
Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases


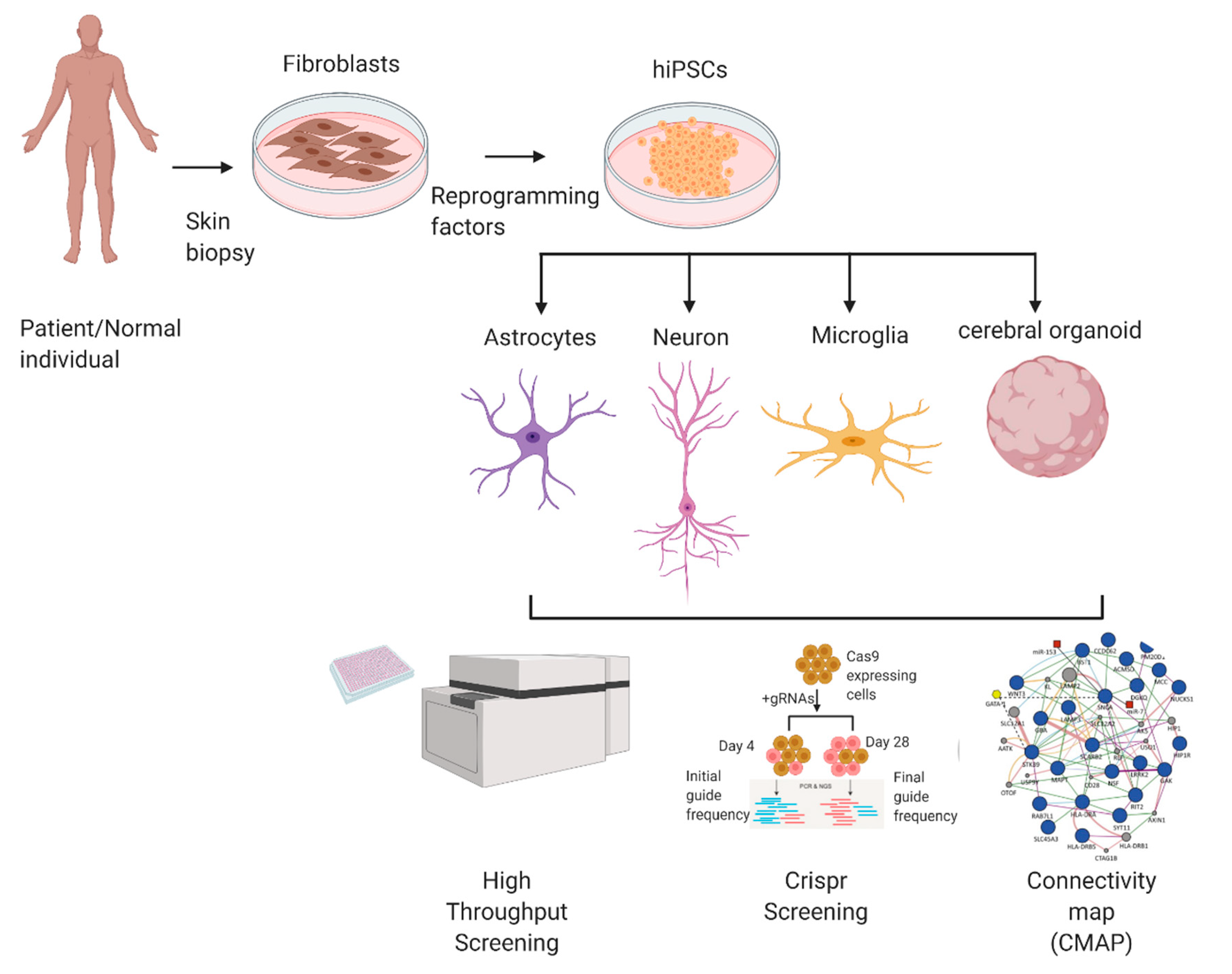{kind=link}
Abstract
1. Introduction
2. Methods for Disease Modeling
2.1. 2-Dimentional (2D) Models
2.2. Three-Dimentional (3D) Models
3. Tools for Drug Discovery
3.1. High-Throughput Screening
3.2. Connectivity Map
4. Disease Modeling Using hiPSCs
4.1. Alzheimer’s Disease (AD)
4.2. Familial AD (fAD)
4.3. Sporadic AD (sAD)
4.4. AD hiPSC-Based Models for Drug Discovery
4.5. Parkinson’s Disease (PD)
4.6. Familial PD (fPD) Modeling
4.7. Sporadic PD (sPD) Modeling
4.8. PD hiPSC-Based Models for Drug Discovery
4.9. Amyotrophic Lateral Sclerosis (ALS)
4.10. Familial ALS (fALS) Modeling with hiPSCs
4.11. Sporadic ALS (sALS)
4.12. ALS hiPSC-Based Models for Drug Discovery
5. Limitations of hiPSC-Based Models of Neurodegenerative Disorders
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Li, X.J.; Zhang, X.; Johnson, M.A.; Wang, Z.B.; Lavaute, T.; Zhang, S.C. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development 2009, 136, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Pasca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Fuzik, J.; Zeisel, A.; Mate, Z.; Calvigioni, D.; Yanagawa, Y.; Szabo, G.; Linnarsson, S.; Harkany, T. Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat. Biotechnol. 2016, 34, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Nakatani, T.; Sakamoto, Y.; Mizuhara, E.; Minaki, Y.; Kumai, M.; Hamaguchi, A.; Nishimura, M.; Inoue, Y.; Hayashi, H.; et al. Differences in neurogenic potential in floor plate cells along an anteroposterior location: Midbrain dopaminergic neurons originate from mesencephalic floor plate cells. Development 2007, 134, 3213–3225. [Google Scholar] [CrossRef]
- Kim, J.H.; Auerbach, J.M.; Rodriguez-Gomez, J.A.; Velasco, I.; Gavin, D.; Lumelsky, N.; Lee, S.H.; Nguyen, J.; Sanchez-Pernaute, R.; Bankiewicz, K.; et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature 2002, 418, 50–56. [Google Scholar] [CrossRef]
- Chung, S.; Hedlund, E.; Hwang, M.; Kim, D.W.; Shin, B.S.; Hwang, D.Y.; Kang, U.J.; Isacson, O.; Kim, K.S. The homeodomain transcription factor Pitx3 facilitates differentiation of mouse embryonic stem cells into AHD2-expressing dopaminergic neurons. Mol. Cell. Neurosci. 2005, 28, 241–252. [Google Scholar] [CrossRef]
- Kim, D.W.; Chung, S.; Hwang, M.; Ferree, A.; Tsai, H.C.; Park, J.J.; Chung, S.; Nam, T.S.; Kang, U.J.; Isacson, O.; et al. Stromal cell-derived inducing activity, Nurr1, and signaling molecules synergistically induce dopaminergic neurons from mouse embryonic stem cells. Stem Cells 2006, 24, 557–567. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef]
- Hyysalo, A.; Ristola, M.; Makinen, M.E.; Hayrynen, S.; Nykter, M.; Narkilahti, S. Laminin α5 substrates promote survival, network formation and functional development of human pluripotent stem cell-derived neurons in vitro. Stem Cell Res. 2017, 24, 118–127. [Google Scholar] [CrossRef]
