
Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
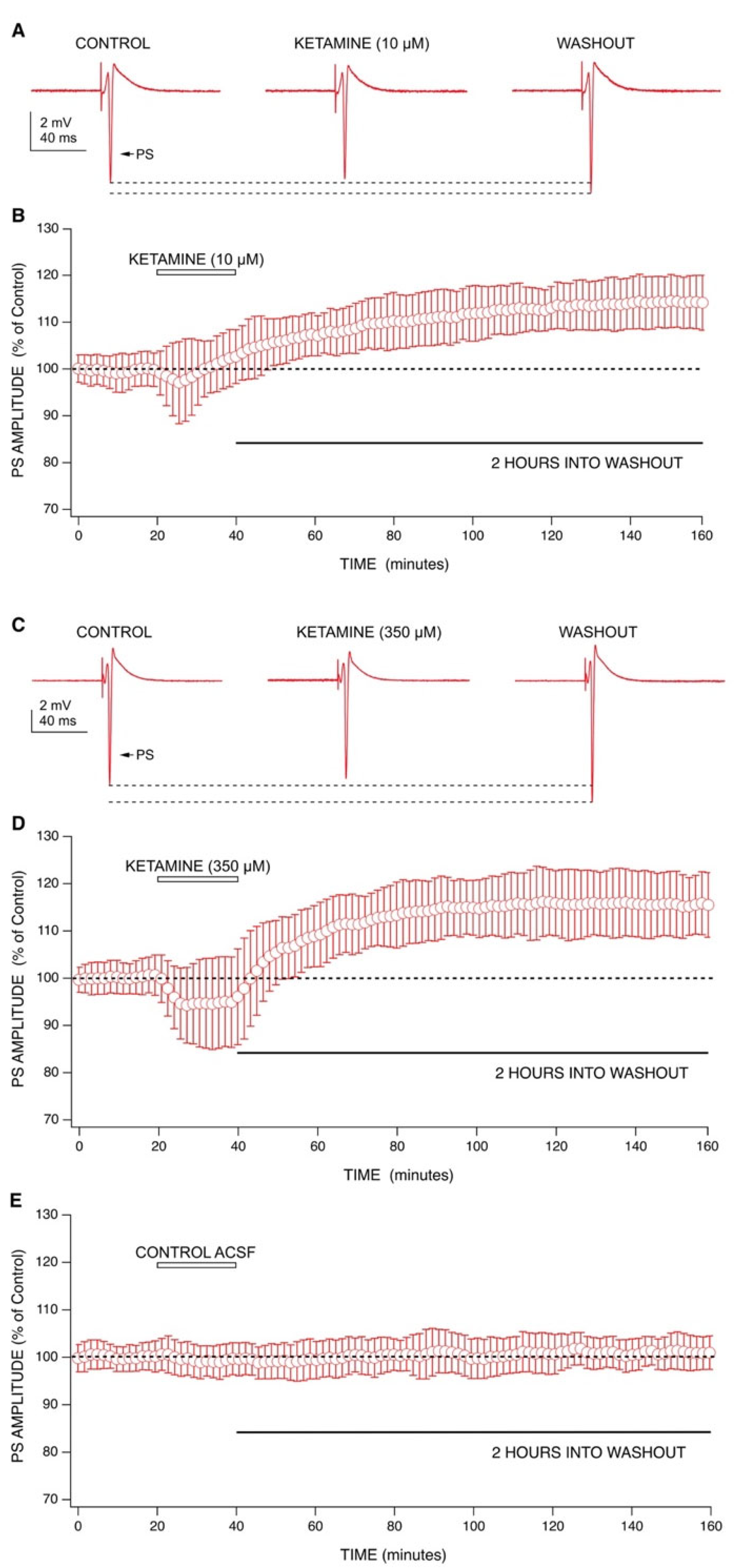
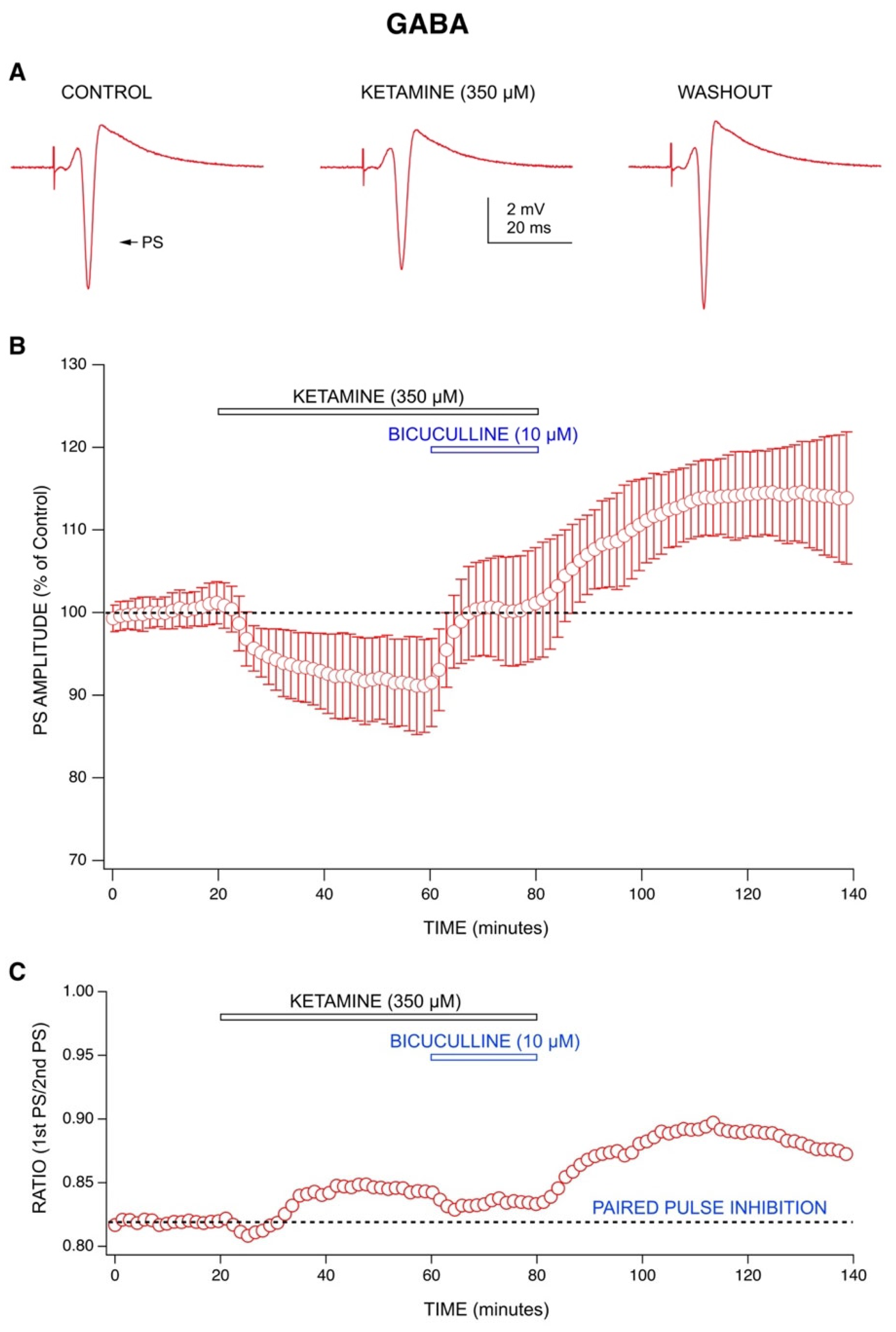
2.1. Ketamine’s Acute Drug Effect: Decrease in Population Spike (PS) Amplitudes with an Enhancement of GABA-Mediated Inhibition
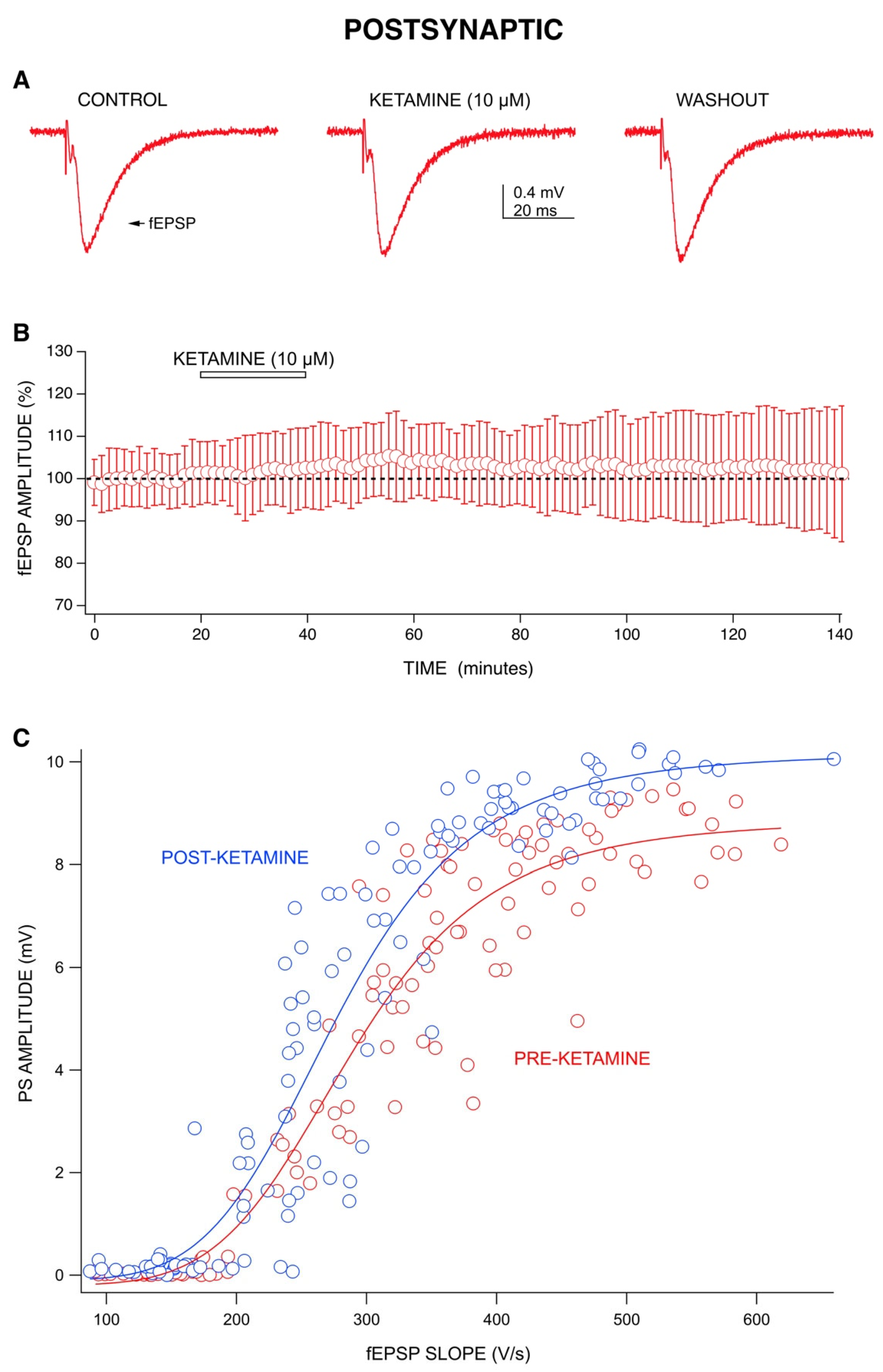
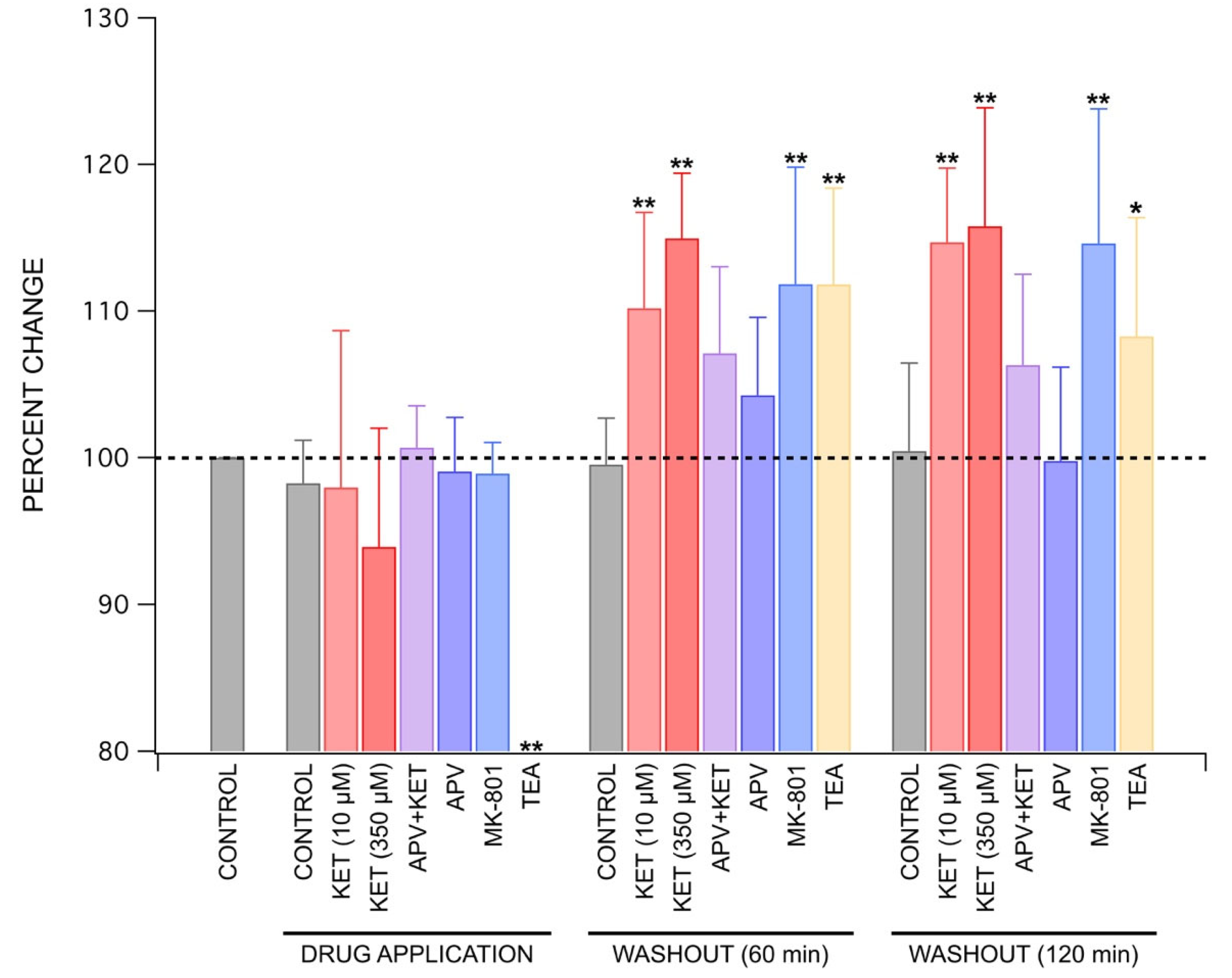
2.2. Ketamine’s Long-Lasting Effect: Increase in PS Amplitudes upon Washout Is Produced by a Post-Synaptic Mechanism
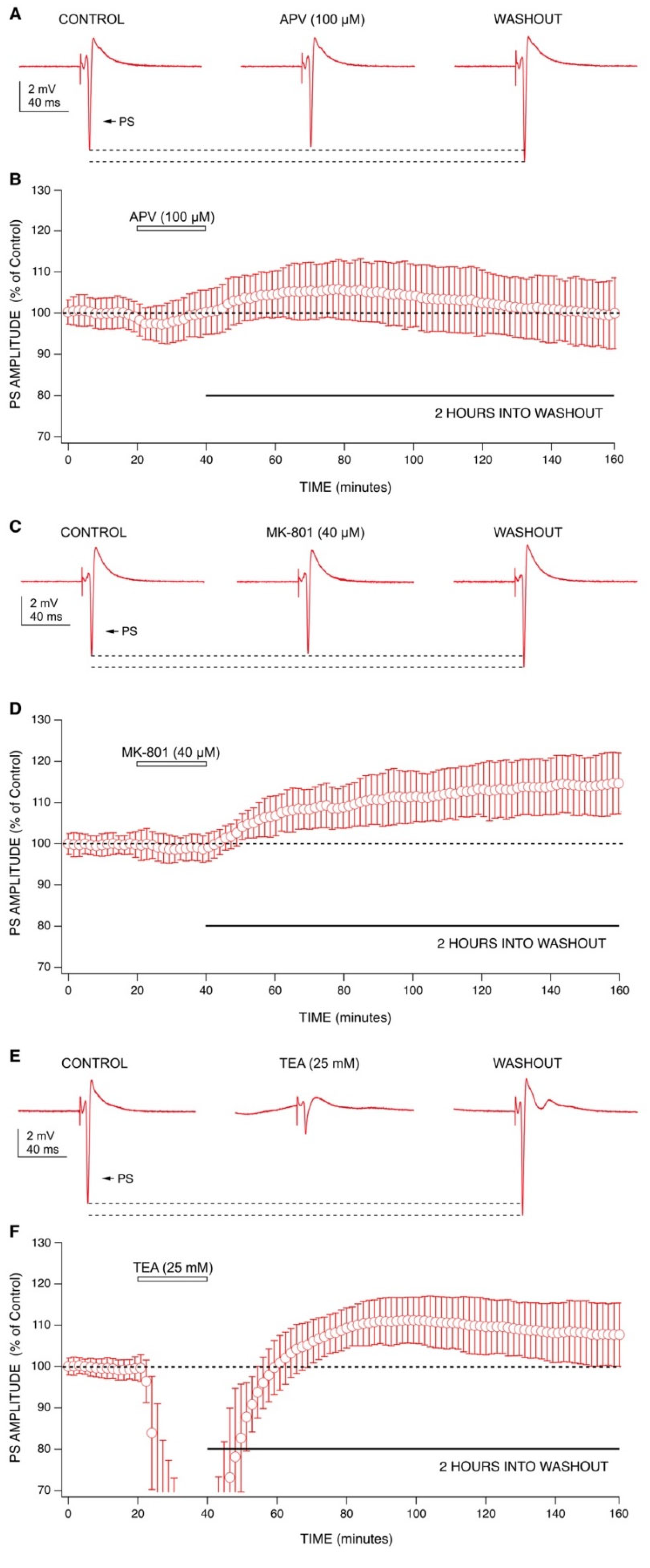
2.3. Mechanisms behind Ketamine’s Long-Lasting Effect May Involve NMDA Receptor Antagonism
3. Discussion
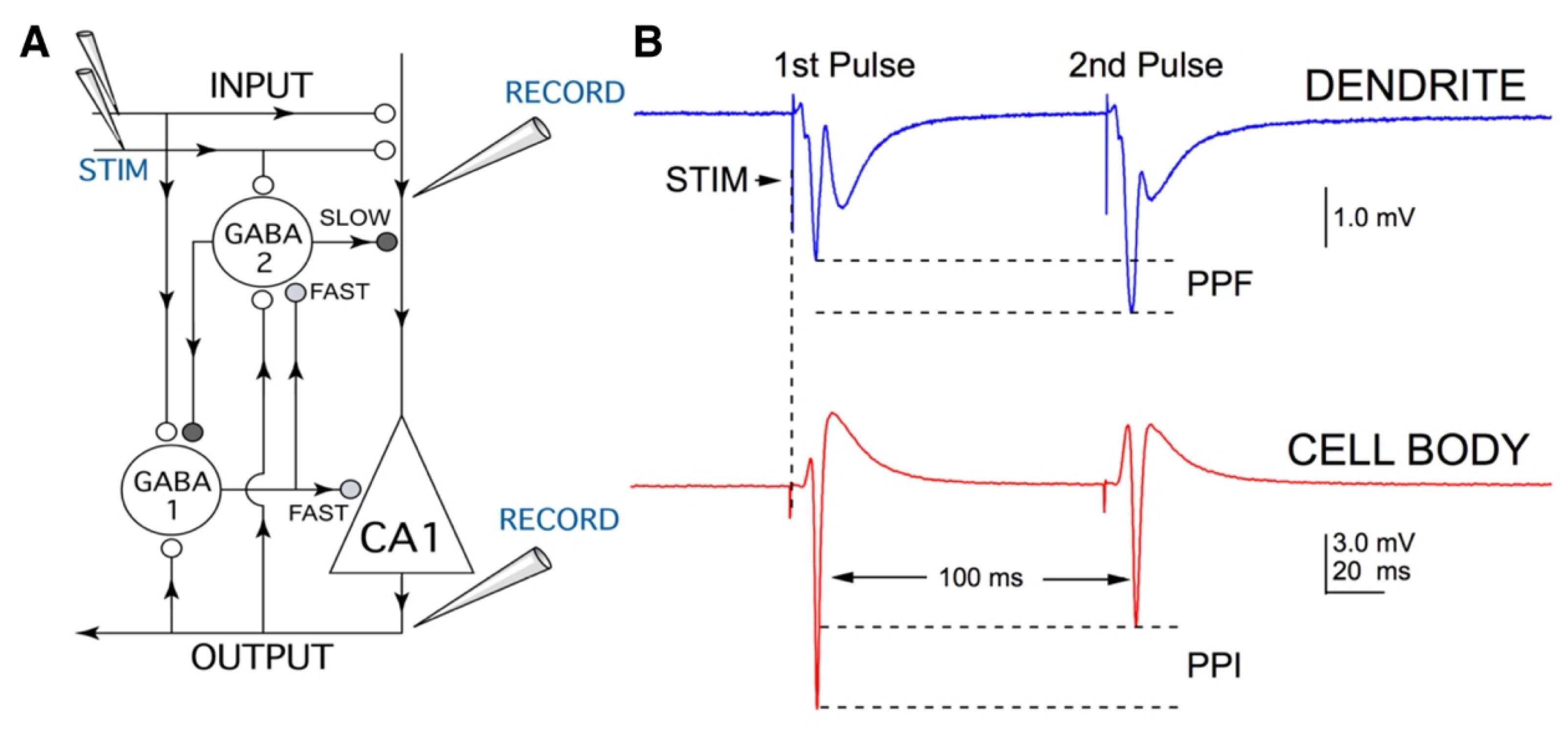
4. Materials and Methods
4.1. Brain Slice Preparation
4.2. Electrophysiological Recordings
4.3. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dundee, J.W.; Knox, J.W.; Black, G.W.; Moore, J.; Pandit, S.K.; Bovill, J.; Clarke, R.S.; Love, S.H.; Elliott, J.; Coppel, D.L. Ketamine as an Induction Agent in Anaesthetics. Lancet 1970, 1, 1370–1371. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.; Kavalali, E.T.; Monteggia, L.M. NMDA Receptor Blockade at Rest Triggers Rapid Behavioural Antidepressant Responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Nosyreva, E.; Szabla, K.; Autry, A.E.; Ryazanov, A.G.; Monteggia, L.M.; Kavalali, E.T. Acute Suppression of Spontaneous Neurotransmission Drives Synaptic Potentiation. J. Neurosci. 