
The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
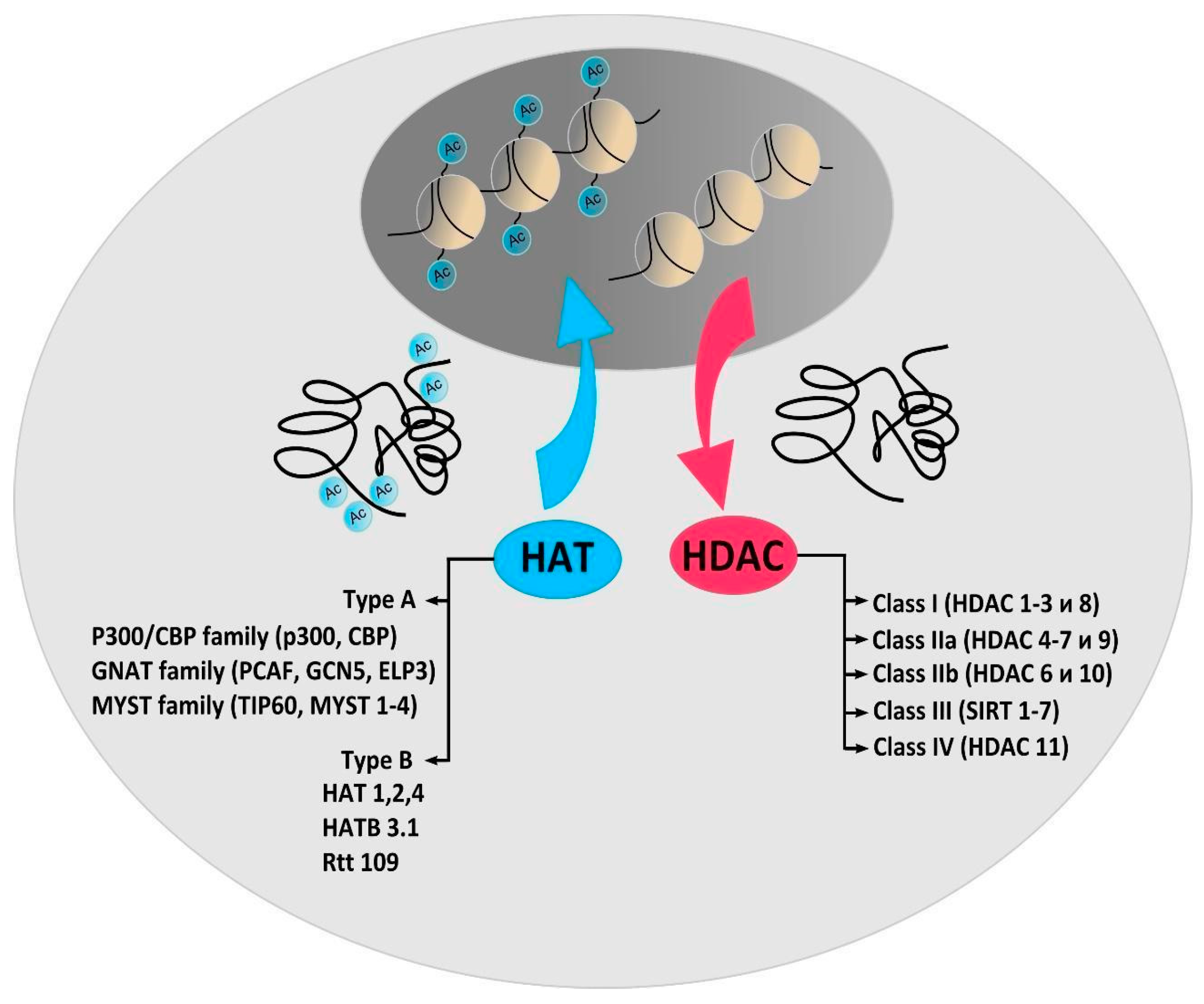
2. Protein Acetylation and Deacetylation Enzymes
3. Histone Acetyltransferases
4. Histone Deacetylases
5. Post-Translational Modifications of HDACs
6. Biological Activity of HDAC Inhibitors
7. Non-Histone Substrates of HAT and HDAC
8. c-Myc
9. E2F1
10. p53
11. ERK1/2
12. Akt
13. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Ethics Approval and Consent to Participate
References
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Kimura, A.; Matsubara, K.; Horikoshi, M. A Decade of Histone Acetylation: Marking Eukaryotic Chromosomes with Specific Codes. J. Biochem. 2005, 138, 647–662. [Google Scholar] [CrossRef]
- Yamauchi, T.; Yamauchi, J.; Kuwata, T.; Tamura, T.; Yamashita, T.; Bae, N.; Westphal, H.; Ozato, K.; Nakatani, Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 11303–11306. [Google Scholar] [CrossRef]
- Demyanenko, S.V.; Dzreyan, V.A.; Uzdensky, A.B. The Expression and Localization of Histone Acetyltransferases HAT1 and PCAF in Neurons and Astrocytes of the Photothrombotic Stroke-Induced Penumbra in the Rat Brain Cortex. Mol. Neurobiol. 2020, 57, 3219–3227. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Valls, E.; Kouzarides, T.; Martínez-Balbás, M. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 2003, 31, 4285–4292. [Google Scholar] [CrossRef]
- Yang, X.; Li, L.; Liang, J.; Shi, L.; Yang, J.; Yi, X.; Zhang, D.; Han, X.; Yu, N.; Shang, Y. Histone Acetyltransferase 1 Promotes Homologous Recombination in DNA Repair by Facilitating Histone Turnover. J. Biol. Chem. 2013, 288, 18271–18282. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone Deacetylase-1 (HDAC1) Is a Molecular Switch between Neuronal Survival and Death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Hiebert, S.W. Role for histone deacetylase 3 in maintenance of genome stability. Cell Cycle 2011, 10, 727–728. [Google Scholar] [CrossRef]
- Demyanenko, S.V.; Dzreyan, V.A.; Neginskaya, M.A.; Uzdensky, A.B. Expression of Histone Deacetylases HDAC1 and HDAC2 and Their Role in Apoptosis in the Penumbra Induced by Photothrombotic Stroke. Mol. Neurobiol. 2020, 57, 226–238. [Google Scholar] [CrossRef]
- Demyanenko, S.; Neginskaya, M.; Berezhnaya, E. Expression of Class I Histone Deacetylases in Ipsilateral and Contralateral Hemispheres after the Focal Photothrombotic Infarction in the Mouse Brain. Transl. Stroke Res. 2018, 9, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Dong, J.; Tang, Y.; Ni, H.-Y.; Zhang, Y.; Su, P.; Liang, H.-Y.; Yao, M.-C.; Yuan, H.-J.; Wang, D.-L.; et al. Opening a New Time Window for Treatment of Stroke by Targeting HDAC2. J. Neurosci. 2017, 37, 6712–6728. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lin, Y.-H.; Ni, H.; Dong, J.; Yuan, H.; Zhang, Y.; Liang, H.; Yao, M.; Zhou, Q.; Wu, H.; et al. Inhibiting Histone Deacetylase 2 (HDAC2) Promotes Functional Recovery From Stroke. J. Am. Hear. Assoc. 2017, 6, e007236. [Google Scholar] [CrossRef] [PubMed]
- Al Shoyaib, A.; Alamri, F.F.; Syeara, N.; Jayaraman, S.; Karamyan, S.T.; Arumugam, T.V.; Karamyan, V.T. The Effect of Histone Deacetylase Inhibitors Panobinostat or Entinostat on Motor Recovery in Mice After Ischemic Stroke. NeuroMolecular Med. 2021, 1–14. [Google Scholar] [CrossRef]