- Ambasudhan, R.; Dolatabadi, N.; Nutter, A.; Masliah, E.; McKercher, S.R.; Lipton, S.A. Potential for cell therapy in Parkinson’s disease using genetically programmed human embryonic stem cell-derived neural progenitor cells. J. Comp. Neurol. 2014, 522, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- El-Akabawy, G.; Medina, L.M.; Jeffries, A.; Price, J.; Modo, M. Purmorphamine increases DARPP-32 differentiation in human striatal neural stem cells through the Hedgehog pathway. Stem Cells Dev. 2011, 20, 1873–1887. [Google Scholar] [CrossRef]
- Arber, C.; Precious, S.V.; Cambray, S.; Risner-Janiczek, J.R.; Kelly, C.; Noakes, Z.; Fjodorova, M.; Heuer, A.; Ungless, M.A.; Rodriguez, T.A.; et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development 2015, 142, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hu, B.; Liu, Y.; Vermilyea, S.C.; Liu, H.; Gao, L.; Sun, Y.; Zhang, X.; Zhang, S.C. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell 2012, 10, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yuan, J.; Sander, B.; Golas, M.M. In vitro differentiation of human neural progenitor cells into striatal GABAergic neurons. Stem Cells Transl. Med. 2015, 4, 775–788. [Google Scholar] [CrossRef]
- Krencik, R.; Zhang, S.C. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Protoc. 2011, 6, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of inflammation-responsive astrocytes from glial progenitors generated from human induced pluripotent stem cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef] [PubMed]
- Palm, T.; Bolognin, S.; Meiser, J.; Nickels, S.; Trager, C.; Meilenbrock, R.L.; Brockhaus, J.; Schreitmuller, M.; Missler, M.; Schwamborn, J.C. Rapid and robust generation of long-term self-renewing human neural stem cells with the ability to generate mature astroglia. Sci. Rep. 2015, 5, 16321. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia response during parkinson’s disease: α-Synuclein intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-Mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Sloan, S.A.; Andersen, J.; Pasca, A.M.; Birey, F.; Pasca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Pasca, S.P. Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of functional human 3D cortico-motor assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef]
- Miura, Y.; Li, M.Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Mantle, J.L.; Lee, K.H. A differentiating neural stem cell-derived astrocytic population mitigates the inflammatory effects of TNF-α and IL-6 in an iPSC-based blood-brain barrier model. Neurobiol. Dis. 2018, 119, 113–120. [Google Scholar] [CrossRef]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An isogenic blood-brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. J. Neurochem. 2017, 140, 874–888. [Google Scholar] [CrossRef]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef]
