
Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners


Abstract
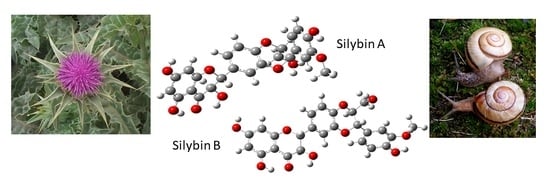
1. Introduction
2. Silymarin and Silymarin Flavonolignans
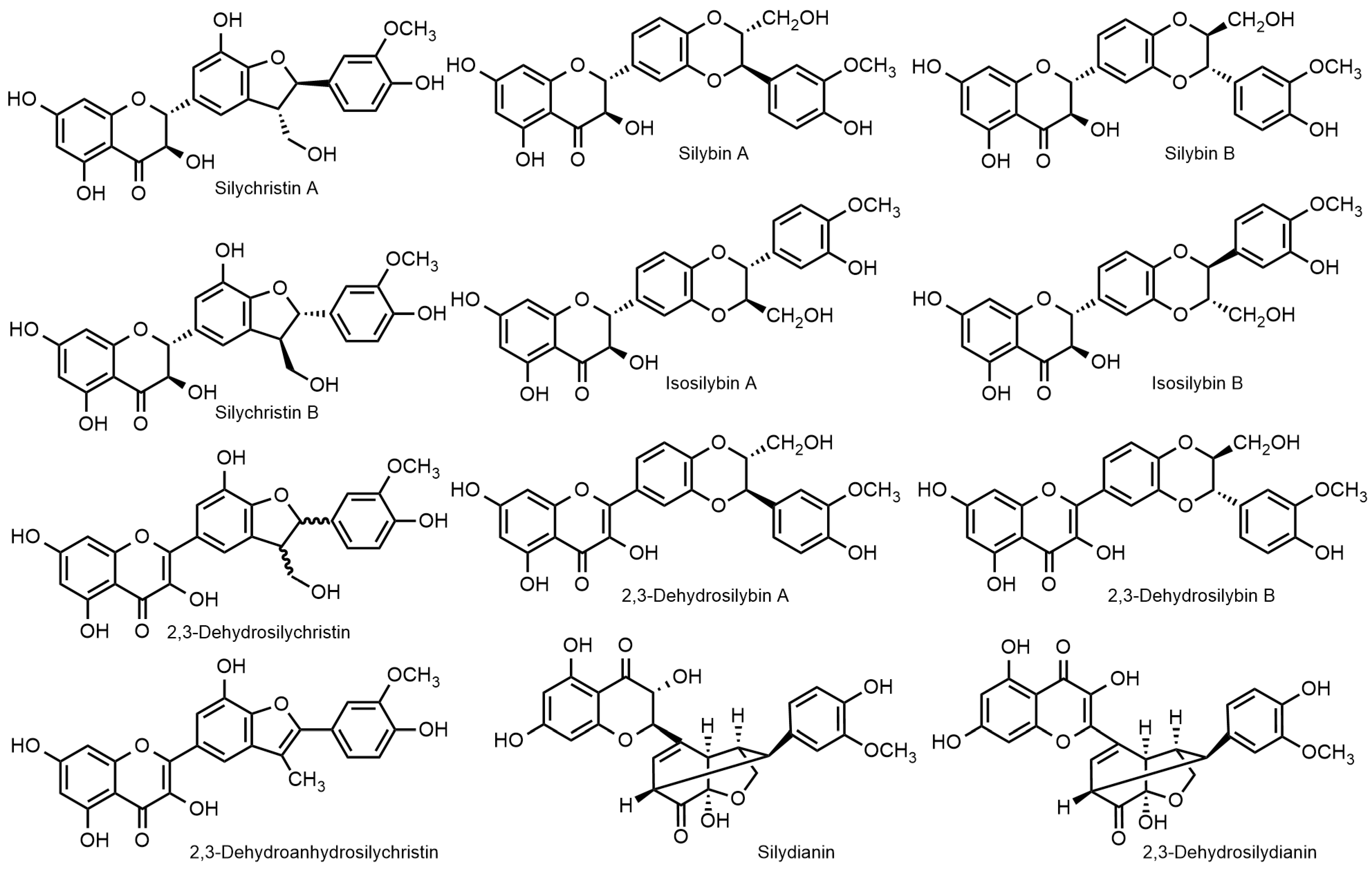
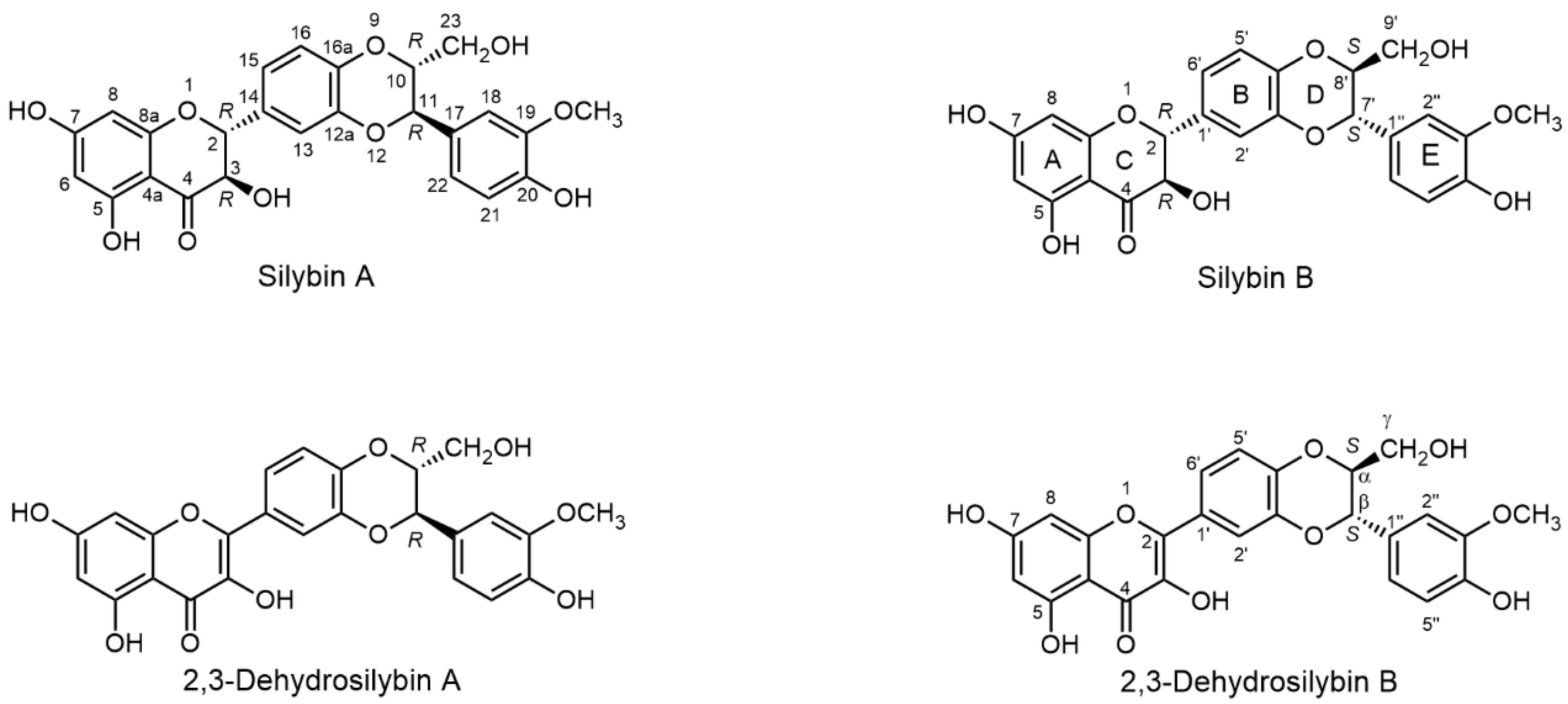
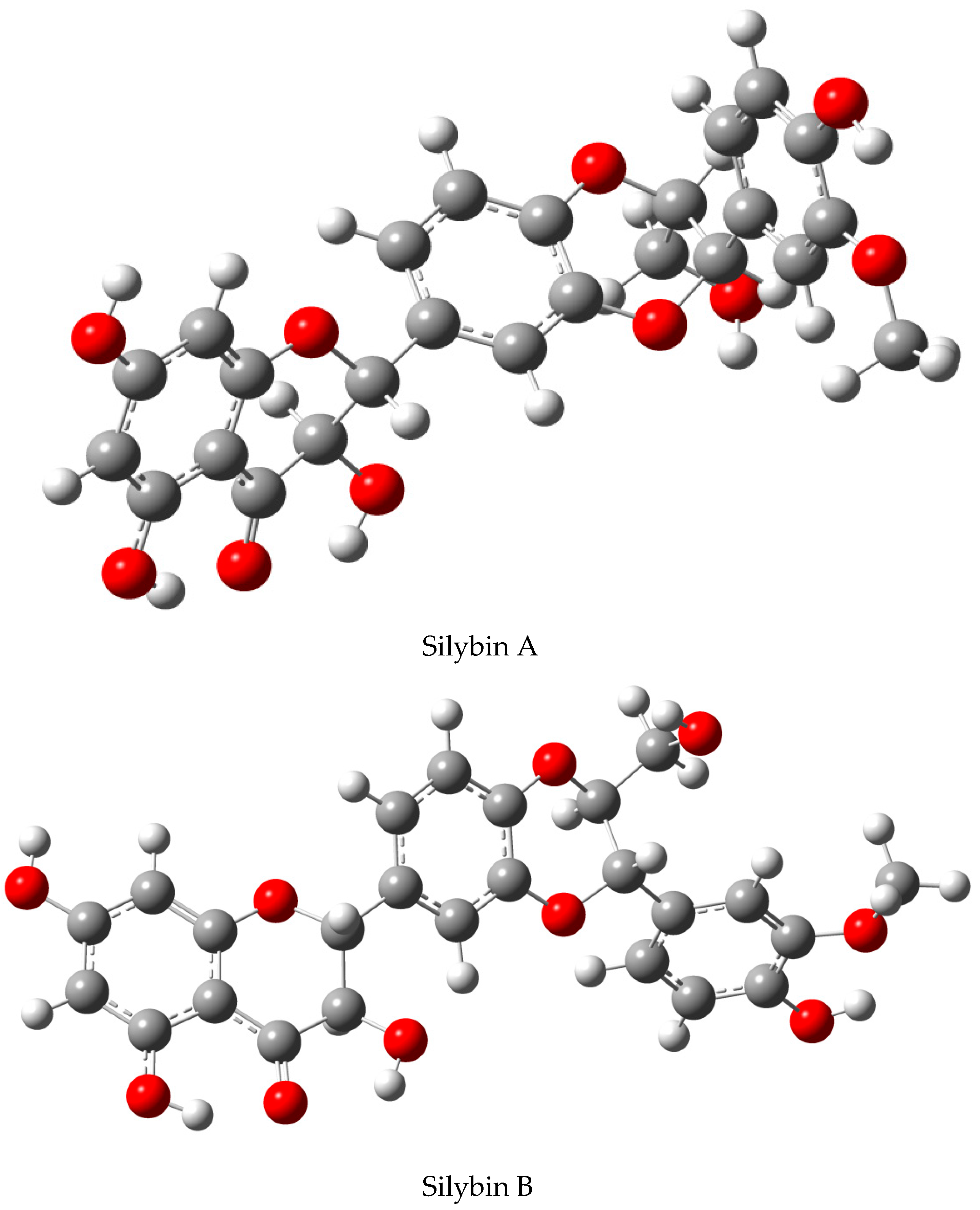
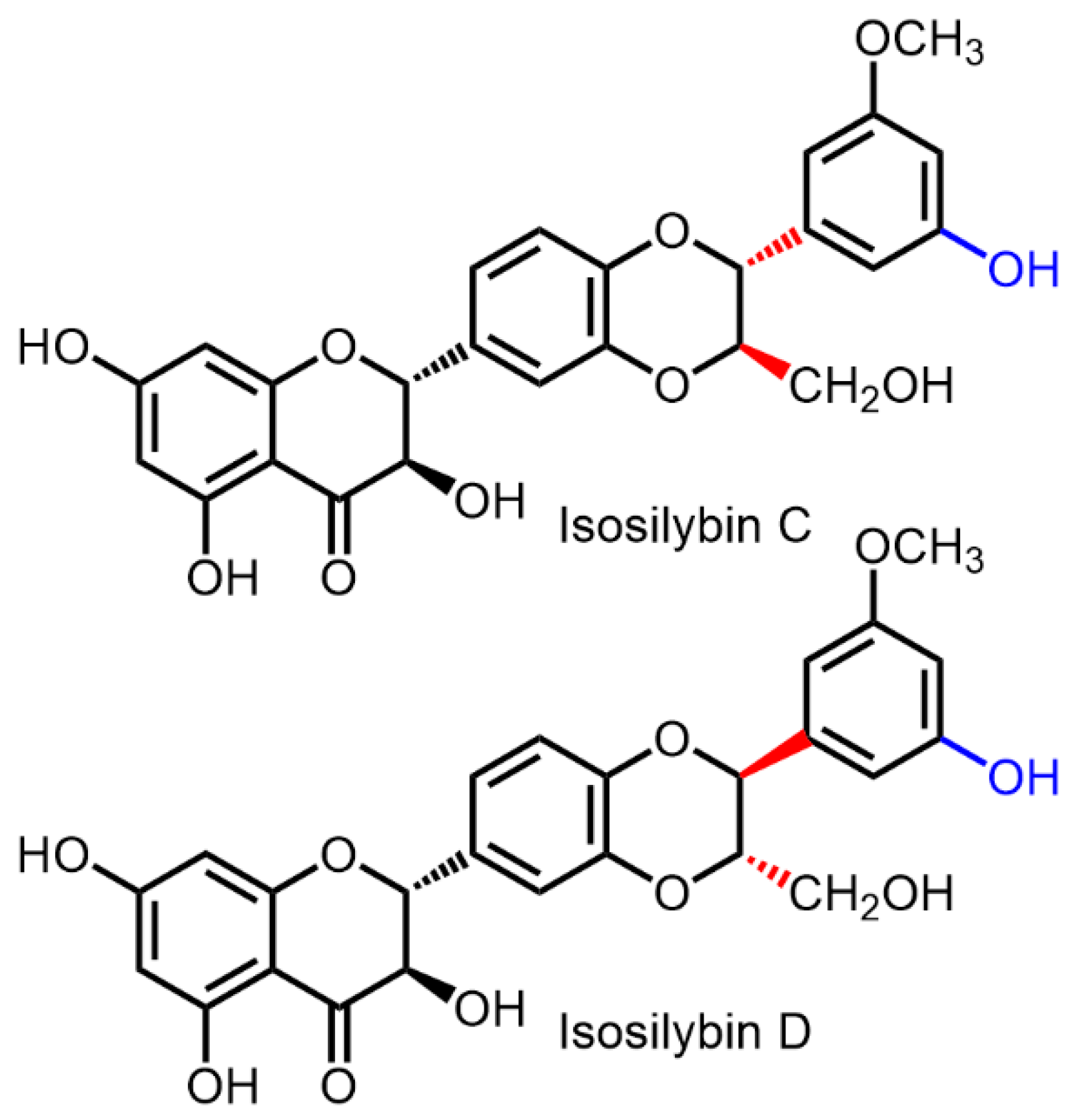
2.1. Nomenclature of Silybin Diastereomers
2.2. Biosynthesis of Silymarin Flavonolignans
2.3. Separation of Flavonolignan Diastereomers
2.4. Analytical Methods for Flavonolignan Diastereomer Determination
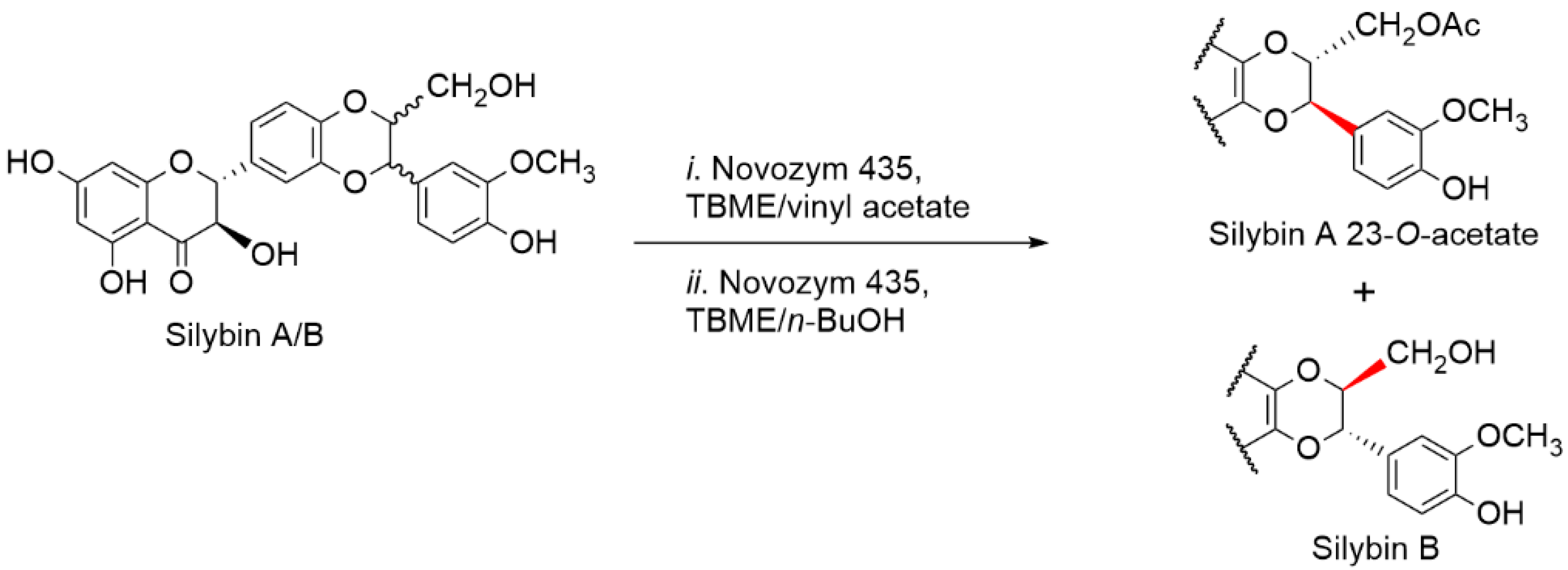
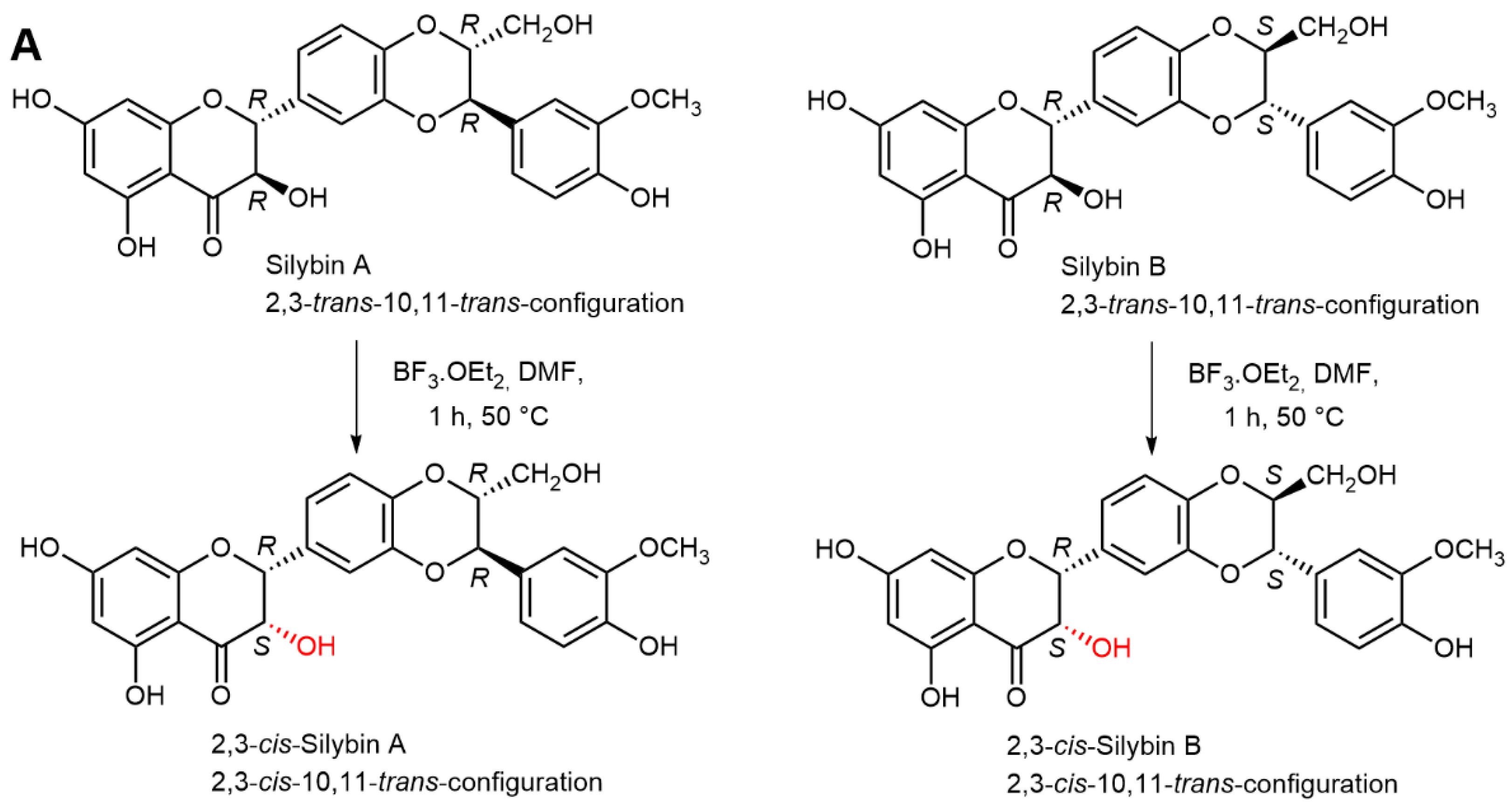
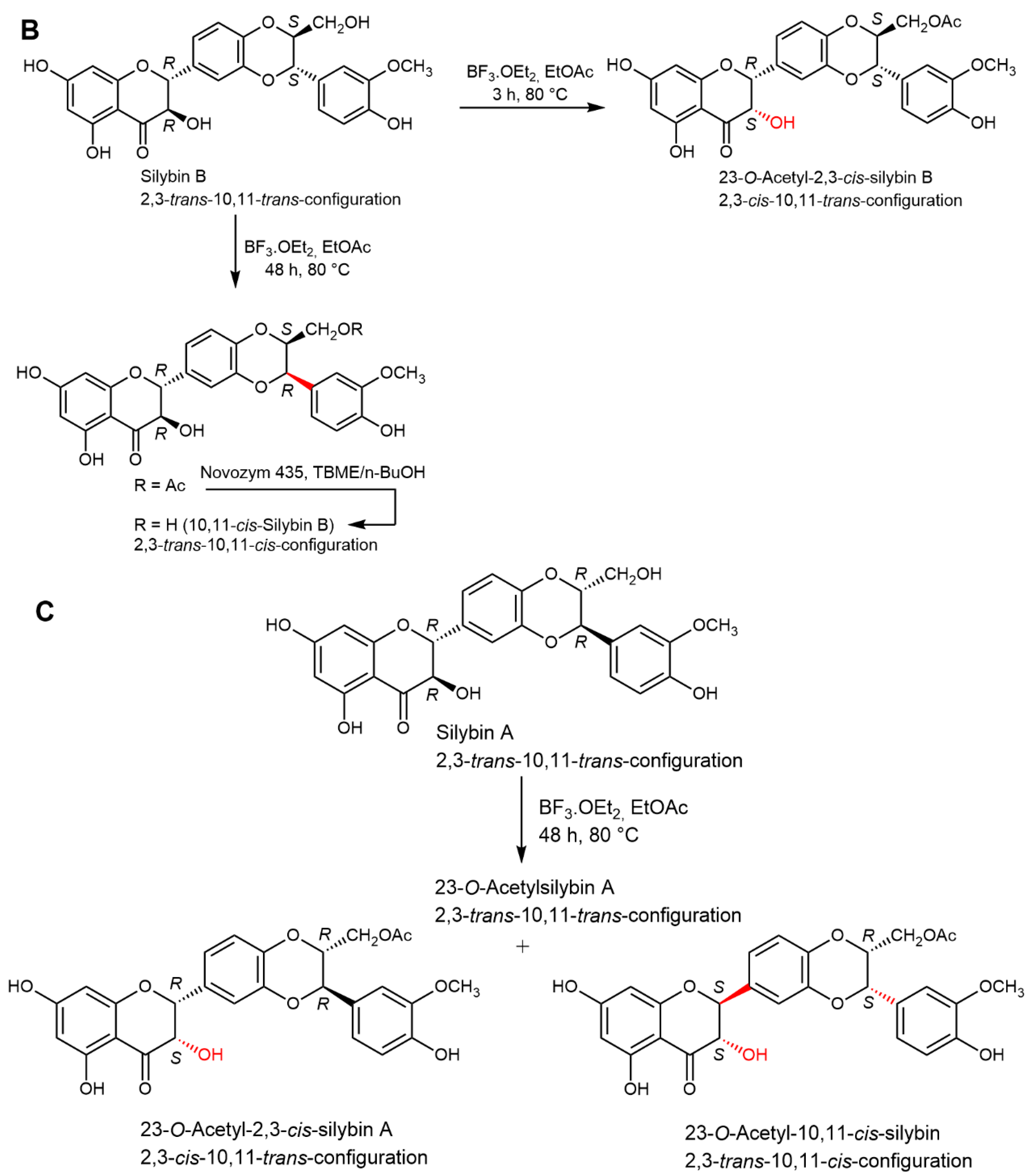
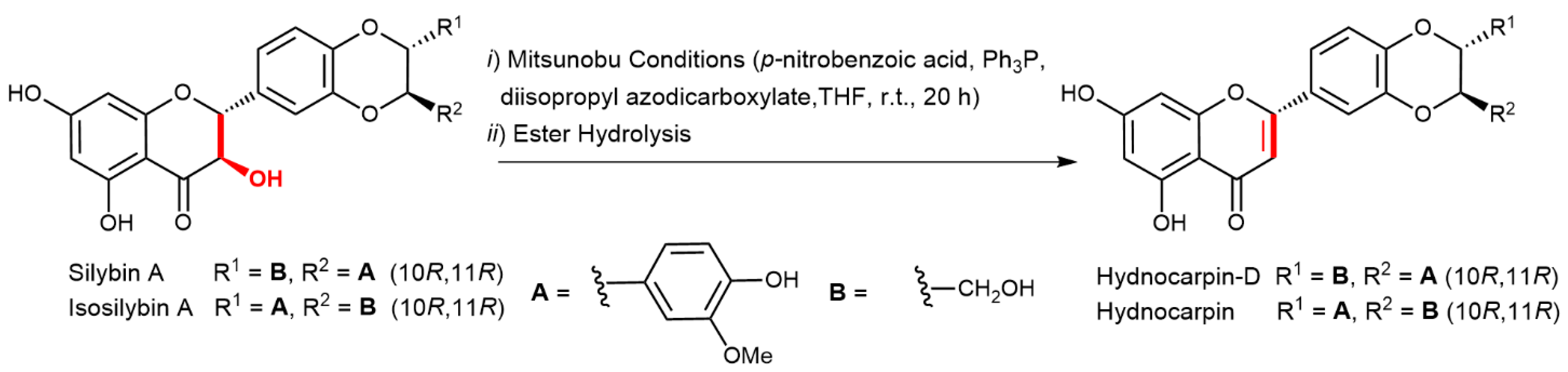
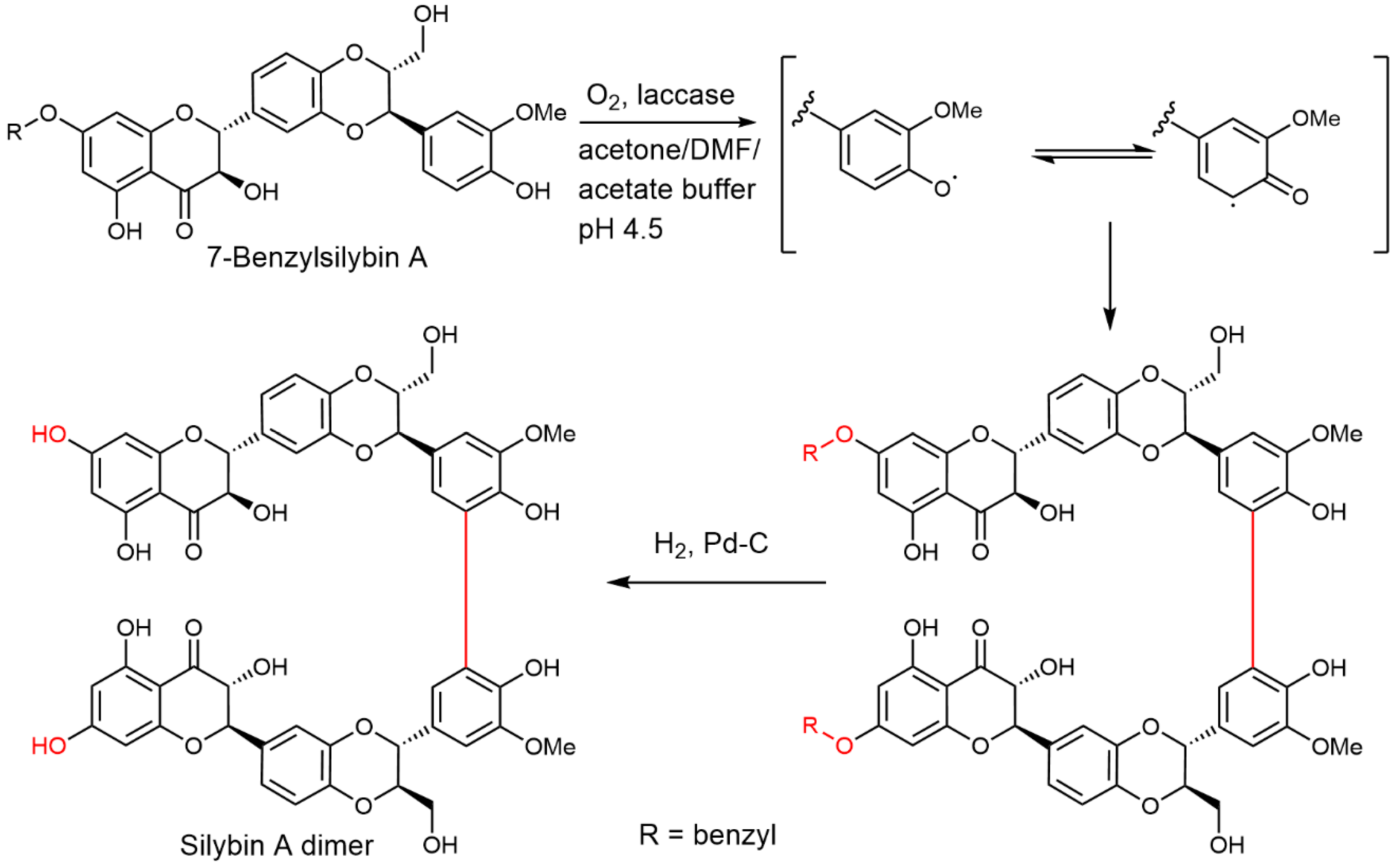
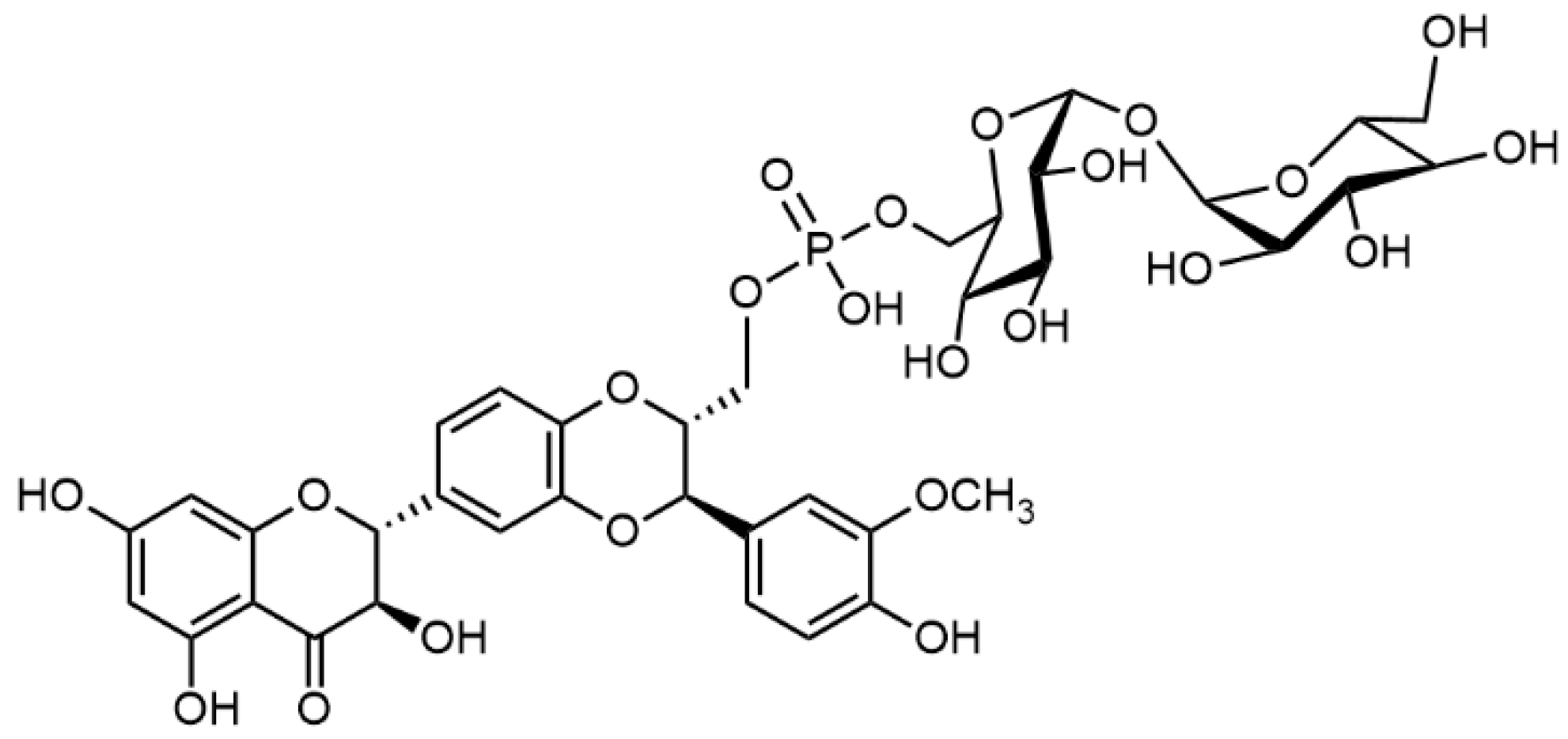
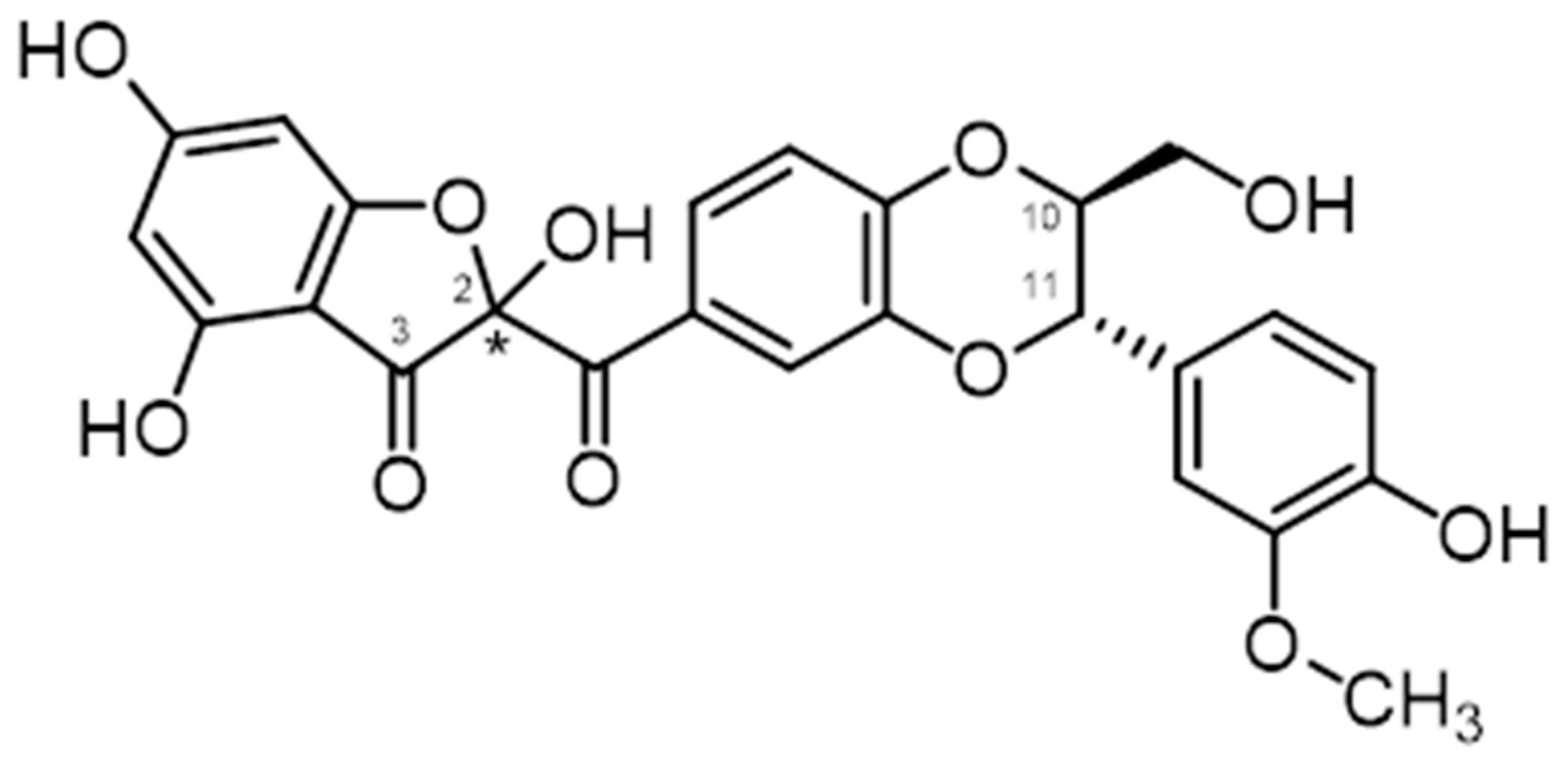
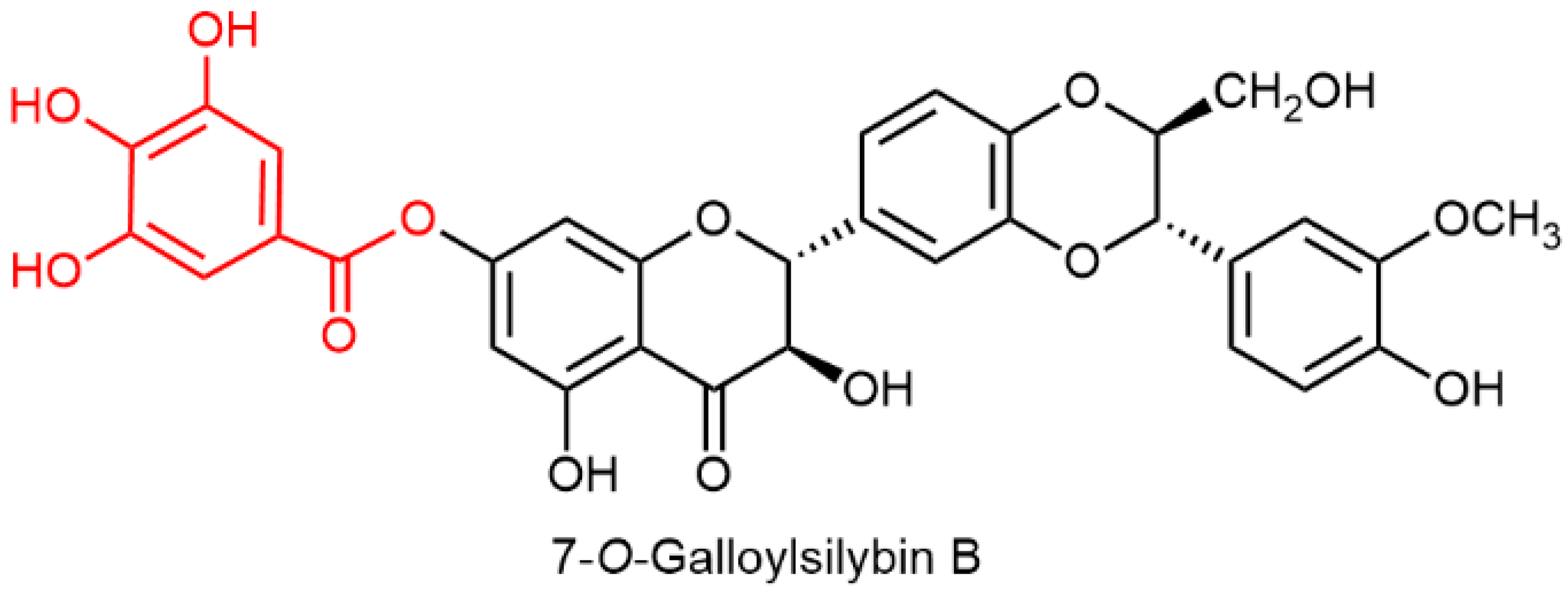
2.5. Chemical and Enzymatic Transformations of Optically Pure Silybin A and B
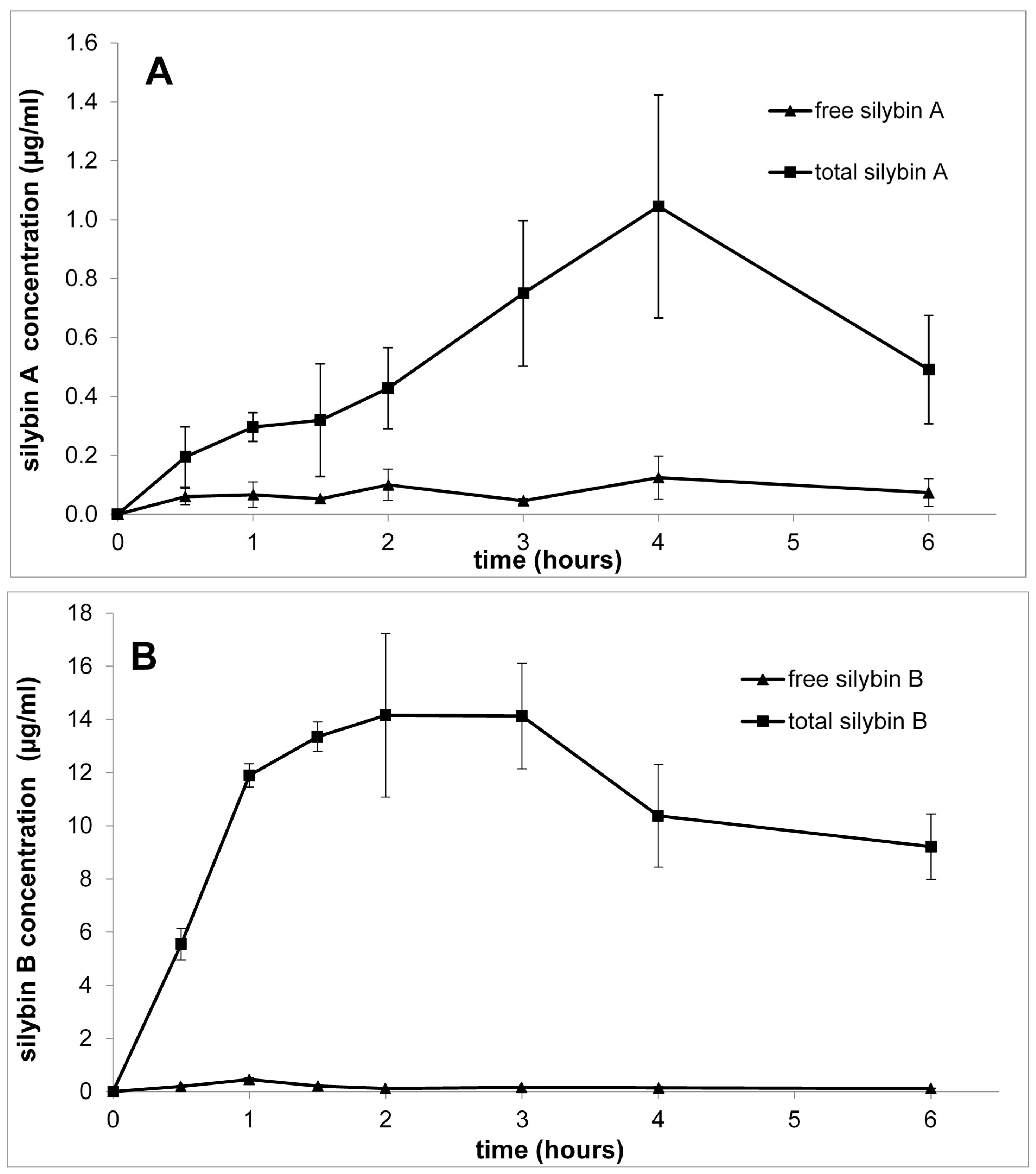
3. Pharmacokinetics of Silybin with Regard to Its Stereochemistry
4. Specific Biological Effects of Silybin A and B and Other Optically Pure Flavonolignans
4.1. Estrogenic Activity
4.2. Antidiabetic and Anticholesterolemic Activity
4.3. Cardiovascular Activity
4.4. Anticancer Activity
4.5. Inhibition of Hepatitis C Virus; Antiviral Activity
4.6. Antiparasitic Activity
4.7. Neurological Activity
4.8. Inhibition of Drug-Metabolizing Enzymes
4.9. Multidrug Resistance Activity
4.10. Other Biological Activities
5. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| ABCA1 | Transmembrane ATP-binding cassette transporter A1; |
| ADME | Absorption, distribution, metabolism, and excretion |
| AhR | Aryl hydrocarbon receptor |
| APX1 | Ascorbate peroxidase |
| AR | Androgen receptor |
| AUC | Area under the curve |
| BRAF | Serine/threonine-protein kinase B-raf |
| Cdk2; Cdk4 | Cyclin-dependent kinases |
| CYP | Cytochrome P-450 |
| d.e. | Diastereomeric excess |
| DM1 | Diabetes mellitus type 1 |
| DM2 | Diabetes mellitus type 2 |
| DMF | Dimethylformamide |
| ECD | Electron circular dichroism |
| EGCG | epi-Gallocatechin-3-gallate |
| EtOAc | Ethyl acetate |
| ER | Estrogen receptors |
| FGT-1 | Facilitative glucose transporter isoform 1 |
| GLP-1 | Glucagon-like peptide-1 production |
| GSH | Glutathione (reduced) |
| HDL | High-density lipoprotein |
| HEK293 | Human embryonic kidney |
| HEWL | Hen egg-white lysozyme |
| HH | Human hepatoma |
| HPC | Human prostate carcinoma |
| HUVEC | Human umbilical vein endothelial cells |
| IR | Infrared spectroscopy |
| IT-TOF | Ion trap time-of-flight |
| IT | Ion trap |
| MCT8 | Monocarboxylate transporter 8 |
| MDCK1 | Madin–Darby canine kidney |
| MDR | Multi-drug resistance |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| Nrf2-HO-1/SOD2 | Estrogen receptor-α-dependent Nrf2-heme oxygenase-1/superoxide Dismutase 2 antioxidative |
| PA | Prostate adenocarcinoma |
| PC12 | Pheochromocytoma of the rat adrenal medulla |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| Q-TOF | Quadrupole time-of-flight |
| SQ | Sector quadrupole |
| SULT | Sulfotransferases |
| TBME | tert-Butyl methyl ether |
| ThT | Thioflavin T |
| TLC | Thin-layer chromatography |
| UDP | Uridine diphosphate |
| UGT | UDP-glucuronosyl transferase. |
| UHPLC | Ultra high performance liquid chromatography |
| VLDL | Very low-density lipoproteins |
References
- Warnholtz, A.; Munzel, T. Why do antioxidants fail to provide clinical benefit? Curr. Control Trials Cardiovasc. Med. 2000, 1, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Rad. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Anthony, K.P.; Saleh, M.A. Free radical scavenging and antioxidant activities of silymarin components. Antioxidants 2013, 2, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Křen, V.; Walterová, D. Silybin and silymarin—New effects and applications. Biomed Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2005, 149, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1156. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, I.; Tetenyi, P.; Antus, S.; Seligmann, O.; Chari, V.M.; Seitz, M.; Wagner, H. Struktur von Silandrin und Silymonin, zwei neuen Flavanolignanen aus einer weißblühenden Silybum marianum Varietät. Planta Med. 1981, 43, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, T.; Whittaker, A.; Benedettelli, S.; Carboni, A.; Andrzejewska, J. The study of flavonolignan association patterns in fruits of diverging Silybum marianum (L.) Gaertn. chemotypes provides new insights into the silymarin biosynthetic pathway. Phytochemistry 2017, 144, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.; Holečková, V.; Petrásková, L.; Biedermann, D.; Valentová, K.; Buchta, M.; Křen, V. The silymarin composition and why does it matter? Food Res. Int. 2017, 100, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Petrásková, L.; Káňová, K.; Valentová, K.; Biedermann, D.; Křen, V. A Simple and rapid HPLC separation and quantification of flavonoid, flavonolignans and 2,3-dehydroflavonolignans in silymarin. Foods 2020, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Fenclová, M.; Stránská-Zachariášová, M.; Beneš, F.; Nováková, A.; Jonatová, P.; Křen, V.; Vítek, L.; Hajšlová, J. Liquid chromatography-drift tube ion mobility-mass spectrometry as a new challenging tool for the separation and characterization of silymarin flavonolignans. Anal. Bioanal. Chem. 2020, 412, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Kroll, D.J.; Shaw, H.S.; Oberlies, N.H. Milk thistle nomenclature: Why it matters in cancer research and pharmacokinetic studies. Integr. Cancer Ther. 2007, 6, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Šimánek, V.; Křen, V.; Ulrichová, J.; Vičar, J.; Cvak, L. Silymarin: “What is in the name...?” An appeal for a change of editorial policy. Hepatology 2000, 32, 442–443. [Google Scholar] [CrossRef]
- Křen, V. Silibinin chirality. J. Photochem. Photobiol. A 2009, 203, 222–223. [Google Scholar] [CrossRef]
- Křen, V.; Gažák, R.; Biedermann, D.; Marhol, P. Silybin (silibinin) structure and chirality. Chromatographia 2010, 71, 167–168. [Google Scholar] [CrossRef]
- O’Neil, M.J. (Ed.) Merck Index, 14th ed.; The Merck Publishing Group: Whitehouse Station, NJ, USA, 2006; pp. 8523–8524. ISBN 978-0-911910-00-1. [Google Scholar]
- Merck Index, 15th ed; O’Neil, M.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2013; entry #8532; ISBN 978-1-84973670-1. [Google Scholar]
- Malan, E.; Swinny, E.; Ferreira, D. A 3-oxygenated flavonolignoid from Distemonanthus benthamianus. Phytochemistry 1994, 37, 1771–1772. [Google Scholar] [CrossRef]
- Yang, L.X.; Huang, K.X.; Li, H.B.; Gong, J.X.; Wang, F.; Feng, Y.B.; Tao, Q.F.; Wu, Y.H.; Li, X.K.; Wu, X.M.; et al. Synthesis, and examination of neuron protective properties of alkenylated and amidated dehydro-silybin derivatives. J. Med. Chem. 2009, 52, 7732–7752. [Google Scholar] [CrossRef]
- Pelter, A.; Hänsel, R. The structure of silybin (silybum substance E6), the first flavonolignan. Tetrahedron Lett. 1968, 25, 2911–2916. [Google Scholar] [CrossRef]
- Kim, C.; Graf, T.N.; Sparacino, C.M.; Wani, M.C.; Wall, M.E. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum). Org. Biomol. Chem. 2003, 1, 1684–1689, Erratum in 2003, 1, 3470. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.N.; Wani, M.C.; Agarwal, R.; Kroll, D.J.; Oberlies, N.H. Gram-scale purification of flavonolignan diastereomes from Silybum marianum (milk thistle) extract in support of preclinical in vivo studies for prostate cancer chemoprevention. Planta Med. 2007, 73, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, J.G.; Lankin, D.C.; Graf, T.N.; Friesen, J.B.; Chen, S.-N.; McAlpine, J.B.; Oberlies, N.H.; Pauli, G.F. HiFSA fingerprinting applied to isomers with near-identical NMR spectra: The silybin/isosilybin case. J. Org. Chem. 2013, 78, 2827–2839. [Google Scholar] [CrossRef]
