
Uncovering the Mechanisms of Adenosine Receptor-Mediated Pain Control: Focus on the A3 Receptor Subtype

 ,
,  ,
,  ,
,  ,
,  ,
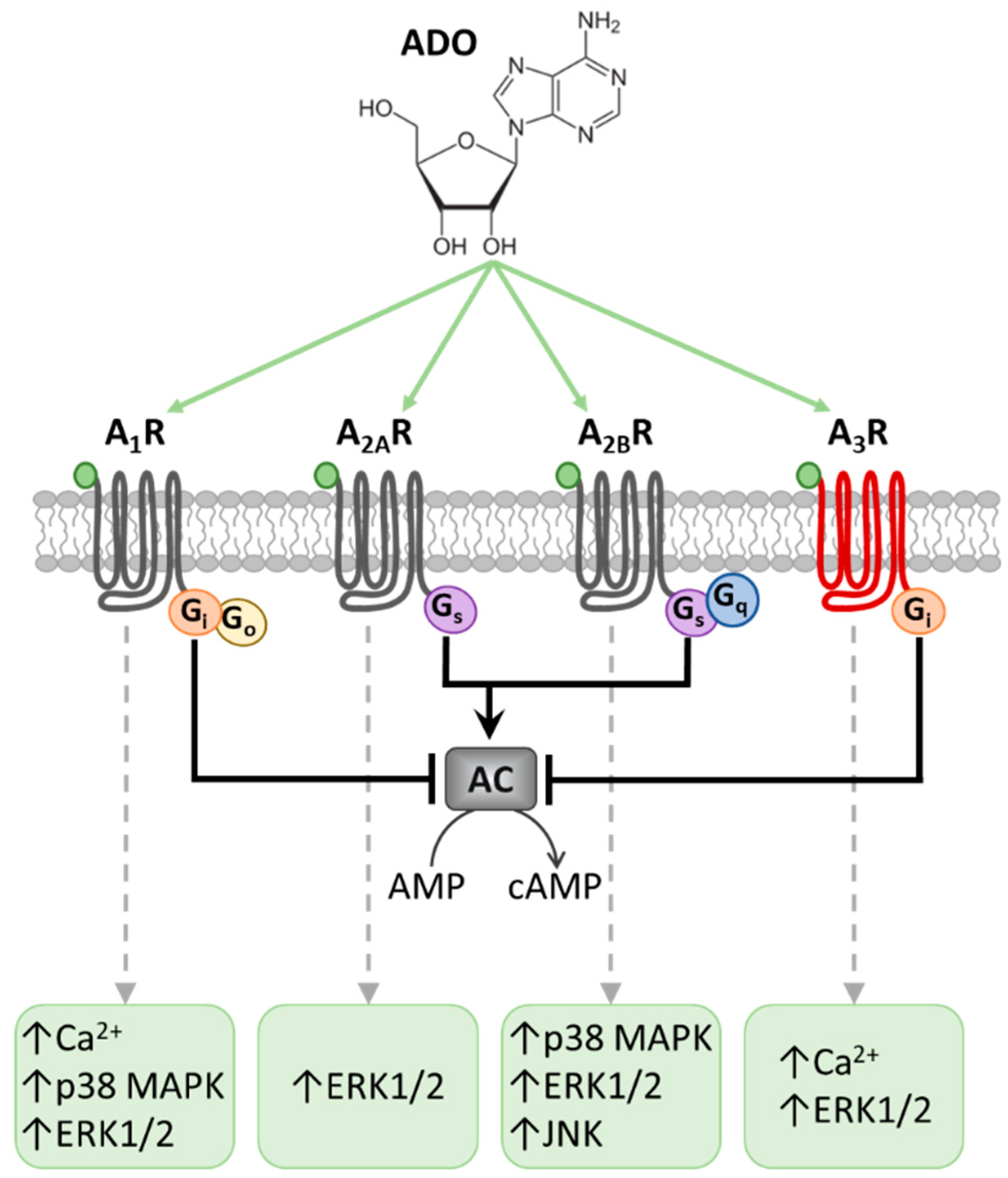
, 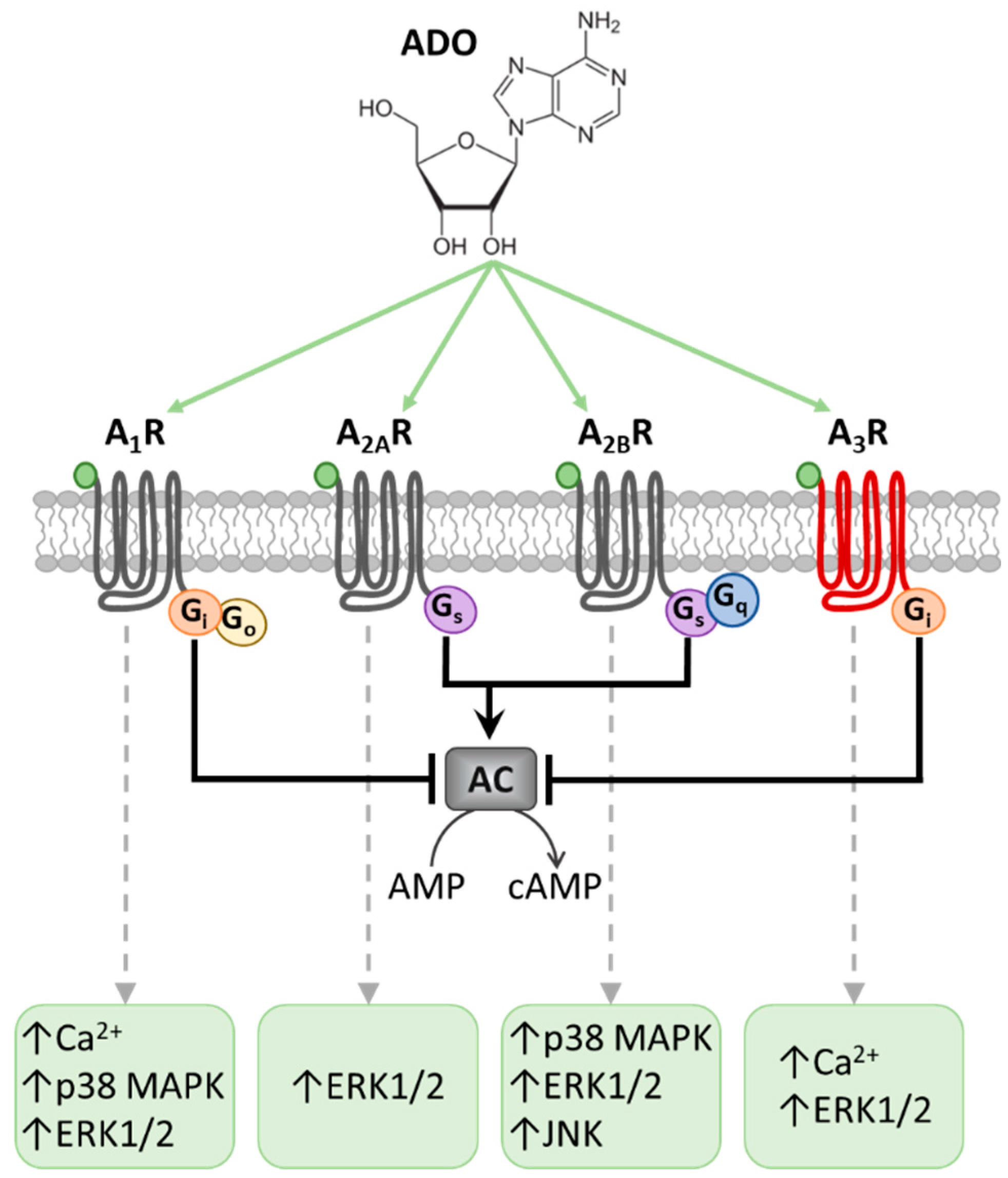{kind=link}
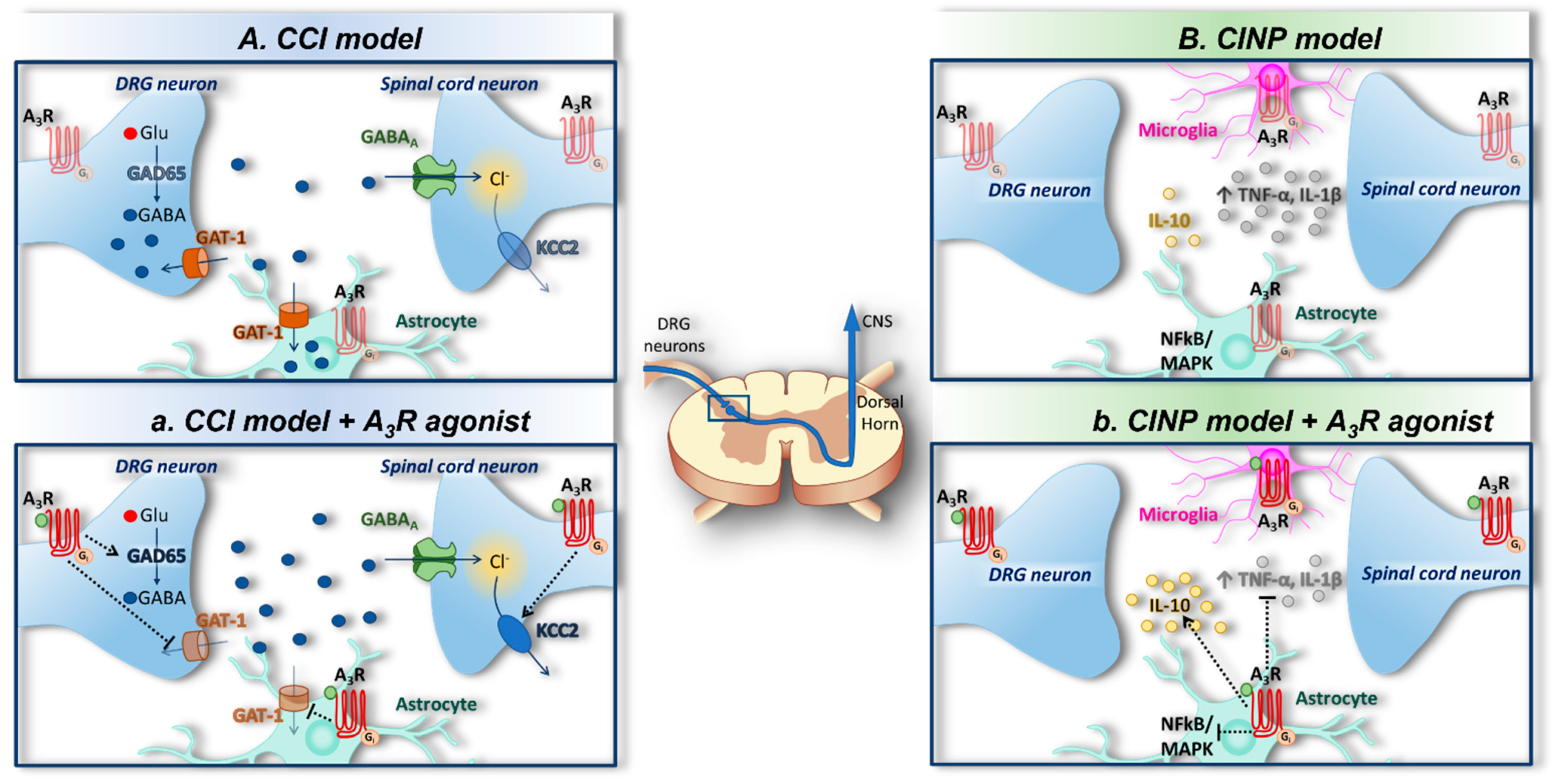{kind=link}
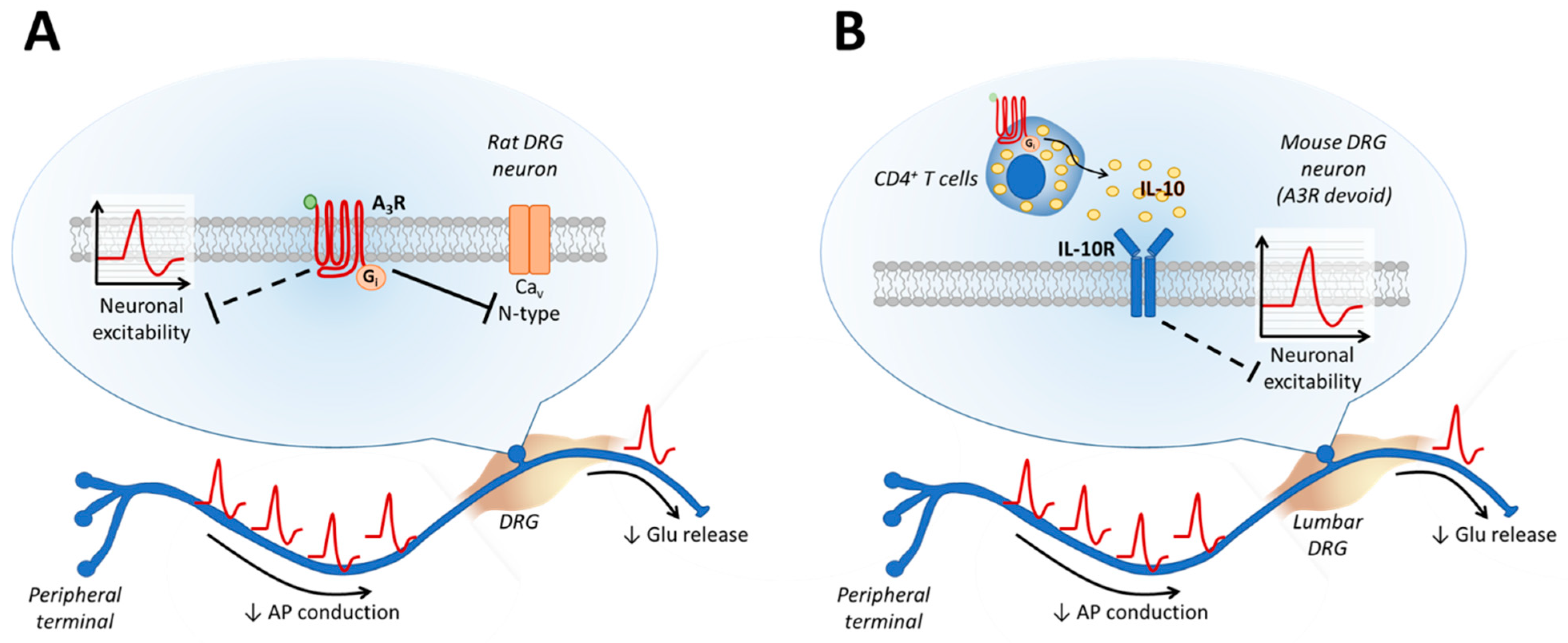{kind=link}
Abstract
:1. Introduction
2. A3R and Pain
3. Mechanisms of A3R-Mediated Pain Control
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A1Rs, A2ARs, A2BRs or A3Rs | A1, A2A, A2B or A3 receptors |
| AC | adenylyl cyclase |
| AK | adenosine kinase |
| AR | adensoine receptor |
| cAMP | cyclic adenosine monophosphate |
| CINP | chemotherapy-induced neuropathic pain |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| DNBS | dinitrobenzenesulfonic acid |
| DRG | dorsal root ganglion |
| ERK1/2 | extracellular signal-regulated kinase ½ |
| GABA | γ-aminobutyric acid |
| GAD65 | Glutamic Acid Decarboxylase 65-kDa |
| GAT-1 | GABA transporter 1 |
| GPCR | G protein-coupled receptors |
| ICa | Ca2+ current |
| IL | interleukin: |
| i.p. | intraperitoneal |
| i.pl | intraplantar |
| i.t. | intrathecal |
| JNK | Jc-Jun-NH2 terminal Kinase JUN N-terminal kinase |
| KCC2 | K+-Cl− co-transporter 2 |
| KO | knock out |
| MAPK | mitogen-activated protein kinase |
| NFκB | nuclear factor k-light-chain-enhancer of activated B cells |
| PKA | protein kinase A |
| PKC | protein kinase C |
| TNFα | tumor necrosis factor-α |
| VDCC | voltage-dependent Ca2+ channel |
References
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef]
- Guieu, R.; Deharo, J.C.; Maille, B.; Crotti, L.; Torresani, E.; Brignole, M.; Parati, G. Adenosine and the Cardiovascular System: The Good and the Bad. J. Clin. Med. 2020, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G.; Krugel, U.; Abbracchio, M.P.; Illes, P. Purinergic signalling: From normal behaviour to pathological brain function. Prog. Neurobiol. 2011, 95, 229–274. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Pedata, F.; Gibb, A.J. P2Y1 receptor modulation of Ca2+-activated K+ currents in medium-sized neurons from neonatal rat striatal slices. J. Neurophysiol. 2012, 107, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Maraula, G.; Traini, C.; Mello, T.; Coppi, E.; Galli, A.; Pedata, F.; Pugliese, A.M. Effects of oxygen and glucose deprivation on synaptic transmission in rat dentate gyrus: Role of A2A adenosine receptors. Neuropharmacology 2013, 67, 511–520. [Google Scholar] [CrossRef]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The Selective Antagonism of Adenosine A2B Receptors Reduces the Synaptic Failure and Neuronal Death Induced by Oxygen and Glucose Deprivation in Rat CA1 Hippocampus in Vitro. Front. Pharmacol. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Fusco, I.; Cherchi, F.; Catarzi, D.; Colotta, V.; Varano, F.; Pedata, F.; Pugliese, A.M.; Coppi, E. Functional characterization of a novel adenosine A2B receptor agonist on short-term plasticity and synaptic inhibition during oxygen and glucose deprivation in the rat CA1 hippocampus. Brain Res. Bull. 2019, 151, 174–180. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Pedata, F.; Pugliese, A.M. New Insight into the Role of Adenosine in Demyelination, Stroke and Neuropathic Pain. Front. Pharmacol. 2021, 11, 2403. [Google Scholar] [CrossRef]
- Cherchi, F.; Pugliese, A.M.; Coppi, E. Oligodendrocyte precursor cell maturation: Role of adenosine receptors. Neural Regen. Res. 2021, 16, 1686–1692. [Google Scholar]
- Muzzi, M.; Coppi, E.; Pugliese, A.M.; Chiarugi, A. Anticonvulsant effect of AMP by direct activation of adenosine A1 receptor. Exp. Neurol. 2013, 250, 189–193. [Google Scholar] [CrossRef]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Lana, D.; Giovannini, M.G.; Pedata, F.; Pugliese, A.M. A2B Adenosine Receptors: When Outsiders May Become an Attractive Target to Treat Brain Ischemia or Demyelination. Int. J. Mol. Sci. 2020, 21, 9697. [Google Scholar] [CrossRef] [PubMed]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Cronstein, B.N. Adenosine, an endogenous antiinflammatory agent. J. Appl. Physiol. 1994, 76, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronstein, B.N. A novel-approach to the development of antiinflammatory agents—Adenosine release at inflamed sites. J. Investig. Med. 1995, 43, 50–57. [Google Scholar] [PubMed]
- Cronstein, B.N.; Sitkovsky, M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef]
- Hasko, G.; Antonioli, L.; Cronstein, B.N. Adenosine metabolism, immunity and joint health. Biochem. Pharmacol. 2018, 151, 307–313. [Google Scholar] [CrossRef]
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338. [Google Scholar] [CrossRef]
- Yoon, M.H.; Bae, H.B.; Choi, J.I. Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth. Analg. 2005, 101, 1417–1421. [Google Scholar] [CrossRef]
- Taiwo, Y.O.; Levine, J.D. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience 1990, 38, 757–762. [Google Scholar] [CrossRef]