- Mansour, A.A.; Goncalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-throughput screening platforms in the discovery of novel drugs for neurodegenerative diseases. Bioengineering 2021, 8, 30. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s biochemical and cell-based approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Janes, J.; Young, M.E.; Chen, E.; Rogers, N.H.; Burgstaller-Muehlbacher, S.; Hughes, L.D.; Love, M.S.; Hull, M.V.; Kuhen, K.L.; Woods, A.K.; et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2018, 115, 10750–10755. [Google Scholar] [CrossRef]
- Sirota, M.; Dudley, J.T.; Kim, J.; Chiang, A.P.; Morgan, A.A.; Sweet-Cordero, A.; Sage, J.; Butte, A.J. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 2011, 3, 96ra77. [Google Scholar] [CrossRef]
- Van Noort, V.; Schölch, S.; Iskar, M.; Zeller, G.; Ostertag, K.; Schweitzer, C.; Werner, K.; Weitz, J.; Koch, M.; Bork, P. Novel drug candidates for the treatment of metastatic colorectal cancer through global inverse gene-expression profiling. Cancer Res. 2014, 74, 5690–5699. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; Gatt, A.; Clarke, E.; Corcoran, J.; Doherty, P.; Chambers, D.; Ballard, C. Drug repurposing for Alzheimer’s disease based on transcriptional profiling of human iPSC-derived cortical neurons. Transl. Psychiatry 2019, 9, 220. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Bancher, C. AD neuropathology. Neurology 1996, 46, 1186–1187. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Liao, M.C.; Muratore, C.R.; Gierahn, T.M.; Sullivan, S.E.; Srikanth, P.; de Jager, P.L.; Love, J.C.; Young-Pearse, T.L. Single-cell detection of secreted Aβ and sAPPα from human iPSC-derived neurons and astrocytes. J. Neurosci. 2016, 36, 1730–1746. [Google Scholar] [CrossRef]
- Konttinen, H.; Cabral-da-Silva, M.E.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernandez, D.; Fazaludeen, M.F.; et al. PSEN1DeltaE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-derived microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs. isogenic control. eLife 2019, 8. [Google Scholar] [CrossRef]
- Ghatak, S.; Dolatabadi, N.; Gao, R.; Wu, Y.; Scott, H.; Trudler, D.; Sultan, A.; Ambasudhan, R.; Nakamura, T.; Masliah, E.; et al. NitroSynapsin ameliorates hypersynchronous neural network activity in Alzheimer hiPSC models. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.; Tessier-Lavigne, M. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 2019, 104, 256–270.e5. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef]