- Lee, D.Y.-W.; Liu, Y. Molecular structure and stereochemistry of silybin A, silybin B, isosilybin A, and isosilybin B, isolated from Silybum marianum (milk thistle). J. Nat. Prod. 2003, 66, 1171–1174, Erratum in 2003, 66, 1632. [Google Scholar] [CrossRef] [PubMed]
- Novotná, M.; Gažák, R.; Biedermann, D.; di Meo, F.; Marhol, P.; Kuzma, M.; Bednárová, L.; Fuksová, K.; Trouillas, P.; Křen, V. Cis-trans isomerization of silybins A and B. Beilstein J. Org. Chem. 2014, 10, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Schrall, R.; Becker, H. Callus- und Suspensionskulturen von Silybum marianum. II. Mitteilung: Umsetzung von Flavonoiden mit Coniferylalkohol zu Flavonolignanen. Planta Med. 1977, 32, 27–32. [Google Scholar] [CrossRef]
- Lv, Y.K.; Xu, S.; Lyu, Y.B.; Zhou, S.H.; Du, G.C.; Chen, J.; Zhou, J.W. Engineering enzymatic cascades for the efficient biotransformation of eugenol and taxifolin to silybin and isosilybin. Green Chem. 2019, 21, 1660–1667. [Google Scholar] [CrossRef]
- Merlini, L.; Zanarotti, A.; Pelter, A.; Rochefort, M.P.; Hansel, R. Biomimetic synthesis of natural silybin. J. Chem. Soc. Chem. Commun. 1979, 16, 695. [Google Scholar] [CrossRef]
- Althagafy, H.S.; Meza-Avina, M.E.; Oberlies, N.H.; Croatt, M.P. Mechanistic study of the biomimetic synthesis of flavonolignan diastereoisomers in milk thistle. J. Org. Chem. 2013, 78, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Lv, Y.K.; Gao, S.; Xu, S.; Du, G.C.; Zhou, J.W.; Chen, J. Spatial organization of silybin biosynthesis in milk thistle [Silybum marianum (L.) Gaertn]. Plant J. 2017, 92, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Corchete, P. Gene expression and flavonolignan production in fruits and cell cultures of Silybum marianum. J. Plant Physiol. 2016, 192, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, T.; Fulvio, F.; Pietrella, M.; Focacci, M.; Lauria, M.; Paris, R. In Silybum marianum Italian wild populations the variability of silymarin profiles results from the combination of only two stable chemotypes. Fitoterapia 2021, 148, 104797. [Google Scholar] [CrossRef] [PubMed]
- Davis-Searles, P.R.; Nakanishi, Y.; Kim, N.-C.; Graf, T.N.; Oberlies, N.H.; Wani, M.C.; Wall, M.E.; Agarwal, R.; Kroll, D.J. Milk thistle and prostate cancer: Differential effects of pure flavonolignans from Silybum marianum on antiproliferative end points in human prostate carcinoma cells. Cancer Res. 2005, 65, 4448–4457. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin B and isosilybin A inhibit growth, induce G1 arrest and cause apoptosis in human prostate cancer LNCaP and 22Rv1 cells. Carcinogenesis 2007, 28, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Trouillas, P.; Biedermann, D.; Fuksová, K.; Marhol, P.; Kuzma, M.; Křen, V. Base-catalyzed oxidation of silybin and isosilybin into 2,3-dehydro-derivatives. Tetrahedron Lett. 2013, 54, 315–317. [Google Scholar] [CrossRef]
- Pyszková, M.; Biler, M.; Biedermann, D.; Valentová, K.; Kuzma, M.; Vrba, J.; Ulrichová, J.; Sokolová, R.; Mojović, M.; Popović-Bijelić, A.; et al. Flavonolignan 2,3-dehydroderivatives: Preparation, antiradical and cytoprotective activity. Free Rad. Biol. Med. 2016, 90, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Svobodová, A.; Psotová, J.; Sedmera, P.; Přikrylová, V.; Walterová, D.; Křen, V. Oxidised derivatives of silybin and their antiradical and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Sasaki, T.; Li, W.; Wang, J.; Zhang, X.; Li, D.; Li, Z.; Cheng, M.; Hua, H.; Koike, K. Identification of flavonolignans from Silybum marianum seeds as allosteric protein tyrosine phosphatase 1B inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Pelter, A.; Hänsel, R. Struktur des Silybins: I. Abbauversuche. Chem. Ber. 1975, 108, 790–802. [Google Scholar] [CrossRef]
- Lotter, H.; Wagner, H. Zur Stereochemie von Silybin. Z. Naturforsch. Sect. C J. Biosci. 1983, 38, 339–341. [Google Scholar] [CrossRef]
- Tittel, G.; Wagner, H. High-performance liquid chromatographic separationof silymarins and their determination in raw extracts of Silybum marianum Gaertn. J. Chromatogr. A 1977, 135, 499–501. [Google Scholar] [CrossRef]