- Karlsten, R.; Gordh, T.; Post, C. Local antinociceptive and hyperalgesic effects in the formalin test after peripheral administration of adenosine analogues in mice. Pharmacol. Toxicol. 1992, 70 Pt 1, 434–438. [Google Scholar] [CrossRef]
- Doak, G.J.; Sawynok, J. Complex role of peripheral adenosine in the genesis of the response to subcutaneous formalin in the rat. Eur. J. Pharmacol. 1995, 281, 311–318. [Google Scholar] [CrossRef]
- Taiwo, Y.O.; Levine, J.D. further confirmation of the role of adenyl-cyclase and of camp-dependent protein-kinase in primary afferent hyperalgesia. Neuroscience 1991, 44, 131–135. [Google Scholar] [CrossRef]
- Khasar, S.G.; Wang, J.F.; Taiwo, Y.O.; Heller, P.H.; Green, P.G.; Levine, J.D. Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat—A G-protein beta-gamma subunit-mediated effect. Neuroscience 1995, 67, 189–195. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Skerritt, J.H.; Werz, M.A. Adenosine agonists reduce voltage-dependent calcium conductance of mouse sensory neurons in cell-culture. J. Physiol. 1986, 370, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Santicioli, P.; Delbianco, E.; Maggi, C.A. Adenosine-a1-receptors mediate the presynaptic inhibition of calcitonin gene-related peptide release by adenosine in the rat spinal-cord. Eur. J. Pharmacol. 1993, 231, 139–142. [Google Scholar] [CrossRef]
- Vincenzi, F.; Pasquini, S.; Borea, P.A.; Varani, K. Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain. Int. J. Mol. Sci. 2020, 21, 8710. [Google Scholar] [CrossRef] [PubMed]
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Kwilasz, A.J.; Ellis, A.; Wieseler, J.; Loram, L.; Favret, J.; McFadden, A.; Springer, K.; Falci, S.; Rieger, J.; Maier, S.F.; et al. Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A2A receptor agonists. Brain Behav. Immun. 2018, 69, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Falsini, M.; Catarzi, D.; Varano, F.; Ceni, C.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Lucarini, E.; et al. Antioxidant-Conjugated 1,2,4-Triazolo 4,3-a pyrazin-3-one Derivatives: Highly Potent and Selective Human A(2A) Adenosine Receptor Antagonists Possessing Protective Efficacy in Neuropathic Pain. J. Med. Chem. 2019, 62, 8511–8531. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Riccio, D.; Ceruti, S. Tackling Chronic Pain and Inflammation through the Purinergic System. Curr. Med. Chem. 2018, 25, 3830–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, S.J.; Nascimento, F.P.; Luiz-Cerutti, M.; Santos, A.R.S. The role of peripheral adenosine receptors in glutamate-induced pain nociceptive behavior. Purinergic Signal. 2021, 17, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Adebiyi, M.G.; Luo, J.L.; Sun, K.Q.; Le, T.T.T.; Zhang, Y.J.; Wu, H.Y.; Zhao, S.S.; Karmouty-Quintana, H.; Liu, H.; et al. Sustained Elevated Adenosine via ADORA2B Promotes Chronic Pain through Neuro-immune Interaction. Cell Rep. 2016, 16, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotanska, M.; Szafarz, M.L.; Mika, K.; Dziubina, A.; Bednarski, M.; Muller, C.E.; Sapa, J.; Kiec-Kononowicz, K. PSB 603—A known selective adenosine A2B receptor antagonist—Has anti-inflammatory activity in mice. Biomed. Pharmacother. 2021, 135, 111164. [Google Scholar] [CrossRef]
- Wei, W.; Du, C.S.; Lv, J.; Zhao, G.X.; Li, Z.X.; Wu, Z.Y.; Hasko, G.; Xie, X. Blocking A2B Adenosine Receptor Alleviates Pathogenesis of Experimental Autoimmune Encephalomyelitis via Inhibition of IL-6 Production and Th17 Differentiation. J. Immunol. 2013, 190, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loram, L.C.; Taylor, F.R.; Strand, K.A.; Harrison, J.A.; RzasaLynn, R.; Sholar, P.; Rieger, J.; Maier, S.F.; Watkins, L.R. Intrathecal injection of adenosine 2A receptor agonists reversed neuropathic allodynia through protein kinase (PK)A/PKC signaling. Brain Behav. Immun. 2013, 33, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, P.K.; Deka, S.; Borah, P.; Abed, S.N.; Klotz, K.N. Medicinal Chemistry and Therapeutic Potential of Agonists, Antagonists and Allosteric Modulators of A1 Adenosine Receptor: Current Status and Perspectives. Curr. Pharm. Des. 2019, 25, 2697–2715. [Google Scholar] [CrossRef]
- Luongo, L.; Salvemini, D. Targeting metabotropic adenosine receptors for neuropathic pain: Focus on A2A. Brain Behav. Immun. 2018, 69, 60–61. [Google Scholar] [CrossRef]
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J. Rheumatol. 2008, 35, 41–48. [Google Scholar]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawynok, J.; Reid, A. Role of G-proteins and adenylate-cyclase in antinociception produced by intrathecal purines. Eur. J. Pharmacol. 1988, 156, 25–34. [Google Scholar] [CrossRef]
- Sawynok, J. Adenosine receptor activation and nociception. Eur. J. Pharmacol. 1998, 347. [Google Scholar] [CrossRef]