- Antonarakis, S.E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 2017, 18, 147–163. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 2012, 4, 124ra129. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Chen, S.-M.; Lu, H.-E.; Lai, S.-M.; Lai, P.-S.; Shen, P.-W.; Chen, P.-Y.; Shen, C.-I.; Harn, H.-J.; Lin, S.-Z.; et al. N-butylidenephthalide attenuates Alzheimer’s disease-like cytopathy in down syndrome induced pluripotent stem cell-derived neurons. Sci. Rep. 2015, 5, 8744. [Google Scholar] [CrossRef]
- Dashinimaev, E.B.; Artyuhov, A.S.; Bolshakov, A.P.; Vorotelyak, E.A.; Vasiliev, A.V. Neurons Derived from Induced Pluripotent Stem Cells of Patients with Down Syndrome Reproduce Early Stages of Alzheimer’s Disease Type Pathology in vitro. J. Alzheimer’s Dis. 2017, 56, 835–847. [Google Scholar] [CrossRef]
- Ovchinnikov, D.A.; Korn, O.; Virshup, I.; Wells, C.A.; Wolvetang, E.J. The impact of APP on Alzheimer-like pathogenesis and gene expression in down syndrome iPSC-derived neurons. Stem Cell Rep. 2018, 11, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Sposito, T.; Preza, E.; Mahoney, C.J.; Seto-Salvia, N.; Ryan, N.S.; Morris, H.R.; Arber, C.; Devine, M.J.; Houlden, H.; Warner, T.T.; et al. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum. Mol. Genet. 2015, 24, 5260–5269. [Google Scholar] [CrossRef]
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018, 97, 1284–1298.e7. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, H.; Ma, Y.; Shi, G.; Song, J.; Tang, Y.; Li, S.; Li, T.; Liu, N.; Tang, F.; et al. Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 2017, 8, 7900–7913. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; D’Avanzo, C.; Aronson, J.; Tanzi, R.E.; Kim, D.Y. Recapitulating amyloid β and tau pathology in human neural cell culture models: Clinical implications. US Neurol. 2015, 11, 102–105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Teglasi, A.; Bock, I.; Giudice, M.L.; Tancos, Z.; Molnar, K.; Laszlo, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Lee, H.K.; Velazquez Sanchez, C.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schroter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar] [CrossRef]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Bohnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Apolipoprotein e sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-based compound screening and in vitro trials identify a dynergistic snti-amyloid β combination for Alzheimer’s disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef]
- Jin, M.; O’Nuallain, B.; Hong, W.; Boyd, J.; Lagomarsino, V.N.; O’Malley, T.T.; Liu, W.; Vanderburg, C.R.; Frosch, M.P.; Young-Pearse, T.; et al. An in vitro paradigm to assess potential anti-Aβ antibodies for Alzheimer’s disease. Nat. Commun. 2018, 9, 2676. [Google Scholar] [CrossRef] [PubMed]
- Cavazzoni, P. FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 8 June 2021).