2013, 33, 6990–7002. [Google Scholar] [CrossRef]
- Wang, D.-S.; Penna, A.; Orser, B.A. Ketamine Increases the Function of γ-Aminobutyric Acid Type A Receptors in Hippocampal and Cortical Neurons. Anesthesiology 2017, 126, 666–677. [Google Scholar] [CrossRef]
- Kamiyama, H.; Matsumoto, M.; Otani, S.; Kimura, S.-I.; Shimamura, K.-I.; Ishikawa, S.; Yanagawa, Y.; Togashi, H. Mechanisms Underlying Ketamine-Induced Synaptic Depression in Rat Hippocampus-Medial Prefrontal Cortex Pathway. Neuroscience 2011, 177, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Durieux, M.E. Inhibition by Ketamine of Muscarinic Acetylcholine Receptor Function. Anesth. Analg. 1995, 81, 57–62. [Google Scholar] [PubMed]
- Hustveit, O.; Maurset, A.; Oye, I. Interaction of the Chiral Forms of Ketamine with Opioid, Phencyclidine, Sigma and Muscarinic Receptors. Pharmacol. Toxicol. 1995, 77, 355–359. [Google Scholar] [CrossRef]
- Chen, X.; Shu, S.; Bayliss, D.A. HCN1 Channel Subunits Are a Molecular Substrate for Hypnotic Actions of Ketamine. J. Neurosci. 2009, 29, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Voss, L.J.; Karalus, S.; Englund, V.; Sleigh, J.W. Ketamine Action in the In Vitro Cortical Slice Is Mitigated by Potassium Channel Blockade. Anesthesiology 2018, 128, 1167–1174. [Google Scholar] [CrossRef]
- Schnoebel, R.; Wolff, M.; Peters, S.C.; Bräu, M.E.; Scholz, A.; Hempelmann, G.; Olschewski, H.; Olschewski, A. Ketamine Impairs Excitability in Superficial Dorsal Horn Neurones by Blocking Sodium and Voltage-Gated Potassium Currents. Br. J. Pharmacol. 2005, 146, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Friederich, P.; Benzenberg, D.; Urban, B.W. Ketamine and Propofol Differentially Inhibit Human Neuronal K(+) Channels. Eur J. Anaesthesiol. 2001, 18, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.M.; Han, M.-H. HCN Channel Targets for Novel Antidepressant Treatment. Neurotherapeutics 2017, 14, 698–715. [Google Scholar] [CrossRef]
- Brau, M.E.; Sander, F.; Vogel, W.; Hempelmann, G. Blocking Mechanisms of Ketamine and Its Enantiomers in Enzymatically Demyelinated Peripheral Nerve as Revealed by Single-Channel Experiments. Anesthesiology 1997, 86, 394–404. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR Inhibition-Independent Antidepressant Actions of Ketamine Metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, E.W.; Troppoli, T.A.; Myers, S.J.; Zanos, P.; Aracava, Y.; Kehr, J.; Lovett, J.; Kim, S.; Wang, F.