
Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword

 and
and
Abstract
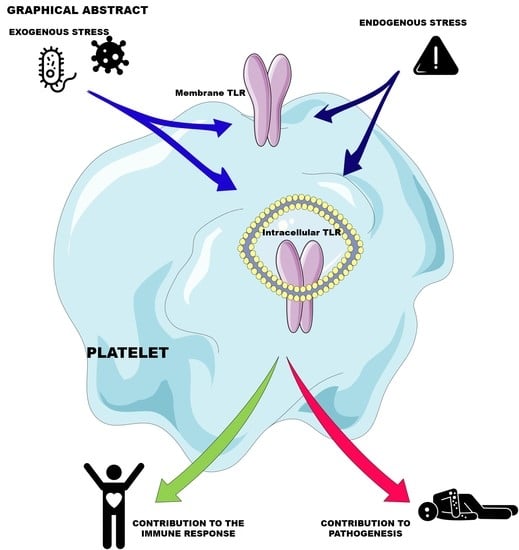
1. Introduction
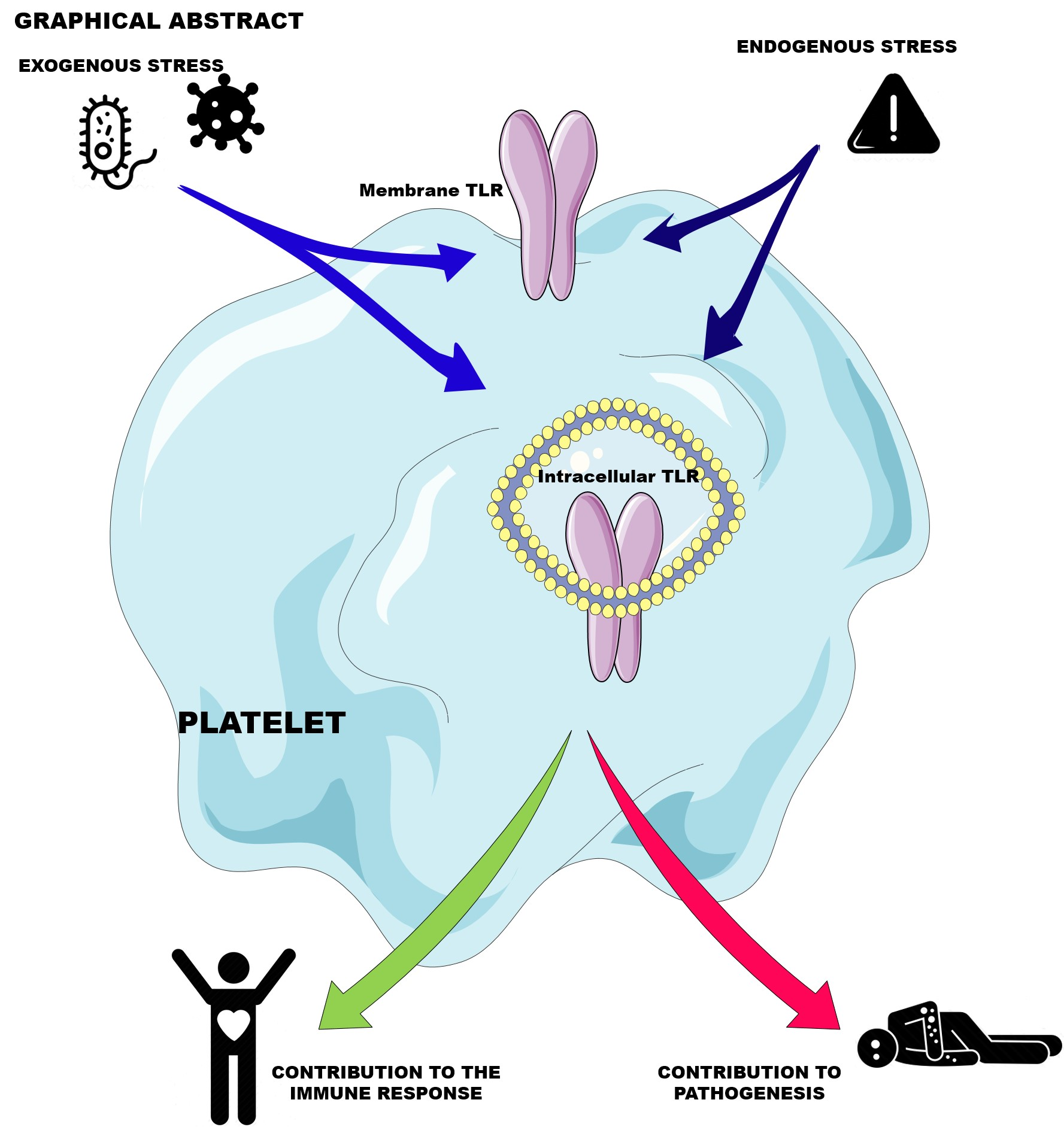
2. The Central Role of Platelets in Hemostasis
3. Platelet Response to Stress
3.1. Apoptosis
3.2. Necrosis
3.3. Autophagy
4. Platelets as Immune Cells
4.1. Release of Platelet Immunomodulatory and Antimicrobial Soluble Factors
4.2. Modulation of Immune Cell Response
4.2.1. Platelet/Neutrophil Interactions
4.2.2. Platelet/Monocyte Interactions
4.2.3. Platelets/Dendritic Cell Interactions
4.2.4. Platelets/B and T Cell Interactions
4.3. Platelets as Pathogen Sensors

4.4. A Focus on Platelet TLRs
4.5. TLR Structure
4.6. TLR Signaling Pathways in Platelets
4.7. Expression and Functional Response of Platelet TLRs
4.7.1. TLR2
4.7.2. TLR4
4.7.3. TLR3
4.7.4. TLR7
4.7.5. TLR9
4.7.6. TLR5, 8, and 10
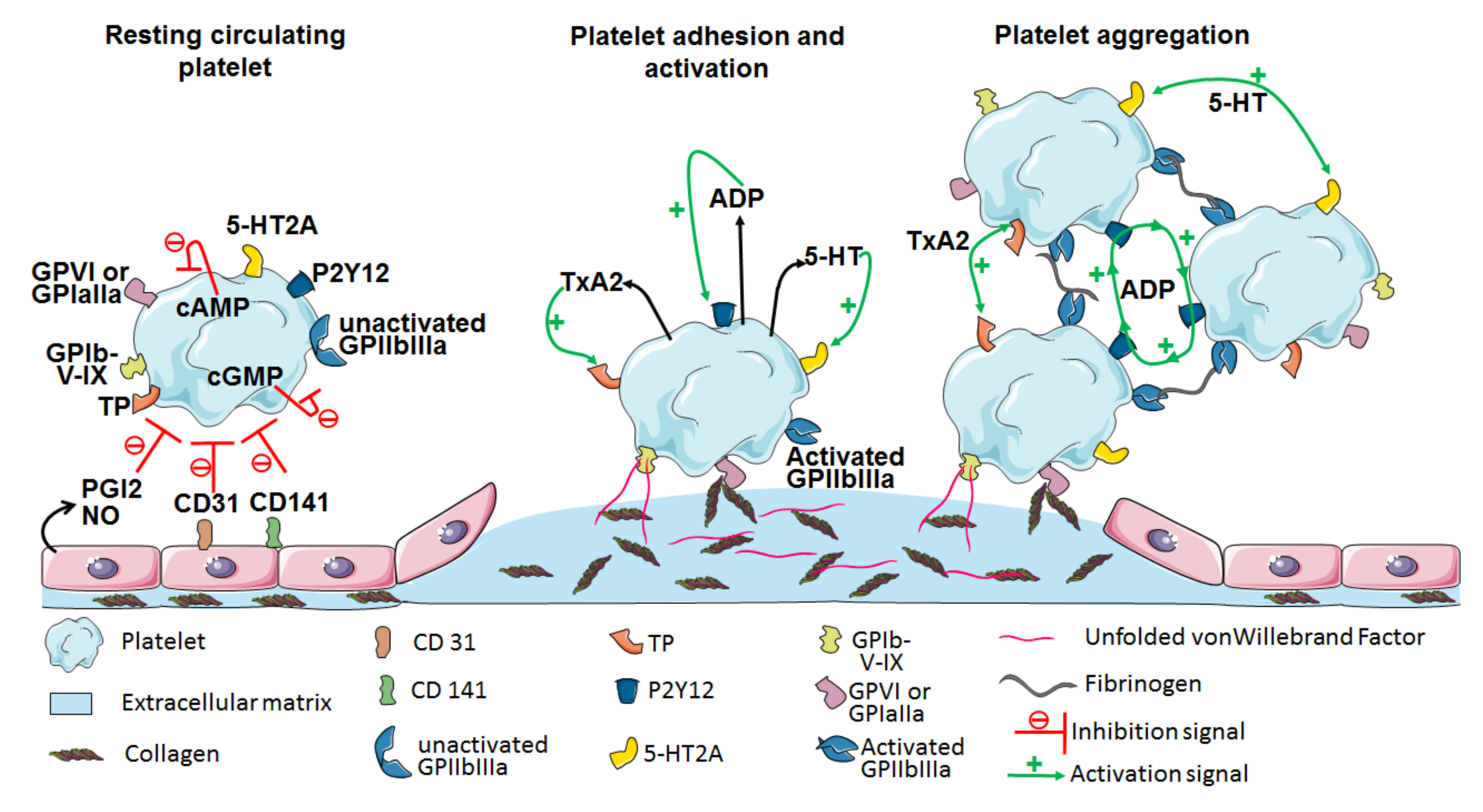
5. Platelet Contribution to Pathogenesis through Their Innate Immunity Receptors
5.1. Sepsis
5.2. COVID-19
5.3. HIV-1 Infection
5.4. Dengue
5.5. Atherosclerosis
5.6. Cancer
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Bennett, J.S. Platelet-fibrinogen interactions. Ann. N. Y. Acad. Sci. 2001, 936, 340–354. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Cognasse, H.; Cognasse, F.; Palle, S.; Chavarin, P.; Olivier, T.; Delezay, O.; Pozzetto, B.; Garraud, O. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol. 2008, 9, 54. [Google Scholar] [CrossRef]
- Hottz, E.D.; Lopes, J.F.; Freitas, C.; Valls-de-Souza, R.; Oliveira, M.F.; Bozza, M.T.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Chaipan, C.; Soilleux, E.J.; Simpson, P.; Hofmann, H.; Gramberg, T.; Marzi, A.; Geier, M.; Stewart, E.A.; Eisemann, J.; Steinkasserer, A.; et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J. Virol. 2006, 80, 8951–8960. [Google Scholar] [CrossRef] [PubMed]
- Arman, M.; Krauel, K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 2015, 13, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.P.; Barry, C.; Tran, A.N.; Traxler, E.A.; Wannemacher, K.M.; Tang, H.Y.; Speicher, K.D.; Blair, I.A.; Speicher, D.W.; Grosser, T.; et al. Deciphering the human platelet sheddome. Blood 2011, 117, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Ford, I.; Douglas, C.W. The role of platelets in infective endocarditis. Platelets 1997, 8, 285–294. [Google Scholar]
- Weber, C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef]
- Schmied, L.; Höglund, P.; Meinke, S. Platelet-Mediated Protection of Cancer Cells From Immune Surveillance—Possible Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 640578. [Google Scholar] [CrossRef]
- Gawaz, M.; Fateh-Moghadam, S.; Pilz, G.; Gurland, H.J.; Werdan, K. Platelet activation and interaction with leucocytes in patients with sepsis or multiple organ failure. Eur J. Clin. Investig. 1995, 25, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; Van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, V.; Gianesello, L.; Pazzi, M.; Stera, C.; Meconi, T.; Frigieri, F.C. Evaluation of coagulation function by rotation thromboelastometry in critically ill patients with severe COVID-19 pneumonia. J. Thromb. Thrombolysis 2020, 50, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2975–2989. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Félétou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef] [PubMed]
- Makhoul, S.; Walter, E.; Pagel, O.; Walter, U.; Sickmann, A.; Gambaryan, S.; Smolenski, A.; Zahedi, R.P.; Jurk, K. Effects of the NO/soluble guanylate cyclase/cGMP system on the functions of human platelets. Nitric Oxide 2018, 76, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, W.S.; Kulkarni, S.; Giuliano, S.; Goncalves, I.; Dopheide, S.M.; Yap, C.L.; Harper, I.S.; Salem, H.H.; Jackson, S.P. Distinct glycoprotein Ib/V/IX and integrin alpha IIbbeta 3-dependent calcium signals cooperatively regulate platelet adhesion under flow. J. Biol. Chem. 2002, 277, 2965–2972. [Google Scholar] [CrossRef] [PubMed]
- May, F.; Hagedorn, I.; Pleines, I.; Bender, M.; Vögtle, T.; Eble, J.; Elvers, M.; Nieswandt, B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009, 114, 3464–3472. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Herbert, J.M.; Pollitt, A.Y. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 2010, 8, 1456–1467. [Google Scholar] [CrossRef]
- Jackson, S.P. Arterial thrombosis--insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Maclean, J.A.; Schoenwaelder, S.M. Chapter 5—Serotonin in Platelets. In Serotonin; Pilowsky, P.M., Ed.; Academic Press: Boston, MA, USA, 2019; pp. 91–119. [Google Scholar]
- Schattner, M. Platelets and galectins. Ann. Transl. Med. 2014, 2, 85. [Google Scholar] [PubMed]
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Investig. 1999, 103, 879–887. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Cornelissen, I.; Coughlin, S.R. Impaired hemostasis and protection against thrombosis in protease-activated receptor 4-deficient mice is due to lack of thrombin signaling in platelets. J. Thromb. Haemost. 2004, 2, 1429–1435. [Google Scholar] [CrossRef]