- Takase, K.; Oda, S.; Kuroda, M.; Funato, H. Monoaminergic and Neuropeptidergic Neurons Have Distinct Expression Profiles of Histone Deacetylases. PLoS ONE 2013, 8, e58473. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Zang, X.-F.; Pan, J.; Zhu, X.-L.; Chen, F.; Chen, Z.-B.; Xu, Y. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin. Exp. Pharmacol. Physiol. 2012, 39, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Demyanenko, S.V.; Dzreyan, V.A.; Uzdensky, A.B. Overexpression of HDAC6, but not HDAC3 and HDAC4 in the penumbra after photothrombotic stroke in the rat cerebral cortex and the neuroprotective effects of α-phenyl tropolone, HPOB, and sodium valproate. Brain Res. Bull. 2020, 162, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Leng, Y.; Wang, J.; Liao, H.-M.; Bergman, J.; Leeds, P.; Kozikowski, A.; Chuang, D.-M. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: Potential roles of α-tubulin acetylation and FGF-21 up-regulation. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bertos, N.R.; Gilquin, B.; Chan, G.; Yen, T.; Khochbin, S.; Yang, X.-J. Role of the Tetradecapeptide Repeat Domain of Human Histone Deacetylase 6 in Cytoplasmic Retention. J. Biol. Chem. 2004, 279, 48246–48254. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Girdwood, D.; Bumpass, D.; Vaughan, O.A.; Thain, A.; Anderson, L.A.; Snowden, A.W.; Garcia-Wilson, E.; Perkins, N.D.; Hay, R.T. p300 Transcriptional Repression Is Mediated by SUMO Modification. Mol. Cell 2003, 11, 1043–1054. [Google Scholar] [CrossRef]
- Yang, C.-J.; Liu, Y.-P.; Dai, H.-Y.; Shiue, Y.-L.; Tsai, C.-J.; Huang, M.-S.; Yeh, Y.-T. Nuclear HDAC6 inhibits invasion by suppressing NF-κB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget 2015, 6, 30263–30276. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Majdzadeh, N.; Wang, L.; Morrison, B.E.; Bassel-Duby, R.; Olson, E.N.; D’Mello, S.R. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev. Neurobiol. 2008, 68, 1076–1092. [Google Scholar] [CrossRef]
- Chen, B.; Cepko, C.L. HDAC4 Regulates Neuronal Survival in Normal and Diseased Retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef]
- Price, V.; Wang, L.; D’Mello, S.R. Conditional deletion of histone deacetylase-4 in the central nervous system has no major effect on brain architecture or neuronal viability. J. Neurosci. Res. 2013, 91, 407–415. [Google Scholar] [CrossRef]
- Bolger, T.A.; Yao, T.P. Intracellular Trafficking of Histone Deacetylase 4 Regulates Neuronal Cell Death. J. Neurosci. 2005, 25, 9544–9553. [Google Scholar] [CrossRef]
- Yuan, H.; Denton, K.; Liu, L.; Li, X.-J.; Benashski, S.; McCullough, L.; Li, J. Nuclear translocation of histone deacetylase 4 induces neuronal death in stroke. Neurobiol. Dis. 2016, 91, 182–193. [Google Scholar] [CrossRef]
- Kassis, H.; Shehadah, A.; Chopp, M.; Roberts, C.; Zhang, Z.G. Stroke Induces Nuclear Shuttling of Histone Deacetylase 4. Stroke 2015, 46, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, B.; Wei, X.; Wang, Z.; Fan, B.; Du, P.; Zhang, Y.; Jian, W.; Chen, L.; Wang, L.; et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xiao, F.; Huang, W.; Wei, Q.; Li, X. MicroRNA-29a-3p strengthens the effect of dexmedetomidine on improving neurologic damage in newborn rats with hypoxic-ischemic brain damage by inhibiting HDAC4. Brain Res. Bull. 2021, 167, 71–79. [Google Scholar] [CrossRef]
- Chawla, S.; Vanhoutte, P.; Arnold, F.J.L.; Huang, C.L.-H.; Bading, H. Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 2003, 85, 151–159. [Google Scholar] [CrossRef]