-H.; Schmidt, S.; et al. Antidepressant-Relevant Concentrations of the Ketamine Metabolite (2 R,6 R)-Hydroxynorketamine Do Not Block NMDA Receptor Function. Proc. Natl. Acad. Sci. USA 2019, 116, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Bliss, T.V.P.; Skrede, K.K. Lamellar Organization of Hippocampal Excitatory Pathways. Exp. Brain Res. 1971, 13, 222–238. [Google Scholar] [CrossRef]
- Andersen, P.; Eccles, J.C.; Lyning, Y. Pathway of Postsynaptic Inhibition in the Hippocampus. J. Neurophysiol. 1964, 27, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.F.; Sanford, R.L.; Lee, W.; Andersen, O.S.; Hemmings, H.C. Clinical Concentrations of Chemically Diverse General Anesthetics Minimally Affect Lipid Bilayer Properties. Proc. Natl. Acad. Sci. USA 2017, 114, 3109–3114. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Skiteva, O.; Zhang, X.; Svenningsson, P.; Chergui, K. Ketamine and Its Metabolite (2R,6R)-Hydroxynorketamine Induce Lasting Alterations in Glutamatergic Synaptic Plasticity in the Mesolimbic Circuit. Mol. Psychiatry 2018, 23, 2066–2077. [Google Scholar] [CrossRef]