- Chi, L.; Li, Y.; Stehno-Bittel, L.; Gao, J.; Morrison, D.C.; Stechschulte, D.J.; Dileepan, K.N. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2001, 21, 231–240. [Google Scholar] [CrossRef]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 2002, 296, 1880–1882. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; Chap, H.; Payrastre, B. Platelet membrane phospholipid asymmetry: From the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J. Thromb. Haemost. 2011, 9, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef]
- McArthur, K.; Chappaz, S.; Kile, B.T. Apoptosis in megakaryocytes and platelets: The life and death of a lineage. Blood 2018, 131, 605–610. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Sut, C.; Tariket, S.; Aubron, C.; Aloui, C.; Hamzeh-Cognasse, H.; Berthelot, P.; Laradi, S.; Greinacher, A.; Garraud, O.; Cognasse, F. The non-hemostatic aspects of transfused platelets. Front. Med. 2018, 5, 42. [Google Scholar] [CrossRef]
- Brown, S.B.; Clarke, M.C.; Magowan, L.; Sanderson, H.; Savill, J. Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J. Biol. Chem. 2000, 275, 5987–5996. [Google Scholar] [CrossRef]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-α-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef]
- Bergmeier, W.; Burger, P.C.; Piffath, C.L.; Hoffmeister, K.M.; Hartwig, J.H.; Nieswandt, B.; Wagner, D.D. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood 2003, 102, 4229–4235. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, J.; He, C.; Yan, R.; Zhou, K.; Cui, Q.; Meng, X.; Li, X.; Zhang, Y.; Nie, Y.; et al. Protein kinase A determines platelet life span and survival by regulating apoptosis. J. Clin. Investig. 2017, 127, 4338–4351. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.A.; Hamzeh-Cognasse, H.; Palle, S.; Anselme-Bertrand, I.; Arthaud, C.A.; Chavarin, P.; Pozzetto, B.; Garraud, O.; Cognasse, F. Role of Siglec-7 in apoptosis in human platelets. PLoS ONE 2014, 9, e106239. [Google Scholar] [CrossRef]
- Gambim, M.H.; Do Carmo Ade, O.; Marti, L.; Veríssimo-Filho, S.; Lopes, L.R.; Janiszewski, M. Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: Experimental evidence for a novel mechanism of septic vascular dysfunction. Crit. Care 2007, 11, R107. [Google Scholar] [CrossRef]
- Nomura, S.; Shimizu, M. Clinical significance of procoagulant microparticles. J. Intensive Care 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, R.I.; Reichenbach, F.; Kraft, P.; Kumar, A.; Lescan, M.; Todt, F.; Göbel, K.; Hilgendorf, I.; Geisler, T.; Bauer, A.; et al. Platelets induce apoptosis via membrane-bound FasL. Blood 2015, 126, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Goelz, N.; Eekels, J.J.M.; Pantic, M.; Kamber, C.T.; Speer, O.; Franzoso, F.D.; Schmugge, M. Platelets express adaptor proteins of the extrinsic apoptosis pathway and can activate caspase-8. PLoS ONE 2021, 16, e0244848. [Google Scholar] [CrossRef]
- Leytin, V.; Gyulkhandanyan, A.V.; Freedman, J. Platelet Apoptosis Can Be Triggered Bypassing the Death Receptors. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619853641. [Google Scholar] [CrossRef] [PubMed]
- Leytin, V. Apoptosis in the anucleate platelet. Blood Rev. 2012, 26, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gyulkhandanyan, A.V.; Mutlu, A.; Freedman, J.; Leytin, V. Selective triggering of platelet apoptosis, platelet activation or both. Br. J. Haematol. 2013, 161, 245–254. [Google Scholar] [CrossRef]
- Yeh, J.J.; Tsai, S.; Wu, D.C.; Wu, J.Y.; Liu, T.C.; Chen, A. P-selectin-dependent platelet aggregation and apoptosis may explain the decrease in platelet count during Helicobacter pylori infection. Blood 2010, 115, 4247–4253. [Google Scholar] [CrossRef]
- Gafter-Gvili, A.; Mansur, N.; Bivas, A.; Zemer-Wassercug, N.; Bishara, J.; Leibovici, L.; Paul, M. Thrombocytopenia in Staphylococcus aureus bacteremia: Risk factors and prognostic importance. Mayo Clin. Proc. 2011, 86, 389–396. [Google Scholar] [CrossRef]
- Mesquita, E.C.; Hottz, E.D.; Amancio, R.T.; Carneiro, A.B.; Palhinha, L.; Coelho, L.E.; Grinsztejn, B.; Zimmerman, G.A.; Rondina, M.T.; Weyrich, A.S.; et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci. Rep. 2018, 8, 14999. [Google Scholar] [CrossRef]
- Towhid, S.T.; Nega, M.; Schmidt, E.M.; Schmid, E.; Albrecht, T.; Munzer, P.; Borst, O.; Gotz, F.; Lang, F. Stimulation of platelet apoptosis by peptidoglycan from Staphylococcus aureus 113. Apoptosis 2012, 17, 998–1008. [Google Scholar] [CrossRef]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Franks, Z.G.; Vieira de Abreu, A.; Grundler, K.; Kile, B.T.; Dhakal, B.K.; Rondina, M.T.; Kahr, W.H.; et al. Bacteria differentially induce degradation of Bcl-xL, a survival protein, by human platelets. Blood 2012, 120, 5014–5020. [Google Scholar] [CrossRef]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef]
- Hua, V.M.; Abeynaike, L.; Glaros, E.; Campbell, H.; Pasalic, L.; Hogg, P.J.; Chen, V.M. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 2015, 126, 2852–2862. [Google Scholar] [CrossRef]
- Yuan, Y.; Alwis, I.; Wu, M.C.L.; Kaplan, Z.; Ashworth, K.; Bark, D., Jr.; Pham, A.; McFadyen, J.; Schoenwaelder, S.M.; Josefsson, E.C.; et al. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Jobe, S.M.; Wilson, K.M.; Leo, L.; Raimondi, A.; Molkentin, J.D.; Lentz, S.R.; Di Paola, J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008, 111, 1257–1265. [Google Scholar] [CrossRef]
- Remenyi, G.; Szasz, R.; Friese, P.; Dale, G.L. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 467–471. [Google Scholar] [CrossRef]