- Park, J.-C.; Jang, S.-Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.-H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid β and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, S.H.; D’Avanzo, C.; Hebisch, M.; Sliwinski, C.; Bylykbashi, E.; Washicosky, K.J.; Klee, J.B.; Brustle, O.; Tanzi, R.E.; et al. A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat. Protoc. 2015, 10, 985–1006. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Stuber, K.; Wunderlich, P.; Ladewig, J.; Kesavan, J.C.; Vandenberghe, R.; Vandenbulcke, M.; van Damme, P.; Walter, J.; Brustle, O.; et al. APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based γ-secretase modulation. Stem Cell Rep. 2013, 1, 491–498. [Google Scholar] [CrossRef]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Wang, M.; Li, A.; Sekiya, M.; Beckmann, N.D.; Quan, X.; Schrode, N.; Fernando, M.B.; Yu, A.; Zhu, L.; Cao, J.; et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron 2021, 109, 257–272.e14. [Google Scholar] [CrossRef] [PubMed]
- Biundo, R.; Weis, L.; Antonini, A. Cognitive decline in Parkinson’s disease: The complex picture. NPJ Parkinsons Dis. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Diederich, N.J.; Fenelon, G.; Stebbins, G.; Goetz, C.G. Hallucinations in Parkinson disease. Nat. Rev. Neurol. 2009, 5, 331–342. [Google Scholar] [CrossRef]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Trevor, A.J.; Castagnoli, N.; Singer, T.P. The formation of reactive intermediates in the MAO-catalyzed oxidation of the nigrostriatal toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 513–519. [Google Scholar] [CrossRef]
- Ungerstedt, U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 1968, 5, 107–110. [Google Scholar] [CrossRef]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef]
- Blandini, F.; Armentero, M.-T.; Martignoni, E. The 6-hydroxydopamine model: News from the past. Parkinsonism Relat. Disord. 2008, 14, S124–S129. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Parkinson’s Dis. 2020, 6, 8. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; de Bock, P.J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grunewald, A.; Seibler, P.; et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Lachenmayer, M.L.; Yue, Z. Genetic animal models for evaluating the role of autophagy in etiopathogenesis of Parkinson disease. Autophagy 2012, 8, 1837–1838. [Google Scholar] [CrossRef]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Treatment of the later stages of Parkinson’s disease—Pharmacological approaches now and in the future. Transl. Neurodegener. 2015, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K.; Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Zabierowski, S.; Dubose, B.N.; Hill, E.J.; Navare, M.; Claros, N.; Rosen, S.; Ramnarine, K.; Horn, C.; Fredrickson, C.; et al. Preclinical efficacy and safety of a human embryonic stem cell-derived midbrain dopamine progenitor product, MSK-DA01. Cell Stem Cell 2021, 28, 217–229.e7. [Google Scholar] [CrossRef]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 6, e26159. [Google Scholar] [CrossRef]
- Oliveira, L.M.; Falomir-Lockhart, L.J.; Botelho, M.G.; Lin, K.H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated α-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994. [Google Scholar] [CrossRef]
- Flierl, A.; Oliveira, L.M.A.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an α-csnuclein gene triplication. PLoS ONE 2014, 9, e112413. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Bjorklund, A. Modeling Parkinson’s disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. Alpha-Synuclein (alphaSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef]
- Gribaudo, S.; Tixador, P.; Bousset, L.; Fenyi, A.; Lino, P.; Melki, R.; Peyrin, J.M.; Perrier, A.L. Propagation of alpha-Synuclein Strains within Human Reconstructed Neuronal Network. Stem Cell Rep. 2019, 12, 230–244. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Pina-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. α-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef]