- Sawynok, J.; Zarrindast, M.R.; Reid, A.R.; Doak, G.J. Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur. J. Pharmacol. 1997, 333. [Google Scholar] [CrossRef]
- Ramkumar, V.; Stiles, G.L.; Beaven, M.A.; Ali, H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast-cells. J. Biol. Chem. 1993, 268, 16887–16890. [Google Scholar] [CrossRef]
- Leung, C.T.; Li, A.; Banerjee, J.; Gao, Z.G.; Kambayashi, T.; Jacobson, K.A.; Civan, M.M. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 2014, 10, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.M.; Janes, K.; Chen, C.; Doyle, T.; Bryant, L.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012, 26, 1855–1865. [Google Scholar] [CrossRef] [Green Version]
- Little, J.W.; Ford, A.; Symons-Liguori, A.M.; Chen, Z.M.; Janes, K.; Doyle, T.; Xie, J.; Luongo, L.; Tosh, D.K.; Maione, S.; et al. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. [Google Scholar] [CrossRef]
- Ford, A.; Castonguay, A.; Cottet, M.; Little, J.W.; Chen, Z.; Symons-Liguori, A.M.; Doyle, T.; Egan, T.M.; Vanderah, T.W.; De Koninck, Y.; et al. Engagement of the GABA to KCC2 Signaling Pathway Contributes to the Analgesic Effects of A3AR Agonists in Neuropathic Pain. J. Neurosci. 2015, 35, 6057–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, K.; Esposito, E.; Doyle, T.; Cuzzocrea, S.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain 2014, 155, 2560–2567. [Google Scholar] [CrossRef] [Green Version]
- Janes, K.; Wahlman, C.; Little, J.W.; Doyle, T.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav. Immun. 2015, 44, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Paoletta, S.; Ford, A.; Janes, K.; Salvemini, D.; Jacobson, K.A. Discovery of next generation A3 adenosine receptor selective agonists for treatment of chronic neuropathic pain. In Abstracts of Papers of the American Chemical Society; 1155 16TH ST, NW; Amer Chemical Soc: Washington, DC, USA, 2014; Volume 247, p. 53. [Google Scholar]
- Paoletta, S.; Tosh, D.K.; Finley, A.; Gizewski, E.T.; Moss, S.M.; Gao, Z.G.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. Rational Design of Sulfonated A3 Adenosine Receptor-Selective Nucleosides as Pharmacological Tools to Study Chronic Neuropathic Pain. J. Med. Chem. 2013, 56, 5949–5963. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Ciancetta, A.; Warnick, E.; O’Connor, R.; Chen, Z.M.; Gizewski, E.; Crane, S.; Gao, Z.G.; Auchampach, J.A.; Salvemini, D.; et al. Purine (N)-Methanocarba Nucleoside Derivatives Lacking an Exocyclic Amine as Selective A3 Adenosine Receptor Agonists. J. Med. Chem. 2016, 59, 3249–3263. [Google Scholar] [CrossRef]