- Denorme, F.; Manne, B.K.; Portier, I.; Eustes, A.S.; Kosaka, Y.; Kile, B.T.; Rondina, M.T.; Campbell, R.A. Platelet necrosis mediates ischemic stroke outcome in mice. Blood 2020, 135, 429–440. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef]
- Vogel, S.; Rath, D.; Borst, O.; Mack, A.; Loughran, P.; Lotze, M.T.; Neal, M.D.; Billiar, T.R.; Gawaz, M. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem. Biophys. Res. Commun. 2016, 478, 143–148. [Google Scholar] [CrossRef]
- Moujalled, D.; Gangatirkar, P.; Kauppi, M.; Corbin, J.; Lebois, M.; Murphy, J.M.; Lalaoui, N.; Hildebrand, J.M.; Silke, J.; Alexander, W.S.; et al. The necroptotic cell death pathway operates in megakaryocytes, but not in platelet synthesis. Cell Death Dis. 2021, 12, 133. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Yan, R.; Tian, J.; Chen, M.; Cui, Q.; Zhao, L.; Hu, R.; Jiang, M.; Li, Z.; et al. Receptor-interacting protein kinase 3 promotes platelet activation and thrombosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, N.; Walczak, H. Cell Death and Inflammation—A Vital but Dangerous Liaison. Trends Immunol. 2019, 40, 387–402. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Kaleağasıoğlu, F.; Ali, D.M.; Berger, M.R. Multiple Facets of Autophagy and the Emerging Role of Alkylphosphocholines as Autophagy Modulators. Front. Pharmacol. 2020, 11, 547. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Chang, C.; Luo, D.; Su, H.; Yu, S.; Hua, W.; Chen, Z.; Hu, H.; Liu, W. Dissection of autophagy in human platelets. Autophagy 2014, 10, 642–651. [Google Scholar] [CrossRef]
- Ouseph, M.M.; Huang, Y.; Banerjee, M.; Joshi, S.; MacDonald, L.; Zhong, Y.; Liu, H.; Li, X.; Xiang, B.; Zhang, G.; et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 2015, 126, 1224–1233. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, M.; Luo, D.; Yue, M.; Wang, S.; Chen, X.; Zhou, Y.; Wang, Y.; Cai, Y.; Hu, X.; et al. Class III PI3K Positively Regulates Platelet Activation and Thrombosis via PI(3)P-Directed Function of NADPH Oxidase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2075–2086. [Google Scholar] [CrossRef]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G.; et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Ma, S.; Bi, S.J.; Su, L.; Huang, S.Y.; Miao, J.Y.; Ma, C.H.; Gao, C.J.; Hou, M.; Peng, J. Enhancing autophagy protects platelets in immune thrombocytopenia patients. Ann. Transl. Med. 2019, 7, 134. [Google Scholar] [CrossRef]
- Tang, H.; Gao, M.; Fu, Y.; Gui, R.; Ma, X. The Effect of Autophagic Activity on the Function of Apheresis Platelets and on the Efficacy of Clinical Platelet Transfusion. Transfus. Med. Hemother. 2020, 47, 302–313. [Google Scholar] [CrossRef]
- Krijgsveld, J.; Zaat, S.A.; Meeldijk, J.; Van Veelen, P.A.; Fang, G.; Poolman, B.; Brandt, E.; Ehlert, J.E.; Kuijpers, A.J.; Engbers, G.H.; et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J. Biol. Chem. 2000, 275, 20374–20381. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel Anti-bacterial Activities of beta-defensin 1 in Human Platelets: Suppression of Pathogen Growth and Signaling of Neutrophil Extracellular Trap Formation. PLoS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef]
- Selvadurai, M.V.; Hamilton, J.R. Structure and function of the open canalicular system—The platelet’s specialized internal membrane network. Platelets 2018, 29, 319–325. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Huang, Z.; Zhang, W.; Jiang, L.; Hultenby, K.; Zhu, L.; Hu, H.; Nilsson, G.P.; Li, N. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood 2011, 117, 3907–3911. [Google Scholar] [CrossRef]
- Heijnen, H.; Van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T granules in human platelets function in TLR9 organization and signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Blann, A.D.; Lip, G.Y. Soluble CD40 ligand, soluble P-selectin, interleukin-6, and tissue factor in diabetes mellitus: Relationships to cardiovascular disease and risk factor intervention. Circulation 2004, 109, 2524–2528. [Google Scholar] [CrossRef]
- Cognasse, F.; Payrat, J.M.; Corash, L.; Osselaer, J.C.; Garraud, O. Platelet components associated with acute transfusion reactions: The role of platelet-derived soluble CD40 ligand. Blood 2008, 112, 4779–4780. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets Mediate Host Defense against Staphylococcus aureus through Direct Bactericidal Activity and by Enhancing Macrophage Activities. J. Immunol. 2017, 198, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C.; Kubes, P. Platelets and infection. Semin. Immunol. 2016, 28, 536–545. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial peptides from human platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Bayer, A.S. Antimicrobial peptides from platelets. Drug Resist. Updat. 1999, 2, 116–126. [Google Scholar] [CrossRef]
- Ojha, A.; Bhasym, A.; Mukherjee, S.; Annarapu, G.K.; Bhakuni, T.; Akbar, I.; Seth, T.; Vikram, N.K.; Vrati, S.; Basu, A.; et al. Platelet factor 4 promotes rapid replication and propagation of Dengue and Japanese encephalitis viruses. EBioMedicine 2019, 39, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, D.J.; Lin, Y.; Miao, H.; Cimbro, R.; Difiore, M.J.; Gianolini, M.E.; Furci, L.; Biswas, P.; Fauci, A.S.; Lusso, P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 9569–9574. [Google Scholar] [CrossRef] [PubMed]