- Sanders, L.H.; Laganiere, J.; Cooper, O.; Mak, S.K.; Vu, B.J.; Huang, Y.A.; Paschon, D.E.; Vangipuram, M.; Sundararajan, R.; Urnov, F.D.; et al. LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol. Dis. 2014, 62, 381–386. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Carrion, M.D.P.; Marsicano, S.; Daniele, F.; Marte, A.; Pischedda, F.; Di Cairano, E.; Piovesana, E.; von Zweydorf, F.; Kremmer, E.; Gloeckner, C.J.; et al. The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci. Rep. 2017, 7, 5377. [Google Scholar] [CrossRef]
- Ma, D.; Ng, E.Y.; Zeng, L.; Lim, C.Y.; Zhao, Y.; Tan, E.K. Development of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient carrying the N551K variant in LRRK2 gene. Stem Cell Res. 2017, 18, 51–53. [Google Scholar] [CrossRef]
- Ma, D.; Zhou, W.; Ng, E.Y.; Zeng, L.; Zhao, Y.; Tan, E.K. Reprogramming of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient with a R1628P variant in the LRRK2 gene. Stem Cell Res. 2017, 18, 45–47. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, H.; Hu, Z.; Fan, K.; Wang, J.; Janoschka, S.; Wang, X.; Ge, S.; Feng, J. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 2015, 33, 68–78. [Google Scholar] [CrossRef][Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-Synuclein accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef]
- Oh, C.K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 attenuates PINK1/Parkin-eependent mitophagy in hiPSC-based parkinson’s disease models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef]
- Panagiotakopoulou, V.; Ivanyuk, D.; de Cicco, S.; Haq, W.; Arsić, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schöndorf, D.C.; Perez, M.-J.; et al. Interferon-γ signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef] [PubMed]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in parkinson’s disease models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef]
- Fernández-Santiago, R.; Carballo-Carbajal, I.; Castellano, G.; Torrent, R.; Richaud, Y.; Sánchez-Danés, A.; Vilarrasa-Blasi, R.; Sánchez-Pla, A.; Mosquera, J.L.; Soriano, J.; et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 2015, 7, 1529–1546. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Santiago, R.; Merkel, A.; Castellano, G.; Heath, S.; Raya, A.; Tolosa, E.; Marti, M.J.; Consiglio, A.; Ezquerra, M. Whole-genome DNA hyper-methylation in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. Clin. Epigenet. 2019, 11, 108. [Google Scholar] [CrossRef]
- Schulze, M.; Sommer, A.; Plötz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type calcium channels determine the vulnerability of dopaminergic neurons to mitochondrial stress in familial parkinson disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Ishikawa, K.I.; Inoshita, T.; Shiba-Fukushima, K.; Saiki, S.; Hatano, T.; Mori, A.; Oji, Y.; Okuzumi, A.; Li, Y.; et al. Identifying therapeutic agents for amelioration of mitochondrial clearance disorder in neurons of familial Parkinson disease. Stem Cell Rep. 2020, 14, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, N.; Prodromidou, K.; Kouroupi, G.; Samiotaki, M.; Panayotou, G.; Xilouri, M.; Stefanis, L.; Grailhe, R.; Taoufik, E.; Matsas, R. High content screening and proteomic analysis identify the kinase Inhibitor BX795 as a potent neuroprotective compound in a patient-derived model of parkinson’s disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Björklund, A.; et al. Long-term Clinical Outcome of Fetal Cell Transplantation for Parkinson Disease: Two Case Reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Morizane, A.; Kikuchi, T.; Hayashi, T.; Mizuma, H.; Takara, S.; Doi, H.; Mawatari, A.; Glasser, M.F.; Shiina, T.; Ishigaki, H.; et al. MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nat. Commun. 2017, 8, 385. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Serio, A.; Patani, R. Concise Review: The Cellular Conspiracy of Amyotrophic Lateral Sclerosis. Stem Cells 2018, 36, 293–303. [Google Scholar] [CrossRef]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.T.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human Motor Neurons With SOD1-G93A Mutation Generated From CRISPR/Cas9 Gene-Edited iPSCs Develop Pathological Features of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2020, 14, 604171. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Hall, C.E.; Sibley, C.R.; Cymes, T.; Forostyak, S.; Carlino, G.; Meyer, I.F.; Schiavo, G.; Zhang, S.-C.; Gibbons, G.M.; et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat. Commun. 2017, 8, 1164. [Google Scholar] [CrossRef]