- Stringer, J.L.; Guyenet, P.G. Elimination of Long-Term Potentiation in the Hippocampus by Phencyclidine and Ketamine. Brain Res. 1983, 258, 159–164. [Google Scholar] [CrossRef]
- Franks, N.P.; Lieb, W.R. Molecular and Cellular Mechanisms of General Anaesthesia. Nature 1994, 367, 607–614. [Google Scholar] [CrossRef]
- Nishikawa, K.; MacIver, M.B. Excitatory Synaptic Transmission Mediated by NMDA Receptors Is More Sensitive to Isoflurane than Are Non-NMDA Receptor-Mediated Responses. Anesthesiology 2000, 92, 228. [Google Scholar] [CrossRef]
- Song, X.; Jensen, M.Ø.; Jogini, V.; Stein, R.A.; Lee, C.-H.; Mchaourab, H.S.; Shaw, D.E.; Gouaux, E. Mechanism of NMDA Receptor Channel Block by MK-801 and Memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef]
- Kleinhaus, A.L.; Prichard, J.W. Calcium Dependent Action Potentials Produced in Leech Retzius Cells by Tetraethylammonium Chloride. J. Physiol. 1975, 246, 351–369. [Google Scholar] [CrossRef][Green Version]
- Peet, M.J.; McLennan, H. Pre-and Postsynaptic Actions of Baclofen: Blockade of the Late Synaptically-Evoked Hyperpolarization of CA1 Hippocampal Neurones. Exp. Brain Res. 1986, 61, 567–574. [Google Scholar] [CrossRef]