- Love, M.S.; Millholland, M.G.; Mishra, S.; Kulkarni, S.; Freeman, K.B.; Pan, W.; Kavash, R.W.; Costanzo, M.J.; Jo, H.; Daly, T.M.; et al. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe 2012, 12, 815–823. [Google Scholar] [CrossRef]
- Schwartzkopff, F.; Grimm, T.A.; Lankford, C.S.; Fields, K.; Wang, J.; Brandt, E.; Clouse, K.A. Platelet factor 4 (CXCL4) facilitates human macrophage infection with HIV-1 and potentiates virus replication. Innate Immun. 2009, 15, 368–379. [Google Scholar] [CrossRef]
- Von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Marx, C.; Novotny, J.; Salbeck, D.; Zellner, K.R.; Nicolai, L.; Pekayvaz, K.; Kilani, B.; Stockhausen, S.; Bürgener, N.; Kupka, D.; et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood 2019, 134, 1859–1872. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef] [PubMed]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The balancing act of neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef]
- Gardiner, E.E.; De Luca, M.; McNally, T.; Michelson, A.D.; Andrews, R.K.; Berndt, M.C. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood 2001, 98, 1440–1447. [Google Scholar] [CrossRef]
- Hidari, K.I.; Weyrich, A.S.; Zimmerman, G.A.; McEver, R.P. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J. Biol. Chem. 1997, 272, 28750–28756. [Google Scholar] [CrossRef]
- Santoso, S.; Sachs, U.J.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef]
- Herster, F.; Bittner, Z.; Codrea, M.C.; Archer, N.K.; Heister, M.; Löffler, M.W.; Heumos, S.; Wegner, J.; Businger, R.; Schindler, M.; et al. Platelets Aggregate With Neutrophils and Promote Skin Pathology in Psoriasis. Front. Immunol. 2019, 10, 1867. [Google Scholar] [CrossRef]
- Kornerup, K.N.; Salmon, G.P.; Pitchford, S.C.; Liu, W.L.; Page, C.P. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J. Appl. Physiol. 2010, 109, 758–767. [Google Scholar] [CrossRef]
- Pan, D.; Amison, R.T.; Riffo-Vasquez, Y.; Spina, D.; Cleary, S.J.; Wakelam, M.J.; Page, C.P.; Pitchford, S.C.; Welch, H.C. P-Rex and Vav Rac-GEFs in platelets control leukocyte recruitment to sites of inflammation. Blood 2015, 125, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsdottir, S.; Hasselbalch, H.C.; Nielsen, C.H. Activated platelets enhance IL-10 secretion and reduce TNF-α secretion by monocytes. J. Immunol. 2013, 191, 4059–4067. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Davidson, D.C.; Jackson, J.W.; Singh, V.B.; Silva, J.; Ramirez, S.H.; Maggirwar, S.B. Characterization of platelet-monocyte complexes in HIV-1-infected individuals: Possible role in HIV-associated neuroinflammation. J. Immunol. 2014, 192, 4674–4684. [Google Scholar] [CrossRef]
- Gawaz, M.; Neumann, F.J.; Dickfeld, T.; Koch, W.; Laugwitz, K.L.; Adelsberger, H.; Langenbrink, K.; Page, S.; Neumeier, D.; Schömig, A.; et al. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation 1998, 98, 1164–1171. [Google Scholar] [CrossRef]
- Czapiga, M.; Kirk, A.D.; Lekstrom-Himes, J. Platelets deliver costimulatory signals to antigen-presenting cells: A potential bridge between injury and immune activation. Exp. Hematol. 2004, 32, 135–139. [Google Scholar] [CrossRef]
- Perros, A.J.; Christensen, A.M.; Flower, R.L.; Dean, M.M. Soluble Mediators in Platelet Concentrates Modulate Dendritic Cell Inflammatory Responses in an Experimental Model of Transfusion. J. Interferon Cytokine Res. 2015, 35, 821–830. [Google Scholar] [CrossRef]
- Nishat, S.; Wuescher, L.M.; Worth, R.G. Platelets Enhance Dendritic Cell Responses against Staphylococcus aureus through CD40-CD40L. Infect. Immun. 2018, 86, e00186-18. [Google Scholar] [CrossRef]
- Saris, A.; Steuten, J.; Schrijver, D.P.; Van Schijndel, G.; Zwaginga, J.J.; Van Ham, S.M.; Ten Brinke, A. Inhibition of Dendritic Cell Activation and Modulation of T Cell Polarization by the Platelet Secretome. Front. Immunol. 2021, 12, 631285. [Google Scholar] [CrossRef]
- Rondina, M.T.; Garraud, O. Emerging evidence for platelets as immune and inflammatory effector cells. Front. Immunol. 2014, 5, 653. [Google Scholar] [CrossRef]
- Srivastava, K.; Cockburn, I.A.; Swaim, A.; Thompson, L.E.; Tripathi, A.; Fletcher, C.A.; Shirk, E.M.; Sun, H.; Kowalska, M.A.; Fox-Talbot, K.; et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe 2008, 4, 179–187. [Google Scholar] [CrossRef]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M., 3rd; Morrell, C.N. Platelets present antigen in the context of MHC class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef]
- Guo, L.; Shen, S.; Rowley, J.W.; Tolley, N.D.; Jia, W.; Manne, B.K.; McComas, K.N.; Bolingbroke, B.; Kosaka, Y.; Krauel, K.; et al. Platelet MHC Class I Mediates CD8+ T Cell Suppression During Sepsis. Blood 2021. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh-Cognasse, H.; Lafarge, S.; Chavarin, P.; Cogne, M.; Richard, Y.; Garraud, O. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp. Hematol. 2007, 35, 1376–1387. [Google Scholar] [CrossRef]
- Sprague, D.L.; Elzey, B.D.; Crist, S.A.; Waldschmidt, T.J.; Jensen, R.J.; Ratliff, T.L. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 2008, 111, 5028–5036. [Google Scholar] [CrossRef]
