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi-prostaglandin F2 alpha, in the rat. Evidence for interaction with thromboxane A2 receptors. J. Clin. Investig. 1992, 90, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Michael Gibson, C.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.; et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: The AEGIS-I trial (ApoA-I event reducing in ischemic syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Duffy, D.; Tortorici, M.A.; Wright, S.D.; Deckelbaum, L.I.; D’Andrea, D.M. Moderate renal impairment does not impact the ability of CSL112 (apolipoprotein A-I [human]) to enhance cholesterol efflux capacity. J. Clin. Pharmacol. 2019, 59, 427–436. [Google Scholar] [CrossRef]
- Hung, A.M.; Tsuchida, Y.; Nowak, K.L.; Sarkar, S.; Chonchol, M.; Whitfield, V.; Salas, N.; Dikalova, A.; Yancey, P.G.; Huang, J.; et al. IL-1 inhibition and function of the HDL-containing fraction of plasma in patients with stages 3 to 5 CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 702–711. [Google Scholar] [CrossRef]
- Kaseda, R.T.Y.; Gamboa, J.L.; Zhong, J.; Zhang, L.; Yang, H.; Dikalova, A.; Bian, A.; Davies, S.; Fogo, A.F.; Linton, M.F.; et al. Angiotensin receptor blocker vs ACE inhibitor effects on HDL functionality in patients on maintenance hemodialysis. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 582–591. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kon, V.; Yang, H.-C.; Smith, L.E.; Vickers, K.C.; Linton, M.F. High-Density Lipoproteins in Kidney Disease. Int. J. Mol. Sci. 2021, 22, 8201. https://doi.org/10.3390/ijms22158201
Kon V, Yang H-C, Smith LE, Vickers KC, Linton MF. High-Density Lipoproteins in Kidney Disease. International Journal of Molecular Sciences. 2021; 22(15):8201. https://doi.org/10.3390/ijms22158201
Chicago/Turabian StyleKon, Valentina, Hai-Chun Yang, Loren E. Smith, Kasey C. Vickers, and MacRae F. Linton. 2021. "High-Density Lipoproteins in Kidney Disease" International Journal of Molecular Sciences 22, no. 15: 8201. https://doi.org/10.3390/ijms22158201
APA StyleKon, V., Yang, H.-C., Smith, L. E., Vickers, K. C., & Linton, M. F. (2021). High-Density Lipoproteins in Kidney Disease. International Journal of Molecular Sciences, 22(15), 8201. https://doi.org/10.3390/ijms22158201