- Tosh, D.K.; Crane, S.; Chen, Z.M.; Paoletta, S.; Gao, Z.G.; Gizewski, E.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. Rigidified A3 Adenosine Receptor Agonists: 1-Deazaadenine Modification Maintains High in Vivo Efficacy. ACS Med. Chem. Lett. 2015, 6, 804–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Giancotti, L.A.; Lauro, F.; Mufti, F.; Salvemini, D. Treatment of chronic neuropathic pain: Purine receptor modulation. Pain 2020, 161, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Finley, A.; Paoletta, S.; Moss, S.M.; Gao, Z.G.; Gizewski, E.T.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. In Vivo Phenotypic Screening for Treating Chronic Neuropathic Pain: Modification of C2-Arylethynyl Group of Conformationally Constrained A3 Adenosine Receptor Agonists. J. Med. Chem. 2014, 57, 9901–9914. [Google Scholar] [CrossRef] [Green Version]
- Tosh, D.K.; Padia, J.; Salvemini, D.; Jacobson, K.A. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 2015, 11, 371–387. [Google Scholar] [CrossRef] [Green Version]
- Wahlman, C.; Doyle, T.M.; Little, J.W.; Luongo, L.; Janes, K.; Chen, Z.; Esposito, E.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 2018, 159, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.R.; Jain, S.; Chen, Z.M.; Tosh, D.K.; Ma, Y.L.; Podszun, M.C.; Rotman, Y.; Salvemini, D.; Jacobson, K.A. Design and in vivo activity of A3 adenosine receptor agonist prodrugs. Purinergic Signal. 2020, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, E.; Coppi, E.; Micheli, L.; Parisio, C.; Vona, A.; Cherchi, F.; Pugliese, A.M.; Pedata, F.; Failli, P.; Palomino, S.; et al. Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 2020, 161, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Naime, D.; Firestein, G. The antiinflammatory effects of an adenosine kinase inhibitor are mediated by adenosine. Arthritis Rheum. 1995, 38, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Wildner, H.; Yevenes, G.E. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev. 2012, 92, 193–235. [Google Scholar] [CrossRef]
- Lozano-Ondoua, A.N.; Hanlon, K.E.; Symons-Liguori, A.M.; Largent-Milnes, T.M.; Havelin, J.J.; Ferland, H.L.; Chandramouli, A.; Owusu-Ankomah, M.; Nikolich-Zugich, T.; Bloom, A.P.; et al. Disease Modification of Breast Cancer-Induced Bone Remodeling by Cannabinoid 2 Receptor Agonists. J. Bone Miner. Res. 2013, 28, 92–107. [Google Scholar] [CrossRef]