- Li, J.L.; Zarbock, A.; Hidalgo, A. Platelets as autonomous drones for hemostatic and immune surveillance. J. Exp. Med. 2017, 214, 2193–2204. [Google Scholar] [CrossRef]
- Boilard, E.; Paré, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Lévesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Moriarty, R.D.; Cox, A.; McCall, M.; Smith, S.G.; Cox, D. Escherichia coli induces platelet aggregation in an FcgammaRIIa-dependent manner. J. Thromb. Haemost. 2016, 14, 797–806. [Google Scholar] [CrossRef]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of bacteria-induced platelet activation is triggered by FcgammaRIIA, integrin alphaIIbbeta3, and platelet factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef]
- Riaz, A.H.; Tasma, B.E.; Woodman, M.E.; Wooten, R.M.; Worth, R.G. Human platelets efficiently kill IgG-opsonized E. coli. FEMS Immunol. Med. MicroBiol. 2012, 65, 78–83. [Google Scholar] [CrossRef]
- Wolff, M.; Handtke, S.; Palankar, R.; Wesche, J.; Kohler, T.P.; Kohler, C.; Gruel, Y.; Hammerschmidt, S.; Greinacher, A. Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae-The role of FcγRIIa and platelet factor 4/heparinantibodies. J. Thromb. Haemost. 2020, 18, 1459–1468. [Google Scholar] [CrossRef]
- Palankar, R.; Kohler, T.P.; Krauel, K.; Wesche, J.; Hammerschmidt, S.; Greinacher, A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA. J. Thromb. Haemost. 2018, 16, 1187–1197. [Google Scholar] [CrossRef]
- Cox, D.; Kerrigan, S.W.; Watson, S.P. Platelets and the innate immune system: Mechanisms of bacterial-induced platelet activation. J. Thromb. Haemost. 2011, 9, 1097–1107. [Google Scholar] [CrossRef]
- Bensing, B.A.; López, J.A.; Sullam, P.M. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect. Immun. 2004, 72, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef]
- Hottz, E.D.; Oliveira, M.F.; Nunes, P.C.; Nogueira, R.M.; Valls-de-Souza, R.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, P.T.; Bozza, F.A. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J. Thromb. Haemost. 2013, 11, 951–962. [Google Scholar] [CrossRef]
- Tomo, S.; Mohan, S.; Ramachandrappa, V.S.; Samadanam, D.M.; Suresh, S.; Pillai, A.B.; Tamilarasu, K.; Ramachandran, R.; Rajendiran, S. Dynamic modulation of DC-SIGN and FcΥR2A receptors expression on platelets in dengue. PLoS ONE 2018, 13, e0206346. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.S.; Huang, T.F.; Hsieh, S.L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef]
- Quirino-Teixeira, A.C.; Rozini, S.V.; Barbosa-Lima, G.; Coelho, D.R.; Carneiro, P.H.; Mohana-Borges, R.; Bozza, P.T.; Hottz, E.D. Inflammatory signaling in dengue-infected platelets requires translation and secretion of nonstructural protein 1. Blood Adv. 2020, 4, 2018–2031. [Google Scholar] [CrossRef]
- Hashimoto, C.; Hudson, K.L.; Anderson, K.V. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 1988, 52, 269–279. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Shiraki, R.; Inoue, N.; Kawasaki, S.; Takei, A.; Kadotani, M.; Ohnishi, Y.; Ejiri, J.; Kobayashi, S.; Hirata, K.; Kawashima, S.; et al. Expression of Toll-like receptors on human platelets. Thromb. Res. 2004, 113, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like receptor molecules on human platelets. Immunol. Cell. Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Arthaud, C.A.; Eyraud, M.A.; Zeni, F.; Pozzetto, B.; McNicol, A.; Garraud, O.; Cognasse, F. Human platelets can discriminate between various bacterial LPS isoforms via TLR4 signaling and differential cytokine secretion. Clin. Immunol. 2012, 145, 189–200. [Google Scholar] [CrossRef]
- D’Atri, L.; Schattner, M. Platelet toll-like receptors in thromboinflammation. Front. BioSci. 2017, 22, 1867–1883. [Google Scholar]
- Spinelli, S.L.; Casey, A.E.; Pollock, S.J.; Gertz, J.M.; McMillan, D.H.; Narasipura, S.D.; Mody, N.A.; King, M.R.; Maggirwar, S.B.; Francis, C.W.; et al. Platelets and megakaryocytes contain functional nuclear factor-kappaB. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 591–598. [Google Scholar] [CrossRef]
- Vallance, T.M.; Zeuner, M.T.; Williams, H.F.; Widera, D.; Vaiyapuri, S. Toll-Like Receptor 4 Signalling and Its Impact on Platelet Function, Thrombosis, and Haemostasis. Mediat. Inflamm. 2017, 2017, 9605894. [Google Scholar] [CrossRef]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Pozzetto, B.; Garraud, O.; Cognasse, F. Toll-like receptor 4 signal transduction in platelets: Novel pathways. Br. J. Haematol. 2010, 151, 89–92. [Google Scholar] [CrossRef]
- Lu, W.J.; Lin, K.H.; Hsu, M.J.; Chou, D.S.; Hsiao, G.; Sheu, J.R. Suppression of NF-κB signaling by andrographolide with a novel mechanism in human platelets: Regulatory roles of the p38 MAPK-hydroxyl radical-ERK2 cascade. BioChem. Pharmacol. 2012, 84, 914–924. [Google Scholar] [CrossRef]
- Damien, P.; Cognasse, F.; Payrastre, B.; Spinelli, S.L.; Blumberg, N.; Arthaud, C.A.; Eyraud, M.A.; Phipps, R.P.; McNicol, A.; Pozzetto, B.; et al. NF-kappaB links TLR2 and PAR1 to soluble immunomodulator factor secretion in human platelets. Front. Immunol. 2017, 8, 85. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef]