- MacIver, M.B. Anesthetic Agent-Specific Effects on Synaptic Inhibition. Anesth. Analg. 2014, 119, 558–569. [Google Scholar] [CrossRef]
- Ali, F.; Gerhard, D.M.; Sweasy, K.; Pothula, S.; Pittenger, C.; Duman, R.S.; Kwan, A.C. Ketamine Disinhibits Dendrites and Enhances Calcium Signals in Prefrontal Dendritic Spines. Nat. Commun. 2020, 11, 72. [Google Scholar] [CrossRef]
- Björkholm, C.; Monteggia, L.M. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Takeyama, K.; Yoshikawa, M.; Oka, T.; Kawaguchi, M.; Suzuki, T.; Hashimoto, A. Ketamine Enhances the Expression of Serine Racemase and D-Amino Acid Oxidase MRNAs in Rat Brain. Eur. J. Pharmacol. 2006, 540, 82–86. [Google Scholar] [CrossRef]
- Panatier, A.; Theodosis, D.T.; Mothet, J.-P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H.R. Glia-Derived d-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, D.; Lamas, J.A. Metabotropic Modulation of Potassium Channels During Synaptic Plasticity. Neuroscience 2020. [Google Scholar] [CrossRef]
- Huang, M.-H.; Lin, K.-H.; Chen, S.-J.; Shen, A.-Y.; Wu, F.-T.; Wu, S.-N. Effects of Ketamine and Its Metabolites on Ion Currents in Differentiated Hippocampal H19-7 Neuronal Cells and in HEK293T Cells Transfected with α-Hslo Subunit. Neurotoxicology 2012, 33, 1058–1066. [Google Scholar] [CrossRef]