- Arnone, A.; Merlini, L.; Zanarotti, A. Constituents of Silybum marianum. Structure of isosilybin and stereochemistry of silybin. J. Chem. Soc. Chem. Commun. 1979, 696–697. [Google Scholar] [CrossRef]
- Sy-Cordero, A.; Day, C.S.; Oberlies, N.H. Absolute configuration of isosilybin A by X-ray crystallography of the heavy atom analogue 7-(4-bromobenzoyl)isosilybin A. J. Nat. Prod. 2012, 75, 1879–1881. [Google Scholar] [CrossRef][Green Version]
- Zanarotti, A. Stereochemistry of silychristin. Mild dehydrogenation of flavonols. Heterocycles 1982, 19, 1585–1586. [Google Scholar] [CrossRef]
- Smith, W.A.; Lauren, D.R.; Burgess, E.J.; Perry, N.B.; Martin, R.J. A silychristin isomer and variation of flavonolignan levels in milk thistle (Silybum marianum) fruits. Planta Med. 2005, 71, 877–880. [Google Scholar] [CrossRef]
- Biedermann, D.; Buchta, M.; Holečková, V.; Sedlák, D.; Valentová, K.; Cvačka, J.; Bednárova, L.; Křenková, A.; Kuzma, M.; Skuta, C.; et al. Křen V. Silychristin: Skeletal alterations and biological activities. J. Nat. Prod. 2016, 79, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Takagi, S.; Rosenstein, R.; Shiono, R.; Wagner, H.; Hörhammer, L.; Seligmann, O.; Farnsworth, N. Structure of silydianin, an isomer of silymarin (silybin), by X-ray analysis. Tetrahedron Lett. 1970, 11, 2675–2678. [Google Scholar] [CrossRef]
- Wagner, H.; Seligmann, O.; Seitz, M.; Abraham, D.; Sonnenbichler, J. Silydianin and silychristin, two isomeric silymarins from Silybum marianum L. Gaertn. (milk thistle). Z. Naturforsch. B 1976, 31, 876–884. [Google Scholar] [CrossRef]
- Biedermann, D.; Moravcová, V.; Valentová, K.; Kuzma, M.; Petrásková, L.; Císařová, I.; Křen, V. Oxidation of flavonolignan silydianin to unexpected lactone-acid derivative. Phytochem. Lett. 2019, 30, 14–20. [Google Scholar] [CrossRef]
- Křen, V.; Sedmera, P.; Kubisch, J.; Halada, P.; Přikrylová, V.; Jegorov, A.; Cvak, L.; Gebhardt, R.; Ulrichová, J.; Šimánek, V. Glycosylation of silybin. J. Chem. Soc. Perkin Trans. 1 1997, 2467–2474. [Google Scholar] [CrossRef]
- Křen, V.; Gažák, R.; Purchartová, K.; Marhol, P.; Biedermann, D.; Sedmera, P. Chemoenzymatic preparative separation of silybin A and B. J. Mol. Catal. B Enzymat. 2009, 61, 247–251. [Google Scholar] [CrossRef]
- Sy-Cordero, A.; Graf, T.N.; Nakanishi, Y.; Wani, M.C.; Agarwal, R.; Kroll, D.J.; Oberlies, N.H. Large-scale isolation of flavonolignans from Silybum marianum extract affords new minor constituents and preliminary structure-activity relationships. Planta Med. 2010, 76, 644–647. [Google Scholar] [CrossRef]
- Di Fabio, G.; Romanucci, V.; di Marino, C.; de Napoli, L.; Zarrelli, A. A rapid and simple chromatographic separation of diastereomers of silibinin and their oxidation to produce 2,3-dehydrosilybin enantiomers in an optically pure form. Planta Med. 2013, 79, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Fuksová, K.; Marhol, P.; Kuzma, M.; Agarwal, R.; Křen, V. Preparative method for isosilybin isolation based on enzymatic kinetic resolution of silymarin mixture. Proc. Biochem. 2013, 48, 184–189. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Properties and synthetic applications of enzymes in organic solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Monti, D.; Gažák, R.; Marhol, P.; Biedermann, D.; Purchartová, K.; Fedrigo, M.; Riva, S.; Křen, V. Enzymatic kinetic resolution of silybin diastereoisomers. J. Nat. Prod. 2010, 73, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Gažák, R.; Marhol, P.; Purchartová, K.; Monti, D.; Biedermann, D.; Riva, S.; Cvak, L.; Křen, V. Large-scale separation of silybin diastereoisomers using lipases. Proc. Biochem. 2010, 45, 1657–1663. [Google Scholar] [CrossRef]
- Vavříková, E.; Gavezzotti, P.; Purchartová, K.; Fuksová, K.; Biedermann, D.; Kuzma, M.; Riva, S.; Křen, V. Regioselective alcoholysis of silychristin acetates catalyzed by lipases. Int. J. Mol. Sci. 2015, 16, 11983–11995. [Google Scholar] [CrossRef] [PubMed]