- Panigrahi, S.; Ma, Y.; Hong, L.; Gao, D.; West, X.Z.; Salomon, R.G.; Byzova, T.V.; Podrez, E.A. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ. Res. 2013, 112, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Klarström Engström, K.; Khalaf, H.; Kälvegren, H.; Bengtsson, T. The role of Porphyromonas gingivalis gingipains in platelet activation and innate immune modulation. Mol. Oral MicroBiol. 2015, 30, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.R.; Bingle, L.; Judge, H.M.; Brown, S.B.; Storey, R.F.; Whyte, M.K.; Dower, S.K.; Buttle, D.J.; Sabroe, I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb. Haemost. 2005, 94, 831–838. [Google Scholar] [CrossRef]
- Keane, C.; Tilley, D.; Cunningham, A.; Smolenski, A.; Kadioglu, A.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J. Thromb. Haemost. 2010, 8, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- De Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; Van der Poll, T.; Nieuwland, R.; Van’t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A.; Kral, J.B.; Yaiw, K.C.; Schrottmaier, W.C.; Kurzejamska, E.; Wang, Y.; Mohammad, A.A.; Religa, P.; Rahbar, A.; Schabbauer, G.; et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Mariano, F.; Biancone, L.; De Martino, A.; Bussolati, B.; Montrucchio, G.; Tobias, P.S. Lipopolysaccharide binding protein and CD14 modulate the synthesis of platelet-activating factor by human monocytes and mesangial and endothelial cells stimulated with lipopolysaccharide. J. Immunol. 1995, 155, 316–324. [Google Scholar] [PubMed]
- Damien, P.; Cognasse, F.; Eyraud, M.A.; Arthaud, C.A.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-Ligand. BMC Immunol. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef]
- Saluk, J.; Bijak, M.; Ponczek, M.B.; Nowak, P.; Wachowicz, B. (1→3)-β-D-Glucan reduces the damages caused by reactive oxygen species induced in human platelets by lipopolysaccharides. Carbohydr. Polym. 2013, 97, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Kappelmayer, J.; Beke Debreceni, I.; Vida, A.; Antal-Szalmas, P.; Clemetson, K.J.; Nagy, B., Jr. Distinct effects of Re- and S-forms of LPS on modulating platelet activation. J. Thromb. Haemost. 2013, 11, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Svensson, M.; Mörgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef]
- Prakash, P.; Kulkarni, P.P.; Lentz, S.R.; Chauhan, A.K. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood 2015, 125, 3164–3172. [Google Scholar] [CrossRef]
- Rigg, R.A.; Healy, L.D.; Nowak, M.S.; Mallet, J.; Thierheimer, M.L.; Pang, J.; McCarty, O.J.; Aslan, J.E. Heat shock protein 70 regulates platelet integrin activation, granule secretion and aggregation. Am. J. Physiol. Cell Physiol. 2016, 310, C568-75. [Google Scholar] [CrossRef]
- Carestia, A.; Rivadeneyra, L.; Romaniuk, M.A.; Fondevila, C.; Negrotto, S.; Schattner, M. Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb. Haemost. 2013, 110, 1035–1045. [Google Scholar]
- Jerez-Dolz, D.; Torramade-Moix, S.; Palomo, M.; Moreno-Castaño, A.; Lopez-Vilchez, I.; Hernandez, R.; Badimon, J.J.; Zafar, M.U.; Diaz-Ricart, M.; Escolar, G. Internalization of microparticles by platelets is partially mediated by toll-like receptor 4 and enhances platelet thrombogenicity. Atherosclerosis 2020, 294, 17–24. [Google Scholar] [CrossRef]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018, 2, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van ‘t Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolas, V.S.; Sergio Roberto, A.R. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Blum, P.; Pircher, J.; Merkle, M.; Czermak, T.; Ribeiro, A.; Mannell, H.; Krotz, F.; Hennrich, A.; Spannagl, M.; Koppel, S.; et al. Arterial thrombosis in the context of HCV-associated vascular disease can be prevented by protein C. Cell Mol. Immunol. 2017. [Google Scholar] [CrossRef]
- Kullaya, V.I.; De Mast, Q.; Van der Ven, A.; ElMoussaoui, H.; Kibiki, G.; Simonetti, E.; De Jonge, M.I.; Ferwerda, G. Platelets Modulate Innate Immune Response Against Human Respiratory Syncytial Virus In Vitro. Viral Immunol. 2017, 30, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Mick, E.; Mikhalev, E.; Benjamin, E.J.; Tanriverdi, K.; Freedman, J.E. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1030–1037. [Google Scholar] [CrossRef]
- Zaslavsky, A.; Adams, M.; Cao, X.; Yamaguchi, A.; Henderson, J.; Busch-Østergren, P.; Udager, A.; Pitchiaya, S.; Tourdot, B.; Kasputis, T.; et al. Antisense oligonucleotides and nucleic acids generate hypersensitive platelets. Thromb. Res. 2021, 200, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef]
- Banerjee, M.; Huang, Y.; Joshi, S.; Popa, G.J.; Mendenhall, M.D.; Wang, Q.J.; Garvy, B.A.; Myint, T.; Whiteheart, S.W. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Toll-like receptor 9 expression and activation in acute coronary syndrome patients on dual anti-platelet therapy. Thromb. Res. 2016, 148, 89–95. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yasuoka, H.; Yoshimoto, K.; Suzuki, K.; Takeuchi, T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci. Rep. 2021, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Bortolus, C.; Lecointe, K.; Parny, M.; Charlet, R.; Sendid, B.; Jawhara, S. Fungal Chitin Reduces Platelet Activation Mediated via TLR8 Stimulation. Front. Cell Infect. MicroBiol. 2019, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Marshall, J.C.; Namendys-Silva, S.A.; Francois, B.; Martin-Loeches, I.; Lipman, J.; Reinhart, K.; Antonelli, M.; Pickkers, P.; Njimi, H.; et al. Assessment of the worldwide burden of critical illness: The intensive care over nations (ICON) audit. Lancet. Respir. Med. 2014, 2, 380–386. [Google Scholar] [CrossRef]