- Saland, S.K.; Kabbaj, M. Sex Differences in the Pharmacokinetics of Low-Dose Ketamine in Plasma and Brain of Male and Female Rats. J. Pharmacol. Exp. Ther. 2018, 367, 393–404. [Google Scholar] [CrossRef]
- Thelen, C.; Flaherty, E.; Saurine, J.; Sens, J.; Mohamed, S.; Pitychoutis, P.M. Sex Differences in the Temporal Neuromolecular and Synaptogenic Effects of the Rapid-Acting Antidepressant Drug Ketamine in the Mouse Brain. Neuroscience 2019, 398, 182–192. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Kounelis-Wuillaume, S.K.; Gheidi, A.; Morrow, J.D.; Spencer-Segal, J.L.; Watson, B.O. Sex- and Stress-Dependent Effects of a Single Injection of Ketamine on Open Field and Forced Swim Behavior. Stress 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.-Y.; Wang, Y.-H.; Liu, W.-G.; Yao, J.-Q.; Yuan, J.; Li, Z.-H.; Ran, Y.-H.; Zhang, L.-M.; Li, Y.-F. The Role of the Excitation:Inhibition Functional Balance in the MPFC in the Onset of Antidepressants. Neuropharmacology 2021, 191, 108573. [Google Scholar] [CrossRef]
- Pandya, M.; Altinay, M.; Malone, D.A.; Anand, A. Where in the Brain Is Depression? Curr. Psychiatry Rep. 2012, 14, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Gredell, J.A.; Turnquist, P.A.; MacIver, M.B.; Pearce, R.A. Determination of Diffusion and Partition Coefficients of Propofol in Rat Brain Tissue: Implications for Studies of Drug Action in Vitro. Br. J. Anaesth. 2004, 93, 810–817. [Google Scholar] [CrossRef]
- Hagan, C.E.; Pearce, R.A.; Trudell, J.R.; MacIver, M.B. Concentration Measures of Volatile Anesthetics in the Aqueous Phase Using Calcium Sensitive Electrodes. J. Neurosci. Methods 1998, 81, 177–184. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, G.; MacIver, M.B. Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability. Int. J. Mol. Sci. 2021, 22, 8091. https://doi.org/10.3390/ijms22158091
Jang G, MacIver MB. Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability. International Journal of Molecular Sciences. 2021; 22(15):8091. https://doi.org/10.3390/ijms22158091
Chicago/Turabian StyleJang, Grace, and M. Bruce MacIver. 2021. "Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability" International Journal of Molecular Sciences 22, no. 15: 8091. https://doi.org/10.3390/ijms22158091
APA StyleJang, G., & MacIver, M. B. (2021). Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability. International Journal of Molecular Sciences, 22(15), 8091. https://doi.org/10.3390/ijms22158091