- Křenek, K.; Marhol, P.; Peikerová, Ž.; Křen, V.; Biedermann, D. Preparatory separation of the silymarin flavonolignans by Sephadex LH-20. Food. Res. Internat. 2014, 65, 115–120. [Google Scholar] [CrossRef]
- Csupor, D.; Csorba, A.; Hohmann, J. Recent advances in the analysis of flavonolignans of Silybum marianum. J. Pharm. Biomed. Anal. 2016, 130, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Tittel, G.; Wagner, H. High-performance liquid chromatography of silymarin. II. Quantitative determination of silymarin from Silybum marianum byhigh-performance liquid chromatography. J. Chromatogr. 1978, 153, 227–232. [Google Scholar] [CrossRef]
- Hammouda, F.M.; Ismail, S.I.; Hassan, N.M.; Zaki, A.K.; Kamel, A.; Rimpler, H. Evaluation of the silymarin content in Silybum marianum (L.) Gaertn. cultivated under different agricultural conditions. Phytother. Res. 1993, 7, 90–91. [Google Scholar] [CrossRef]
- Weyhenmeyer, R.; Mascher, H.; Birkmayer, J. Study on dose-linearity of the pharmacokinetics of silibinin diastereomers using a new stereospecific assay. Int. J. Clin. Pharmacol. Ther. Toxicol. 1992, 30, 134–138. [Google Scholar] [PubMed]
- Mascher, H.; Kikuta, C.; Weyhenmeyer, R. Diastereomeric separation of free and conjugated silibinin in plasma by reversed phase HPLC after specific extraction. J. Liq. Chromatogr. 1993, 16, 2777–2789. [Google Scholar] [CrossRef]
- Rickling, B.; Hans, B.; Kramarczyk, R.; Krumbiegel, G.; Weyhenmeyer, R. Two high-performance liquidchromatographic assays for the determination of free and total silibinin diastereomers in plasma using column switching with electrochemical detection and reversed-phase chromatography with ultraviolet detection. J. Chromatogr. B Biomed. Appl. 1995, 670, 267–277. [Google Scholar] [CrossRef]
- Hoh, C.S.L.; Boocock, D.J.; Marczylo, T.H.; Brown, V.A.; Cai, H.; Steward, W.P.; Berry, D.P.; Gescher, A.J. Quantitation of silibinin, a putative cancer chemopreventive agent derived from milk thistle (Silybum marianum), in human plasma by high-performance liquid chromatography and identification of possible metabolites. J. Agric. Food Chem. 2007, 55, 2532–2535. [Google Scholar] [CrossRef] [PubMed]
- Hadad, G.M.; Emara, S.; Abdel-Salam, R.A. Validated optimized method for simultaneous analysis of active silymarin components and dimethyl-4’-dimethoxy-5,6,5’,6’-dimethylenedioxybiphenyl-2,2’-dicarboxylate in a pharmaceutical preparation by use of a monolithic silica. Chromatographia 2009, 70, 217–221. [Google Scholar] [CrossRef]
- Marhol, P.; Gažák, R.; Bednář, P.; Křen, V. Narrow-bore core-shell particles and monolithic columns in the analysis of silybin diastereoisomers. J. Sep. Sci. 2011, 34, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Quaglia, M.G.; Bossu, E.; Donati, E.; Mazzanti, G.; Brandt, A. Determination of silymarine in the extract from the dried Silybum marianum fruits by high performance liquid chromatography and capillary electrophoresis. J. Pharm. Biomed. Anal. 1999, 19, 435–442. [Google Scholar] [CrossRef]
- Fibigr, J.; Šatínský, D.; Solich, P. A new approach to the rapid separation of isomeric compounds in a Silybum marianum extract using UHPLC core-shell column with F5 stationary phase. J. Pharm. Biomed. Anal. 2017, 134, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Wu, H.-F. Analysis of silymarin extracted from a commercialdosage by combining liquid–liquid extraction with negative electrospraytandem mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 2960–2962. [Google Scholar] [CrossRef] [PubMed]
- Kuki, A.; Biri, B.; Nagy, L.; Deak, G.; Kalmar, J.; Mandi, A.; Nagy, M.; Zsuga, M.; Kéki, S. Collision induced dissociation study of the major components of silymarin. Int. J. Mass Spectrom. 2012, 315, 46–54. [Google Scholar] [CrossRef]
- Kuki, A.; Nagy, L.; Deák, G.; Nagy, M.; Zsuga, M.; Kéki, S. Identification of silymarin constituents: An improved HPLC–MS method. Chromatographia 2012, 75, 175–180. [Google Scholar] [CrossRef]