- Cognasse, F.; Lafarge, S.; Chavarin, P.; Acquart, S.; Garraud, O. Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med. 2007, 33, 382–384. [Google Scholar] [CrossRef]
- Claushuis, T.A.; Van Vught, L.A.; Scicluna, B.P.; Wiewel, M.A.; Klein Klouwenberg, P.M.; Hoogendijk, A.J.; Ong, D.S.; Cremer, O.L.; Horn, J.; Franitza, M.; et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood 2016, 127, 3062–3072. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immun. 2018, 49, 740–753. [Google Scholar] [CrossRef]
- Angus, D.C.; Yang, L.; Kong, L.; Kellum, J.A.; Delude, R.L.; Tracey, K.J.; Weissfeld, L. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit. Care Med. 2007, 35, 1061–1067. [Google Scholar] [CrossRef]
- Larkin, C.M.; Hante, N.K.; Breen, E.P.; Tomaszewski, K.A.; Eisele, S.; Radomski, M.W.; Ryan, T.A.; Santos-Martinez, M.J. Role of matrix metalloproteinases 2 and 9, toll-like receptor 4 and platelet-leukocyte aggregate formation in sepsis-associated thrombocytopenia. PLoS ONE 2018, 13, e0196478. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, T.; Magosevich, D.; Moreno, M.C.; Guzman, M.A.; D’Atri, L.P.; Carestia, A.; Fandiño, M.E.; Fondevila, C.; Schattner, M. Nucleosomes and neutrophil extracellular traps in septic and burn patients. Clin. Immunol. 2017, 183, 254–262. [Google Scholar] [CrossRef] [PubMed]
- De Stoppelaar, S.F.; Claushuis, T.A.; Jansen, M.P.; Hou, B.; Roelofs, J.J.; Van ‘t Veer, C.; Van der Poll, T. The role of platelet MyD88 in host response during gram-negative sepsis. J. Thromb. Haemost. 2015, 13, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.J.; Aghakasiri, N.; Rumbaut, R.E. Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PLoS ONE 2012, 7, e41254. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Sheu, J.R.; Lee, C.R.; Lin, C.H.; Hsiao, G.; Ko, W.C.; Chen, Y.C.; Yen, M.H. Mechanisms involved in the antiplatelet activity of Staphylococcus aureus lipoteichoic acid in human platelets. Thromb. Haemost. 2000, 83, 777–784. [Google Scholar]
- Waller, A.K.; Sage, T.; Kumar, C.; Carr, T.; Gibbins, J.M.; Clarke, S.R. Staphylococcus aureus lipoteichoic acid inhibits platelet activation and thrombus formation via the Paf receptor. J. Infect. Dis. 2013, 208, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Neder, J.; Cui, P.; Suen, A.; Tanaka, K.; Zou, L.; Chao, W. Toll-like receptors 2 and 7 mediate coagulation activation and coagulopathy in murine sepsis. J. Thromb. Haemost. 2019, 17, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Rayes, J.; Lax, S.; Wichaiyo, S.; Watson, S.K.; Di, Y.; Lombard, S.; Grygielska, B.; Smith, S.W.; Skordilis, K.; Watson, S.P. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat. Commun. 2017, 8, 2239. [Google Scholar] [CrossRef] [PubMed]
- Ribes, A.; Vardon-Bounes, F.; Mémier, V.; Poette, M.; Au-Duong, J.; Garcia, C.; Minville, V.; Sié, P.; Bura-Rivière, A.; Voisin, S.; et al. Thromboembolic events and Covid-19. Adv. Biol. Regul. 2020, 77, 100735. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Althaus, K.; Marini, I.; Zlamal, J.; Pelzl, L.; Singh, A.; Häberle, H.; Mehrländer, M.; Hammer, S.; Schulze, H.; Bitzer, M.; et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood 2021, 137, 1061–1071. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 2021, 12, 50. [Google Scholar] [CrossRef]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, J.; Fang, Y.; Lu, S.; Wu, J.; Zheng, X.; Deng, F. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J. Hematol. Oncol. 2021, 14, 72. [Google Scholar] [CrossRef]
- Zucker-Franklin, D.; Seremetis, S.; Zheng, Z.Y. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood 1990, 75, 1920–1923. [Google Scholar] [CrossRef]
- Youssefian, T.; Drouin, A.; Masse, J.M.; Guichard, J.; Cramer, E.M. Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002, 99, 4021–4029. [Google Scholar] [CrossRef]
- Boukour, S.; Massé, J.M.; Bénit, L.; Dubart-Kupperschmitt, A.; Cramer, E.M. Lentivirus degradation and DC-SIGN expression by human platelets and megakaryocytes. J. Thromb. Haemost. 2006, 4, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.R.; Singh, M.V.; Dewhurst, S.; Schifitto, G.; Maggirwar, S.B. Platelets function as an acute viral reservoir during HIV-1 infection by harboring virus and T-cell complex formation. Blood Adv. 2020, 4, 4512–4521. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Hamzeh-Cognasse, H.; Berthet, J.; Damien, P.; Lucht, F.; Pozzetto, B.; Garraud, O. Altered release of regulated upon activation, normal T-cell expressed and secreted protein from human, normal platelets: Contribution of distinct HIV-1MN gp41 peptides. AIDS 2009, 23, 2057–2059. [Google Scholar] [CrossRef]
- Van der Heijden, W.A.; Van de Wijer, L.; Jaeger, M.; Grintjes, K.; Netea, M.G.; Urbanus, R.T.; Van Crevel, R.; Van den Heuvel, L.P.; Brink, M.; Rodenburg, R.J.; et al. Long-term treated HIV infection is associated with platelet mitochondrial dysfunction. Sci. Rep. 2021, 11, 6246. [Google Scholar] [CrossRef] [PubMed]