- Kelley, K.W.; Ben Haim, L.; Schirmer, L.; Tyzack, G.E.; Tolman, M.; Miller, J.G.; Tsai, H.-H.; Chang, S.M.; Molofsky, A.V.; Yang, Y.; et al. Kir4.1-dependent astrocyte-fast motor neuron interactions wre required for peak strength. Neuron 2018, 98, 306–319.e7. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Devlin, A.C.; Burr, K.; Borooah, S.; Foster, J.D.; Cleary, E.M.; Geti, I.; Vallier, L.; Shaw, C.E.; Chandran, S.; Miles, G.B. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 2015, 6, 5999. [Google Scholar] [CrossRef] [PubMed]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef]
- Zhang, Z.; Almeida, S.; Lu, Y.; Nishimura, A.L.; Peng, L.; Sun, D.; Wu, B.; Karydas, A.M.; Tartaglia, M.C.; Fong, J.C.; et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS ONE 2013, 8, e76055. [Google Scholar] [CrossRef]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Kefalakes, E.; Kassebaum, C.; Bursch, F.; Lojewski, X.; et al. 4-Aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells 2016, 34, 1563–1575. [Google Scholar] [CrossRef]
- Liu, M.L.; Zang, T.; Zhang, C.L. Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 2016, 14, 115–128. [Google Scholar] [CrossRef]
- Higelin, J.; Demestre, M.; Putz, S.; Delling, J.P.; Jacob, C.; Lutz, A.K.; Bausinger, J.; Huber, A.K.; Klingenstein, M.; Barbi, G.; et al. FUS mislocalization and vulnerability to DNA damage in ALS patients derived hiPSCs and aging motoneurons. Front. Cell. Neurosci. 2016, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; Jensen, B.K.; Wen, X.; Cai, J.; Kropf, E.; Iacovitti, L.; Pasinelli, P.; Trotti, D. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 2016, 17, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Trageser, K.J.; Smith, C.; Herman, F.J.; Ono, K.; Pasinetti, G.M. Mechanisms of Immune Activation by c9orf72-Expansions in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2019, 13, 1298. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Frick, P.; Neumann, M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014, 127, 347–357. [Google Scholar] [CrossRef]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; de Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef]
- Lorenzini, I.; Alsop, E.; Levy, J.; Gittings, L.M.; Rabichow, B.E.; Lall, D.; Moore, S.; Bustos, L.; Pevey, R.; Burciu, C.; et al. Activated iPSC-microglia from C9orf72 ALS/FTD patients exhibit endosomal-lysosomal dysfunction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Noto, Y.; Shibuya, K.; Vucic, S.; Kiernan, M.C. Novel therapies in development that inhibit motor neuron hyperexcitability in amyotrophic lateral sclerosis. Expert Rev. Neurother. 2016, 16, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, M.O.; Heuberger, J.; Sleutjes, B.; Ziagkos, D.; van den Berg, L.H.; Ferguson, T.A.; Franssen, H.; Groeneveld, G.J. Acute effects of Riluzole and Retigabine on axonal excitability in patients with Amyotrophic lateral sclerosis: A randomized, double-blind, placebo-controlled, crossover trial. Clin. Pharmacol. Ther. 2018, 104, 1136–1145. [Google Scholar] [CrossRef]
- De Boer, A.S.; Eggan, K. A perspective on stem cell modeling of amyotrophic lateral sclerosis. Cell Cycle 2015, 14, 3679–3688. [Google Scholar] [CrossRef]
- Huang, X.; Roet, K.C.D.; Zhang, L.; Brault, A.; Berg, A.P.; Jefferson, A.B.; Klug-McLeod, J.; Leach, K.L.; Vincent, F.; Yang, H.; et al. Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 2021, 35, 109224. [Google Scholar] [CrossRef]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Model. Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Yoon, S.J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Velasco, V.; Shariati, S.A.; Esfandyarpour, R. Microtechnology-based methods for organoid models. Microsyst. Nanoeng. 2020, 6, 76. [Google Scholar] [CrossRef]