- Solís-Gómez, A.; Sato-Berrú, R.Y.; Mata-Zamora, M.E.; Saniger, J.M.; Guirado-López, R.A. Characterizing the properties of anticancer silibinin and silybin B complexes with UV-Vis, FT-IR, and Raman spectroscopies: A combined experimental and theoretical study. J. Mol. Struct. 2019, 1182, 109–118. [Google Scholar] [CrossRef]
- Valentová, K.; Purchartová, K.; Rydlová, L.; Roubalová, L.; Biedermann, D.; Petrásková, L.; Křenková, A.; Pelantová, H.; Holečková-Moravcová, V.; Tesařová, E.; et al. Tentative sulfated metabolites of flavonolignans and 2,3-dehydroflavonolignans: Preparation and antioxidant evaluation. Int. J. Mol. Sci. 2018, 19, 2349. [Google Scholar] [CrossRef] [PubMed]
- Tvrdý, V.; Catapano, M.C.; Rawlik, T.; Karlíčková, J.; Biedermann, D.; Křen, V.; Mladěnka, P.; Valentová, K. Interaction of isolated silymarin flavonolignans with iron and copper. J. Inorg. Biochem. 2018, 189, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Křen, V.; Minghetti, A.; Sedmera, P.; Havlíček, V.; Přikrylová, V.; Crespi-Perellino, N. Glucosylation of silybin by plant cell cultures of Papaver somniferum var. setigerum. Phytochemistry 1998, 47, 217–220. [Google Scholar] [CrossRef]
- Křen, V.; Ulrichová, J.; Kosina, P.; Stevenson, D.; Sedmera, P.; Přikrylová, V.; Halada, P.; Šimánek, V. Chemoenzymatic preparation of silybine β-glucuronides and their biological evaluation. Drug Metab. Disp. 2000, 28, 1513–1517. [Google Scholar]
- Charrier, C.; Azerad, R.; Marhol, P.; Purchartová, K.; Kuzma, M.; Křen, V. Preparation of silybin phase II metabolites: Streptomyces catalyzed glucuronidation. J. Mol. Catal. B Enzym. 2014, 102, 167–173. [Google Scholar] [CrossRef]
- Gufford, B.T.; Graf, T.N.; Paguigan, N.D.; Oberlies, N.H.; Paine, M.F. Chemoenzymatic synthesis, characterization, and scale-up of milk thistle flavonolignan glucuronides. Drug Metab. Disp. 2015, 43, 1734–1743. [Google Scholar] [CrossRef]
- Jančová, P.; Anzenbacherová, E.; Šiller, M.; Křen, V.; Anzenbacher, P.; Šimánek, V. Stereoselective metabolism of silybin by UDP glucuronosyltransferases in vitro. Xenobiotica 2011, 41, 743–751. [Google Scholar] [CrossRef]
- Purchartová, K.; Engels, L.; Marhol, P.; Slámová, K.; Šulc, M.; Kuzma, M.; Elling, L.; Křen, V. Enzymatic preparation of silybin phase II metabolites: Sulfation using aryl sulfotransferase from rat liver. Appl. Microbiol. Biotechnol. 2013, 97, 10391–10398. [Google Scholar] [CrossRef] [PubMed]
- Marhol, P.; Bednář, P.; Kolářová, P.; Večeřa, R.; Ulrichová, J.; Tesařová, E.; Vavříková, E.; Kuzma, M.; Křen, V. Pharmacokinetics of pure silybin diastereoisomers and identification of their metabolites in rat plasma. J. Funct. Foods 2015, 14, 570–580. [Google Scholar] [CrossRef]
- Marhol, P.; Hartog, A.F.; van der Horst, M.A.; Wever, R.; Purchartová, K.; Fuksová, K.; Kuzma, M.; Cvačka, J.; Křen, V. Preparation of silybin and isosilybin sulfates by sulfotransferase from Desulfitobacterium hafniense. J. Mol. Catal. B Enzymat. 2013, 89, 24–27. [Google Scholar] [CrossRef]
- Agarwal, C.; Wadhwa, R.; Deep, G.; Biedermann, D.; Gažák, R.; Křen, V.; Agarwal, R. Agarwal: Anti-cancer efficacy of silybin derivatives—A structure-activity relationship. PLoS ONE 2013, 8, e60074. [Google Scholar] [CrossRef] [PubMed]
- Purchartová, K.; Marhol, P.; Gažák, R.; Monti, D.; Riva, S.; Kuzma, M.; Křen, V. Regioselective alcoholysis of silybin A and B acetates with lipases. J. Mol. Catal. B Enzymat. 2011, 71, 119–123. [Google Scholar] [CrossRef]
- Chambers, S.; Valentová, K.; Křen, V. Non-taxifolin derived flavonolignans: Phytochemistry and biology. Curr. Pharm. Design 2015, 21, 5489–5500. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Schramm, S.; Heilmann, J.; Biedermann, D.; Křen, V.; Decker, M. Unconventional application of the Mitsunobu reaction: Selective flavonolignan dehydration yielding hydnocarpins. Beilstein J. Org. Chem. 2016, 12, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Vimberg, V.; Kuzma, M.; Stodůlková, E.; Novák, P.; Bednárová, L.; Šulc, M.; Gažák, R. Hydnocarpin-type flavonolignans: Semisynthesis and inhibitory effects on Staphylococcus aureus biofilm formation. J. Nat. Prod. 2015, 78, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Zarrelli, A.; Romanucci, V.; de Napoli, L.; Previtera, L.; di Fabio, G. Synthesis of new silybin derivatives and evaluation of their antioxidant properties. Helv. Chim. Acta 2015, 98, 399–409. [Google Scholar] [CrossRef]