- Coppi, E.; Cherchi, F.; Fusco, I.; Failli, P.; Vona, A.; Dettori, I.; Gaviano, L.; Lucarini, E.; Jacobson, K.A.; Tosh, D.K.; et al. Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 2019, 160, 1103–1118. [Google Scholar] [CrossRef]
- Gross, R.A.; Macdonald, R.L.; Ryanjastrow, T. 2-Chloroadenosine reduces the n-calcium current of cultured mouse sensory neurons in a pertussis toxin-sensitive manner. J. Physiol. 1989, 411, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Hannon, H.E.; Atchison, W.D. Omega-Conotoxins as Experimental Tools and Therapeutics in Pain Management. Mar. Drugs 2013, 11, 680–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, K.K. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert Opin. Investig. Drugs 2000, 9, 2403–2410. [Google Scholar] [CrossRef]
- McDowell, G.C.; Pope, J.E. Intrathecal Ziconotide: Dosing and Administration Strategies in Patients with Refractory Chronic Pain. Neuromodulation 2016, 19, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Brookes, M.E.; Eldabe, S.; Batterham, A. Ziconotide Monotherapy: A Systematic Review of Randomised Controlled Trials. Curr. Neuropharmacol. 2017, 15, 217–231. [Google Scholar] [CrossRef]
- Durante, M.; Squillace, S.; Lauro, F.; Giancotti, L.A.; Coppi, E.; Cherchi, F.; Di Cesare Mannelli, L.; Ghelardini, C.; Kolar, G.; Wahlman, C.; et al. Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J. Clin. Investig. 2021, 131, e139299. [Google Scholar] [CrossRef]
- McQuay, H.J.; Tramer, M.; Nye, B.A.; Carroll, D.; Wiffen, P.J.; Moore, R.A. Systematic review of antidepressants in neuropathic pain. Pain 1996, 68, 217–227. [Google Scholar] [CrossRef]
- Esser, M.J.; Sawynok, J. Caffeine blockade of the thermal antihyperalgesic effect of acute amitriptyline in a rat model of neuropathic pain. Eur. J. Pharmacol. 2000, 399, 131–139. [Google Scholar] [CrossRef]
- Ulugol, A.; Karadag, H.C.; Tamer, M.; Firat, Z.; Aslantas, A.; Dokmeci, I. Involvement of adenosine in the anti-allodynic effect of amitriptyline in streptozotocin-induced diabetic rats. Neurosci. Lett. 2002, 328, 129–132. [Google Scholar] [CrossRef]