- Damien, P.; Cognasse, F.; Lucht, F.; Suy, F.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. Highly active antiretroviral therapy alters inflammation linked to platelet cytokines in HIV-1-infected patients. J. Infect. Dis. 2013, 208, 868–870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falasca, K.; Lanuti, P.; Ucciferri, C.; Pieragostino, D.; Cufaro, M.C.; Bologna, G.; Federici, L.; Miscia, S.; Pontolillo, M.; Auricchio, A.; et al. Circulating extracellular vesicles as new inflammation marker in HIV infection. Aids 2021, 35, 595–604. [Google Scholar] [CrossRef]
- Hottz, E.D.; Medeiros-de-Moraes, I.M.; Vieira-de-Abreu, A.; De Assis, E.F.; Vals-de-Souza, R.; Castro-Faria-Neto, H.C.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J. Immunol. 2014, 193, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Lien, T.-S.; Chan, H.; Sun, D.-S.; Wu, J.-C.; Lin, Y.-Y.; Lin, G.-L.; Chang, H.-H. Exposure of Platelets to Dengue Virus and Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent Platelet Cell Death and Thrombocytopenia in Mice. Front. Immunol. 2021, 12, 616394. [Google Scholar] [CrossRef]
- Chao, C.H.; Wu, W.C.; Lai, Y.C.; Tsai, P.J.; Perng, G.C.; Lin, Y.S.; Yeh, T.M. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 2019, 15, e1007625. [Google Scholar] [CrossRef]
- Koenen, R.R.; Von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- Lutgens, E.; Daemen, M.J. CD40-CD40L interactions in atherosclerosis. Trends CardioVasc. Med. 2002, 12, 27–32. [Google Scholar] [CrossRef]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef]
- Biswas, S.; Xin, L.; Panigrahi, S.; Zimman, A.; Wang, H.; Yakubenko, V.P.; Byzova, T.V.; Salomon, R.G.; Podrez, E.A. Novel phosphatidylethanolamine derivatives accumulate in circulation in hyperlipidemic ApoE−/− mice and activate platelets via TLR2. Blood 2016, 127, 2618–2629. [Google Scholar] [CrossRef]
- Biswas, S.; Zimman, A.; Gao, D.; Byzova, T.V.; Podrez, E.A. TLR2 Plays a Key Role in Platelet Hyperreactivity and Accelerated Thrombosis Associated With Hyperlipidemia. Circ. Res. 2017, 121, 951–962. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef] [PubMed]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Raslan, Z.; Aburima, A.; Magwenzi, S.; Wraith, K.S.; Spurgeon, B.E.J.; Hindle, M.S.; Law, R.; Febbraio, M.; Naseem, K.M. Atherogenic lipid stress induces platelet hyperactivity through CD36-mediated hyposensitivity to prostacyclin: The role of phosphodiesterase 3A. Haematologica 2020, 105, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Santos, M.M.; Fernandes, C.; Liao, M.; Iamarene, K.; Zhang, J.Y.; Sukhova, G.K.; Shi, G.P. Toll-like receptor 7 deficiency protects apolipoprotein E-deficient mice from diet-induced atherosclerosis. Sci. Rep. 2017, 7, 847. [Google Scholar] [CrossRef]
- Prandoni, P.; Falanga, A.; Piccioli, A. Cancer and venous thromboembolism. Lancet Oncol. 2005, 6, 401–410. [Google Scholar] [CrossRef]
- Tuzovic, M.; Herrmann, J.; Iliescu, C.; Marmagkiolis, K.; Ziaeian, B.; Yang, E.H. Arterial Thrombosis in Patients with Cancer. Curr. Treat. Options CardioVasc. Med. 2018, 20, 40. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, D.; Darbousset, R.; Farge, D.; Debourdeau, P.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 2014, 41, 346–358. [Google Scholar] [CrossRef]
- Shirai, T.; Inoue, O.; Tamura, S.; Tsukiji, N.; Sasaki, T.; Endo, H.; Satoh, K.; Osada, M.; Sato-Uchida, H.; Fujii, H.; et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J. Thromb. Haemost. 2017, 15, 513–525. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Gil-Pulido, J.; Sarukhanyan, E.; Burkard, P.; Shityakov, S.; Schonhart, C.; Stegner, D.; Remer, K.; Nurden, P.; Nurden, A.T.; et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood 2020, 135, 1146–1160. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Yan, L.; Yang, W.; Wu, F.Q.; Ling, Y.; Chen, S.Z.; Tang, L.; Tan, Y.X.; Cao, D.; Wu, M.C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
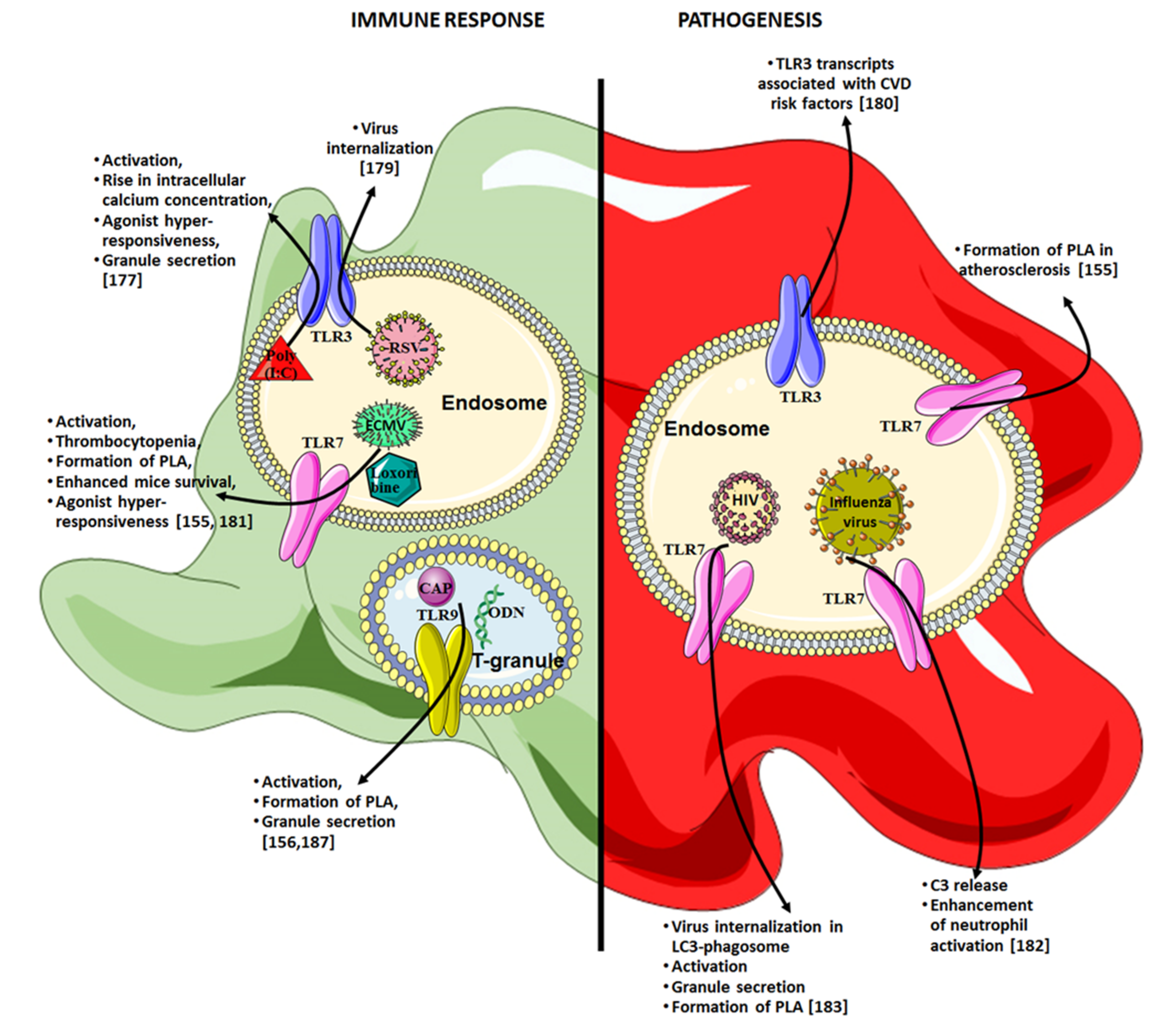{kind=link}
| Platelet Response to Stress | Major Biological Molecules Involved | Mechanisms/Consequences |
|---|---|---|
| Apoptosis |
| |
| ||
| ||
|
| |
|
| |
| ||
| Necrosis |
|
|
|
| |
|
| |
| Autophagy |
|
|
| Immune Cells Interacting with Platelets. | Immunomodulatory Molecules Released by Platelet | Modulation of Immune Response |
|---|---|---|
| Neutrophils |
| |
| Monocytes | ||
| Dendritic cells |
| |
| B and T cells |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebermeyer, T.; Cognasse, F.; Berthelot, P.; Mismetti, P.; Garraud, O.; Hamzeh-Cognasse, H. Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword. Int. J. Mol. Sci. 2021, 22, 7894. https://doi.org/10.3390/ijms22157894
Ebermeyer T, Cognasse F, Berthelot P, Mismetti P, Garraud O, Hamzeh-Cognasse H. Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword. International Journal of Molecular Sciences. 2021; 22(15):7894. https://doi.org/10.3390/ijms22157894
Chicago/Turabian StyleEbermeyer, Théo, Fabrice Cognasse, Philippe Berthelot, Patrick Mismetti, Olivier Garraud, and Hind Hamzeh-Cognasse. 2021. "Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword" International Journal of Molecular Sciences 22, no. 15: 7894. https://doi.org/10.3390/ijms22157894
APA StyleEbermeyer, T., Cognasse, F., Berthelot, P., Mismetti, P., Garraud, O., & Hamzeh-Cognasse, H. (2021). Platelet Innate Immune Receptors and TLRs: A Double-Edged Sword. International Journal of Molecular Sciences, 22(15), 7894. https://doi.org/10.3390/ijms22157894