- Gavezzotti, P.; Vavříková, E.; Valentová, K.; Fronza, G.; Kudanga, T.; Kuzma, M.; Riva, S.; Biedermann, D.; Křen, V. Enzymatic oxidative dimerization of silymarin flavonolignans. J. Mol. Catal. B Enzymat. 2014, 109, 24–30. [Google Scholar] [CrossRef]
- Gažák, R.; Sedmera, P.; Vrbacký, M.; Vostálová, J.; Drahota, Z.; Marhol, P.; Walterová, D.; Křen, V. Molecular mechanisms of silybin and 2,3-dehydrosilybin antiradical activity—Role of individual hydroxyl groups. Free Rad. Biol. Med. 2009, 46, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Marsal, P.; Svobodová, A.; Vostálová, J.; Hrbáč, J.; Gažák, R.; Křen, V.; Lazzaroni, R.; Duroux, J.-L.; Sedmera, P.; et al. Mechanism of the antioxidant action of silybin and 2,3-dehydrosilybin flavonolignans: A joint experimental and theoretical study. J. Phys. Chem. A. 2008, 112, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Džubák, P.; Hajdúch, M.; Gažák, R.; Svobodová, A.; Psotová, J.; Walterová, D.; Sedmera, P.; Křen, V. New derivatives of silybin and 2,3-dehydrosilybin and their cytotoxic and P-glycoprotein modulatory activity. Bioorg. Med. Chem. 2006, 14, 3793–3810. [Google Scholar] [CrossRef] [PubMed]
- Althagafy, H.S.; Graf, T.N.; Sy-Cordero, A.A.; Gufford, B.T.; Paine, M.F.; Wagoner, J.; Polyak, S.J.; Croatt, M.P.; Oberlies, N.H. Semisynthesis, cytotoxicity, antiviral activity, and drug interaction liability of 7-O-methylated analogues of flavonolignans from milk thistle. Bioorg. Med. Chem. 2013, 21, 3919–3926. [Google Scholar] [CrossRef] [PubMed]
- Sy-Cordero, A.; Graf, T.N.; Runyon, S.P.; Wani, M.C.; Kroll, D.J.; Agarwal, R.; Brantley, S.J.; Paine, M.F.; Polyak, S.J.; Oberlies, N.H. Enhanced bioactivity of silybin B methylation products. Bioorg. Med. Chem. 2013, 21, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Hurtová, M.; Biedermann, D.; Kuzma, M.; Křen, V. Mild and selective method of bromination of flavonoids. J. Nat. Prod. 2020, 83, 3324–3331. [Google Scholar] [CrossRef]
- Gažák, R.; Valentová, K.; Fuksová, K.; Marhol, P.; Kuzma, M.; Medina, M.A.; Oborná, I.; Ulrichová, J.; Křen, V. Synthesis and antiangiogenic activity of new silybin galloyl esters. J. Med. Chem. 2011, 54, 7397–7407. [Google Scholar] [CrossRef] [PubMed]
- Schramm, S.; Huang, G.; Gunesch, S.; Lang, F.; Roa, J.; Högger, P.; Sabaté, R.; Maher, P.; Decker, M. Regioselective synthesis of 7-O-esters of the flavonolignan silibinin and SARs lead to compounds with overadditive neuroprotective effects. Eur. J. Med. Chem. 2018, 146, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Aβ amyloid growth and toxicity by silybins: The crucial role of stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- García-Vinñuales, S.; Ahmed, R.; Sciacca, M.F.M.; Lanza, V.; Giuffrida, M.L.; Zimbone, S.; Romanucci, V.; Zarrelli, A.; Bongiorno, C.; Spinella, N.; et al. Trehalose conjugates of silybin as prodrugs for targeting toxic Aβ aggregates. ACS Chem. Neurosci. 2020, 11, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Křen, V.; Marhol, P.; Purchartová, K.; Gabrielová, E.; Modrianský, M. Biotransformation of silybin and its congeners. Curr. Drug. Metab. 2013, 14, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, E.; Purchartová, K.; Stamatis, H.; Kolisis, F.; Křen, V. Bioavailability of silymarin flavonolignans: Drug formulations and biotransformation. Phytochem. Rev. 2014, 13, 1–18. [Google Scholar] [CrossRef]