- Kim, Y.; Kwon, S.Y.; Jung, H.S.; Park, Y.J.; Kim, Y.S.; In, J.H.; Choi, J.W.; Kim, J.A.; Joo, J.D. Amitriptyline inhibits the MAPK/ERK and CREB pathways and proinflammatory cytokines through A3AR activation in rat neuropathic pain models. Korean J. Anesthesiol. 2019, 72, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Stockstill, K.; Wahlman, C.; Braden, K.; Chen, Z.M.; Yosten, G.L.; Tosh, D.K.; Jacobson, K.A.; Doyle, T.M.; Samson, W.K.; Salvemini, D. Sexually dimorphic therapeutic response in bortezomib-induced neuropathic pain reveals altered pain physiology in female rodents. Pain 2020, 161, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.M.; Largent-Milnes, T.M.; Chen, Z.M.; Staikopoulos, V.; Esposito, E.; Dalgarno, R.; Fan, C.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chronic Morphine-Induced Changes in Signaling at the A3 Adenosine Receptor Contribute to Morphine-Induced Hyperalgesia, Tolerance, and Withdrawal. J. Pharmacol. Exp. Ther. 2020, 374, 331–341. [Google Scholar] [CrossRef]
- Leduc-Pessah, H.; Xu, C.; Fan, C.Y.; Dalgarno, R.; Kohro, Y.; Sparanese, S.; Burke, N.N.; Jacobson, K.A.; Altier, C.; Salvemini, D.; et al. Spinal A3 adenosine receptor activation acutely restores morphine antinociception in opioid tolerant male rats. J. Neurosci. Res. 2021. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppi, E.; Cherchi, F.; Lucarini, E.; Ghelardini, C.; Pedata, F.; Jacobson, K.A.; Di Cesare Mannelli, L.; Pugliese, A.M.; Salvemini, D. Uncovering the Mechanisms of Adenosine Receptor-Mediated Pain Control: Focus on the A3 Receptor Subtype. Int. J. Mol. Sci. 2021, 22, 7952. https://doi.org/10.3390/ijms22157952
Coppi E, Cherchi F, Lucarini E, Ghelardini C, Pedata F, Jacobson KA, Di Cesare Mannelli L, Pugliese AM, Salvemini D. Uncovering the Mechanisms of Adenosine Receptor-Mediated Pain Control: Focus on the A3 Receptor Subtype. International Journal of Molecular Sciences. 2021; 22(15):7952. https://doi.org/10.3390/ijms22157952
Chicago/Turabian StyleCoppi, Elisabetta, Federica Cherchi, Elena Lucarini, Carla Ghelardini, Felicita Pedata, Kenneth A. Jacobson, Lorenzo Di Cesare Mannelli, Anna Maria Pugliese, and Daniela Salvemini. 2021. "Uncovering the Mechanisms of Adenosine Receptor-Mediated Pain Control: Focus on the A3 Receptor Subtype" International Journal of Molecular Sciences 22, no. 15: 7952. https://doi.org/10.3390/ijms22157952
APA StyleCoppi, E., Cherchi, F., Lucarini, E., Ghelardini, C., Pedata, F., Jacobson, K. A., Di Cesare Mannelli, L., Pugliese, A. M., & Salvemini, D. (2021). Uncovering the Mechanisms of Adenosine Receptor-Mediated Pain Control: Focus on the A3 Receptor Subtype. International Journal of Molecular Sciences, 22(15), 7952. https://doi.org/10.3390/ijms22157952