- Wei, J.-Y.; Lu, Q.-N.; Li, W.-M.; He, W. Intracellular translocation of histone deacetylase 5 regulates neuronal cell apoptosis. Brain Res. 2015, 1604, 15–24. [Google Scholar] [CrossRef]
- Cho, Y.; Sloutsky, R.; Naegle, K.M.; Cavalli, V. Injury-Induced HDAC5 Nuclear Export Is Essential for Axon Regeneration. Cell 2013, 155, 894–908. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Laudati, G.; Guida, N.; Mascolo, L.; Serani, A.; Cuomo, O.; Cantile, M.; Boscia, F.; Molinaro, P.; Anzilotti, S.; et al. HDAC4 and HDAC5 form a complex with DREAM that epigenetically down-regulates NCX3 gene and its pharmacological inhibition reduces neuronal stroke damage. Br. J. Pharmacol. 2019, 40, 2081–2097. [Google Scholar] [CrossRef] [PubMed]
- She, D.T.; Jo, N.-G.; Arumugam, T. Emerging Roles of Sirtuins in Ischemic Stroke. Transl. Stroke Res. 2017, 8, 405–423. [Google Scholar] [CrossRef]
- Ng, F.; Wijaya, L.; Tang, B.L. SIRT1 in the brain—Connections with aging-associated disorders and lifespan. Front. Cell. Neurosci. 2015, 9, 64. [Google Scholar] [CrossRef]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Jiménez, M.; Hurtado, O.; Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Pradillo, J.M.; McBurney, M.W.; Lizasoain, I.; Moro, M.A. Silent Information Regulator 1 Protects the Brain Against Cerebral Ischemic Damage. Stroke 2013, 44, 2333–2337. [Google Scholar] [CrossRef]
- Hattori, Y.; Okamoto, Y.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Kinoshita, M.; Ihara, M. SIRT1 attenuates severe ischemic damage by preserving cerebral blood flow. Neuroreport 2015, 26, 113–117. [Google Scholar] [CrossRef]
- Li, Z.; Pang, L.; Fang, F.; Zhang, G.; Zhang, J.; Xie, M.; Wang, L. Resveratrol attenuates brain damage in a rat model of focal cerebral ischemia via up-regulation of hippocampal Bcl-2. Brain Res. 2012, 1450, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Hong, Y.; Lu, X.; Zhang, J.; Chen, H.; Li, Y.; Ma, Y.; Ying, W. SIRT2 mediates oxidative stress-induced apoptosis of differentiated PC12 cells. NeuroReport 2014, 25, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Krey, L.; Lühder, F.; Kusch, K.; Czech-Zechmeister, B.; Könnecke, B.; Outeiro, T.F.; Trendelenburg, G. Knockout of Silent Information Regulator 2 (SIRT2) Preserves Neurological Function after Experimental Stroke in Mice. J. Cereb. Blood Flow Metab. 2015, 35, 2080–2088. [Google Scholar] [CrossRef]
- Fan, J.-H.; Song, H.-M.; Zhang, X.; Yan, W.-J.; Han, S.; Yin, Y.-L. Acute cerebral ischemia-induced down-regulation of Sirt3 protein expression contributes to neuronal injury via damaging mitochondrial function. Sheng Li Xue Bao 2021, 73, 17–25. [Google Scholar]
- Yang, X.; Zhang, Y.; Geng, K.; Yang, K.; Shao, J.; Xia, W. Sirt3 Protects Against Ischemic Stroke Injury by Regulating HIF-1α/VEGF Signaling and Blood–Brain Barrier Integrity. Cell. Mol. Neurobiol. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liberale, L.; Gaul, D.S.; Akhmedov, A.; Bonetti, N.R.; Nageswaran, V.; Costantino, S.; Pahla, J.; Weber, J.; Fehr, V.; Vdovenko, D.; et al. Endothelial SIRT6 blunts stroke size and neurological deficit by preserving blood–Brain barrier integrity: A translational study. Eur. Heart J. 2020, 41, 1575–1587. [Google Scholar] [CrossRef]