- Carlson, A.L.; Bennett, N.K.; Francis, N.L.; Halikere, A.; Clarke, S.; Moore, J.C.; Hart, R.P.; Paradiso, K.; Wernig, M.; Kohn, J.; et al. Generation and transplantation of reprogrammed human neurons in the brain using 3D microtopographic scaffolds. Nat. Commun. 2016, 7, 10862. [Google Scholar] [CrossRef]
- McMurtrey, R.J. Patterned and functionalized nanofiber scaffolds in three-dimensional hydrogel constructs enhance neurite outgrowth and directional control. J. Neural Eng. 2014, 11, 066009. [Google Scholar] [CrossRef]
- Carballo-Molina, O.A.; Sánchez-Navarro, A.; López-Ornelas, A.; Lara-Rodarte, R.; Salazar, P.; Campos-Romo, A.; Ramos-Mejía, V.; Velasco, I. Semaphorin 3C released from a biocompatible hydrogel guides and promotes axonal growth of rodent and human dopaminergic neurons. Tissue Eng. Part A 2016, 22, 850–861. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Oksdath, M.; Perrin, S.L.; Bardy, C.; Hilder, E.F.; DeForest, C.A.; Arrua, R.D.; Gomez, G.A. Review: Synthetic scaffolds to control the biochemical, mechanical, and geometrical environment of stem cell-derived brain organoids. APL Bioeng. 2018, 2, 041501. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Hsu, C.-C.; Nguyen, L.T.B.; Ye, H.; Cui, Z. Neural tissue engineering with structured hydrogels in CNS models and therapies. Biotechnol. Adv. 2020, 42, 107370. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, D.S.; Barrasa, M.I.; Shu, J.; Rietjens, R.; Zhang, S.; Mitalipova, M.; Berube, P.; Fu, D.; Shultz, L.D.; Bell, G.W.; et al. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 25293–25303. [Google Scholar] [CrossRef]
- D’Alessio, R.; Koukouli, F.; Blanchard, S.; Catteau, J.; Raïs, C.; Lemonnier, T.; Féraud, O.; Bennaceur-Griscelli, A.; Groszer, M.; Maskos, U. Long-term development of human iPSC-derived pyramidal neurons quantified after transplantation into the neonatal mouse cortex. Dev. Biol. 2020, 461, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Real, R.; Peter, M.; Trabalza, A.; Khan, S.; Smith, M.A.; Dopp, J.; Barnes, S.J.; Momoh, A.; Strano, A.; Volpi, E.; et al. In vivo modeling of human neuron dynamics and Down syndrome. Science 2018, 362. [Google Scholar] [CrossRef]
- Soldner, F.; Jaenisch, R. Stem cells, genome editing, and the path to translational medicine. Cell 2018, 175, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Okuno, H.; Nakabayashi, K.; Abe, K.; Ando, T.; Sanosaka, T.; Kohyama, J.; Akamatsu, W.; Ohyama, M.; Takahashi, T.; Kosaki, K.; et al. Changeability of the fully methylated status of the 15q11.2 region in induced pluripotent stem cells derived from a patient with Prader-Willi syndrome. Congenit. Anom. 2017, 57, 96–103. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Ambasudhan, R.; Talantova, M.; Coleman, R.; Yuan, X.; Zhu, S.; Lipton, S.A.; Ding, S. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 2011, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, D.G.; Kim, W.K.; McCoy, M.J.; Lake, A.M.; Ouwenga, R.; Lee, S.W.; Xing, X.; Li, D.; Lee, H.J.; Heuckeroth, R.O.; et al. MicroRNAs induce a permissive chromatin environment that enables neuronal subtype-specific reprogramming of adult human fibroblasts. Cell Stem Cell 2017, 21, 332–348.e9. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef]
- Bhaduri, A.; Andrews, M.G.; Kriegstein, A.R.; Nowakowski, T.J. Are organoids ready for prime time? Cell Stem Cell 2020, 27, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Todhunter, M.E.; LaBarge, M.A.; Gartner, Z.J. Opportunities for organoids as new models of aging. J. Cell Biol. 2018, 217, 39–50. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trudler, D.; Ghatak, S.; Lipton, S.A. Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 8196. https://doi.org/10.3390/ijms22158196
Trudler D, Ghatak S, Lipton SA. Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(15):8196. https://doi.org/10.3390/ijms22158196
Chicago/Turabian StyleTrudler, Dorit, Swagata Ghatak, and Stuart A. Lipton. 2021. "Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 15: 8196. https://doi.org/10.3390/ijms22158196
APA StyleTrudler, D., Ghatak, S., & Lipton, S. A. (2021). Emerging hiPSC Models for Drug Discovery in Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(15), 8196. https://doi.org/10.3390/ijms22158196