- Tsai, S.-C.; Seto, E. Regulation of Histone Deacetylase 2 by Protein Kinase CK2. J. Biol. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.D.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.-K.; Yoo, E.-K.; Kang, G.; Nguyen, V.H.; Min, J.-J.; Kim, J.-K.; et al. Regulation of Acetylation of Histone Deacetylase 2 by p300/CBP-Associated Factor/Histone Deacetylase 5 in the Development of Cardiac Hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Chen, J.; Ma, Y.; Liu, X. Updating a Strategy for Histone Deacetylases and Its Inhibitors in the Potential Treatment of Cerebral Ischemic Stroke. Dis. Markers 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhong, B.; Tan, J.; Chen, C.; Lei, Q.; Zeng, L. The Emerging Role of Epigenetics in Cerebral Ischemia. Mol. Neurobiol. 2016, 54, 1887–1905. [Google Scholar] [CrossRef]
- Ahmad Ganai, S.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors-emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.B.; Chibane, F.; Wang, Z.; Chuang, D.-M. Potential Roles of HDAC Inhibitors in Mitigating Ischemia-induced Brain Damage and Facilitating Endogenous Regeneration and Recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef]
- Felling, R.J.; Song, H. Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 2015, 268, 37–45. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical Experience With Intravenous and Oral Formulations of the Novel Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid in Patients With Advanced Hematologic Malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Shen, S.; Casaccia-Bonnefil, P. Post-Translational Modifications of Nucleosomal Histones in Oligodendrocyte Lineage Cells in Development and Disease. J. Mol. Neurosci. 2008, 35, 13–22. [Google Scholar] [CrossRef]
- Pedre, X.; Mastronardi, F.; Bruck, W.; López-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed Histone Acetylation Patterns in Normal-Appearing White Matter and Early Multiple Sclerosis Lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Tapiola, T.; Korhonen, P.; Suuronen, T. Neuronal apoptosis induced by histone deacetylase inhibitors. Mol. Brain Res. 1998, 61, 203–206. [Google Scholar] [CrossRef]
- Liu, D.X.; Nath, N.; Chellappan, S.P.; Greene, L.A. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005, 19, 719–732. [Google Scholar] [CrossRef]
- Kim, D.; Frank, C.L.; Dobbin, M.M.; Tsunemoto, R.K.; Tu, W.; Peng, P.L.; Guan, J.-S.; Lee, B.-H.; Moy, L.Y.; Giusti-Rodríguez, P.; et al. Deregulation of HDAC1 by p25/Cdk5 in Neurotoxicity. Neuron 2008, 60, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, A.; Hetman, M. Inhibitors of Histone Deacetylases Enhance Neurotoxicity of DNA Damage. Neuromol. Med. 2014, 16, 727–741. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Wang, X.; Liu, L.; Wang, X. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 2009, 467, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Forgione, N.; Tropepe, V. Histone deacetylase inhibition promotes Caspase-independent cell death of ventral midbrain neurons. Mol. Cell. Neurosci. 2011, 48, 117–128. [Google Scholar] [CrossRef]
- Dincman, T.A.; Beare, J.E.; Ohri, S.S.; Gallo, V.; Hetman, M.; Whittemore, S.R. Histone deacetylase inhibition is cytotoxic to oligodendrocyte precursor cells in vitro and in vivo. Int. J. Dev. Neurosci. 2016, 54, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, Q.; Xia, M.; Chen, J.; Chen, Y.; Cao, X.; Liu, Y.; Yuan, Z.; Wang, X.; Xu, Y. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 2020, 34, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, Z.; Zhang, Y.; Wang, G.; Huang, Z.; Zhang, Q.; Li, J. Design, synthesis and biological evaluation of brain penetrant benzazepine-based histone deacetylase 6 inhibitors for alleviating stroke-induced brain infarction. Eur. J. Med. Chem. 2021, 218, 113383. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Höschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the Acetyltransferases p300 and CBP Reveals a Targetable Function for p300 in the Survival and Invasion Pathways of Prostate Cancer Cell Lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21 CIP1/WAF1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Downey, M. Non-histone protein acetylation by the evolutionarily conserved GCN5 and PCAF acetyltransferases. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2021, 1864, 194608. [Google Scholar] [CrossRef]
- Sikder, S.; Kaypee, S.; Kundu, T.K. Regulation of epigenetic state by non-histone chromatin proteins and transcription factors: Implications in disease. J. Biosci. 2020, 45, 1–16. [Google Scholar] [CrossRef]
- Demyanenko, S.; Uzdensky, A. Profiling of Signaling Proteins in Penumbra After Focal Photothrombotic Infarct in the Rat Brain Cortex. Mol. Neurobiol. 2017, 54, 6839–6856. [Google Scholar] [CrossRef]
- Uzdensky, A.B. Apoptosis regulation in the penumbra after ischemic stroke: Expression of pro- and antiapoptotic proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer. Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- McGahan, L.; Hakim, A.M.; Robertson, G.S. Hippocampal Myc and p53 expression following transient global ischemia. Mol. Brain Res. 1998, 56, 133–145. [Google Scholar] [CrossRef]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.-J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Zhao, G.; Lv, X.; Chen, H.-Z.; Xue, Z.; Yang, B.; Liu, D.-P.; Liang, C.-C. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int. J. Biochem. Cell Biol. 2011, 43, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Carafa, V.; Conte, M.; Tambaro, F.P.; Abbondanza, C.; Martens, J.; Nees, M.; Benedetti, R.; Pallavicini, I.; Minucci, S.; et al. c-Myc Modulation and Acetylation Is a Key HDAC Inhibitor Target in Cancer. Clin. Cancer Res. 2017, 23, 2542–2555. [Google Scholar] [CrossRef]
- Ecker, J.; Thatikonda, V.; Sigismondo, G.; Selt, F.; Valinciute, G.; Oehme, I.; Müller, C.; Buhl, J.L.; Ridinger, J.; Usta, D.; et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro-Oncology 2021, 23, 226–239. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, Y.; Tang, D.; Dorfman, R.G.; Xu, L.; Zhou, Q.; Zhou, L.; Wang, Y.; Li, Y.; Yin, Y.; et al. Low levels of pyruvate induced by a positive feedback loop protects cholangiocarcinoma cells from apoptosis. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Ghosh, R. Transcription addiction: Can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis. 2014, 5, e1360. [Google Scholar] [CrossRef] [PubMed]
- Mathey-Prevot, B.; Parker, B.-T.; Im, C.; Hong, C.; Dong, P.; Yao, G.; You, L. Quantifying E2F1 protein dynamics in single cells. Quant. Biol. 2020, 8, 20–30. [Google Scholar] [CrossRef]
- Folch, J.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pizarro, J.G.; Beas-Zárate, C.; Pallàs, M.; Camins, A. Role of Cell Cycle Re-Entry in Neurons: A Common Apoptotic Mechanism of Neuronal Cell Death. Neurotox. Res. 2011, 22, 195–207. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Camins, A.; Verdaguer, E.; Folch, J.; Beas-Zarate, C.; Canudas, A.M.; Pallas, M. Inhibition of Ataxia Telangiectasia-p53-E2F-1 Pathway in Neurons as a Target for the Prevention of Neuronal Apoptosis. Curr. Drug Metab. 2007, 8, 709–715. [Google Scholar] [CrossRef]
- MacManus, J.P.; Jian, M.; Preston, E.; Rasquinha, I.; Webster, J.; Zurakowski, B. Absence of the Transcription Factor E2F1 Attenuates Brain Injury and Improves Behavior after Focal Ischemia in Mice. J. Cereb. Blood Flow Metab. 2003, 23, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chopp, M.; Zhang, Z.G.; Zaloga, C.; Niewenhuis, L.; Gautam, S. p53-immunoreactive protein and p53 mRNA expression after transient middle cerebral artery occlusion in rats. Stroke 1994, 25, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Dzreyan, V.; Rodkin, S.; Nikul, V.; Pitinova, M.; Uzdensky, A. The Expression of E2F1, p53, and Caspase 3 in the Rat Dorsal Root Ganglia After Sciatic Nerve Transection. J. Mol. Neurosci. 2021, 71, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Abramova, M.V.; Pospelova, T.V.; Nikulenkov, F.P.; Hollander, C.M.; Fornace, A.J.; Pospelov, V.A. G1/S Arrest Induced by Histone Deacetylase Inhibitor Sodium Butyrate in E1A + Ras-transformed Cells Is Mediated through Down-regulation of E2F Activity and Stabilization of β-Catenin. J. Biol. Chem. 2006, 281, 21040–21051. [Google Scholar] [CrossRef]
- Martínez-Balbás, M.; Bauer, U.-M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef]
- Ianari, A.; Gallo, R.; Palma, M.; Alesse, E.; Gulino, A. Specific Role for p300/CREB-binding Protein-associated Factor Activity in E2F1 Stabilization in Response to DNA Damage. J. Biol. Chem. 2004, 279, 30830–30835. [Google Scholar] [CrossRef]
- Galbiati, L.; Mendoza-Maldonado, R.; Gutierrez, M.I.; Giacca, M. Regulation of E2F-1 after DNA Damage by p300-Mediated Acetylation and Ubiquitination. Cell Cycle 2005, 4, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Tao, Y.; Li, M.; Che, T.; Qu, J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp. Ther. Med. 2020, 20, 2923–2940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ji, W.; Liu, X.; Ouyang, G.; Xiao, W. ELL Inhibits E2F1 Transcriptional Activity by Enhancing E2F1 Deacetylation via Recruitment of Histone Deacetylase 1. Mol. Cell. Biol. 2014, 34, 765–775. [Google Scholar] [CrossRef]
- Wu, M.; Seto, E.; Zhang, J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget 2015, 6, 11252–11263. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Qiao, L.; Feng, R.; Xu, Q.; Zhang, Y.; Fang, Z.; Shen, J.; Li, S. IL-6-induced acetylation of E2F1 aggravates oxidative damage of retinal pigment epithelial cell line. Exp. Eye Res. 2020, 200, 108219. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.-Z.; Zhao, X.-Y.; Zhang, H.-L. p53-mediated neuronal cell death in ischemic brain injury. Neurosci. Bull. 2010, 26, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Luo, J.; Brooks, C.L.; Nikolaev, A.; Li, M. Dynamics of the p53 Acetylation Pathway. Novartis Found. Symp. 2004, 259, 197–207. [Google Scholar]
- Juan, L.-J.; Shia, W.-J.; Chen, M.-H.; Yang, W.-M.; Seto, E.; Lin, Y.-S.; Wu, C.-W. Histone Deacetylases Specifically Down-regulate p53-dependent Gene Activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.-I.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of p53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Brandl, A.; Wagner, T.; Uhlig, K.M.; Knauer, S.K.; Stauber, R.H.; Melchior, F.; Schneider, G.; Heinzel, T.; Krämer, O.H. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J. Mol. Cell Biol. 2012, 4, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, H.; Zhu, M.; Hu, P.; Liu, X.; Qing, Y.; Wang, X.; Wang, H.; Wang, Z.; Xu, J.; et al. Involvement of p53 Acetylation in Growth Suppression of Cutaneous T-Cell Lymphomas Induced by HDAC Inhibition. J. Investig. Dermatol. 2020, 140, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Jin, J.; Chen, J.; Li, J.; Yu, X.; Mo, H.; Chen, G. SIRT1 activation by resveratrol reduces brain edema and neuronal apoptosis in an experimental rat subarachnoid hemorrhage model. Mol. Med. Rep. 2017, 16, 9627–9635. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993. [Google Scholar] [CrossRef]
- Kakiuchi, A.; Kakuki, T.; Ohwada, K.; Kurose, M.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; et al. HDAC inhibitors suppress the proliferation, migration and invasiveness of human head and neck squamous cell carcinoma cells via p63-mediated tight junction molecules and p21-mediated growth arrest. Oncol. Rep. 2021, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Warita, H.; Sasaki, C.; Zhang, W.R.; Sakai, K.; Shiro, Y.; Mitsumoto, Y.; Mori, T.; Abe, K. Immunoreactive Akt, PI3-K and ERK protein kinase expression in ischemic rat brain. Neurosci. Lett. 1999, 274, 45–48. [Google Scholar] [CrossRef]
- Liu, B.-N.; Han, B.-X.; Liu, F. Neuroprotective effect of pAkt and HIF-1 α on ischemia rats. Asian Pac. J. Trop. Med. 2014, 7, 221–225. [Google Scholar] [CrossRef]
- Iaconelli, J.; Lalonde, J.; Watmuff, B.; Liu, B.; Mazitschek, R.; Haggarty, S.J.; Karmacharya, R. Lysine Deacetylation by HDAC6 Regulates the Kinase Activity of AKT in Human Neural Progenitor Cells. ACS Chem. Biol. 2017, 12, 2139–2148. [Google Scholar] [CrossRef]
- Hart, J.R.; Vogt, P.K. Phosphorylation of AKT: A Mutational Analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Zalewska, T.; Sypecka, J.; Ziemka-Nalecz, M. Effect of the HDAC Inhibitor, Sodium Butyrate, on Neurogenesis in a Rat Model of Neonatal Hypoxia–Ischemia: Potential Mechanism of Action. Mol. Neurobiol. 2019, 56, 6341–6370. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Chen, C.; Tu, Y.; Sun, H.-T.; Zhao, M.-L.; Cheng, S.-X.; Qu, Y.; Zhang, S. Sirt1 Promotes Axonogenesis by Deacetylation of Akt and Inactivation of GSK3. Mol. Neurobiol. 2013, 48, 490–499. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt Signaling by Sirtuins. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.P. The Deacetylase SIRT1 Promotes Membrane Localization and Activation of Akt and PDK1 During Tumorigenesis and Cardiac Hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3β/Snail signals. Oncogene 2020, 39, 4956–4969. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demyanenko, S.; Sharifulina, S. The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses. Int. J. Mol. Sci. 2021, 22, 7947. https://doi.org/10.3390/ijms22157947
Demyanenko S, Sharifulina S. The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses. International Journal of Molecular Sciences. 2021; 22(15):7947. https://doi.org/10.3390/ijms22157947
Chicago/Turabian StyleDemyanenko, Svetlana, and Svetlana Sharifulina. 2021. "The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses" International Journal of Molecular Sciences 22, no. 15: 7947. https://doi.org/10.3390/ijms22157947
APA StyleDemyanenko, S., & Sharifulina, S. (2021). The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses. International Journal of Molecular Sciences, 22(15), 7947. https://doi.org/10.3390/ijms22157947