- Tvrdý, V.; Pourová, J.; Jirkovský, E.; Křen, V.; Valentová, K.; Mladěnka, P. Systematic review of pharmacokinetics and potential pharmacokinetic interactions of flavonolignans from silymarin. Med. Res. Rev. 2021, 41, 2195–2246. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Lou, H.X.; Ren, D.M.; Sun, L.R.; Ma, B.; Ji, M. Stereoselective metabolism of silybin diastereoisomers in the glucuronidation process. J. Pharm. Biomed. Anal. 2004, 34, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Dumas, T.E.; Schrieber, S.J.; Hawke, R.L.; Fried, M.W.; Smith, P.C. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab. Dispos. 2008, 36, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-J.; Brinda, B.J.; Chavin, K.D.; Bernstein, H.J.; Patrick, K.S.; Markowitz, J.S. An assessment of pharmacokinetics and antioxidant activity of free silymarin flavonolignans in healthy volunteers: A dose escalation study. Drug Metab. Dispos. 2013, 41, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Gunaratna, C.; Zhang, T. Application of liquid chromatography-electrospray ionization-ion trap mass spectrometry to investigate the metabolism of silibinin in human liver microsomes. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 794, 303–310. [Google Scholar] [CrossRef]
- Jančová, P.; Anzenbacherová, E.; Papoušková, B.; Lemr, K.; Lužná, P.; Veinlichová, A.; Anzenbacher, P.; Šimánek, V. Silybin is metabolized by cytochrome P450 2C8 in vitro. Drug Metab. Dispos. 2007, 35, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, D.-H.; Zhang, Y.-T.; Chen, X.-M.; Li, L.-L.; Cai, S.-Q. Biotransformation on the flavonolignan constituents of Silybi Fructus by an intestinal bacterial strain Eubacterium limosum ZL-II. Fitoterapia 2014, 92, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Vrba, J.; Papoušková, B.; Roubalová, L.; Zatloukalová, M.; Biedermann, D.; Křen, V.; Valentová, K.; Ulrichová, J.; Vacek, J. Metabolism of flavonolignans in human hepatocytes. J. Pharm. Biomed. Anal. 2018, 152, 94–101. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, J.; Wang, X.; Li, H.; Mao, X.; Peng, Y.; Zheng, J. Characterization of glutathione conjugates derived from reactive metabolites of seven silymarin isomers. Xenobiotica 2019, 49, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Vrba, J.; Papoušková, B.; Lněničková, K.; Kosina, P.; Křen, V.; Ulrichová, J. Identification of UDP-glucuronosyltransferases involved in the metabolism of silymarin flavonolignans. J. Pharm. Biomed. Anal. 2020, 178, 112972. [Google Scholar] [CrossRef] [PubMed]
- Vrba, J.; Papoušková, B.; Kosina, P.; Lněničková, K.; Valentová, K.; Ulrichová, J. Identification of human sulfotransferases active towards silymarin flavonolignans and taxifolin. Metabolites 2020, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.R.; Lee, J.K.; Brouwer, K.L.; Wen, Z.; Smith, P.C.; Hawke, R.L. Hepatic metabolism and biliary excretion of silymarin flavonolignans in isolated perfused rat livers: Role of multidrug resistance-associated protein 2 (Abcc2). Drug Metab. Dispos. 2008, 36, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Plíšková, M.; Vondráček, J.; Křen, V.; Gažák, R.; Sedmera, P.; Walterová, D.; Psotová, J.; Šimánek, V.; Machala, M. Effects of silymarin flavonolignans and synthetic silybin derivatives on estrogen and aryl hydrocarbon receptor activation. Toxicology 2005, 215, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Pferschy-Wenzig, E.M.; Atanasov, A.G.; Malainer, C.; Noha, S.M.; Kunert, O.; Schuster, D.; Heiss, E.H.; Oberlies, N.H.; Wagner, H.; Bauer, R.; et al. Identification of isosilybin a from milk thistle seeds as an agonist of peroxisome proliferator-activated receptor gamma. J. Nat. Prod. 2014, 77, 842–847. [Google Scholar] [CrossRef]
- Wang, L.; Rotter, S.; Ladurner, A.; Heiss, E.H.; Oberlies, N.H.; Dirsch, V.M.; Atanasov, A.G. Silymarin constituents enhance ABCA1 expression in THP-1 macrophages. Molecules 2016, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Zhang, L.; Yan, T.; Wu, B.; Xuc, F.; Jia, Y. Silychristin A activates Nrf2-HO-1/SOD2 pathway to reduce apoptosis and improve GLP-1 production through upregulation of estrogen receptor α in GLUTag cells. Eur. J. Pharmacol. 2020, 881, 173236. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Hu, X.; Li, S.; Wang, J.; Li, Z.; Li, D.; Xu, F.; Gao, M.; Hua, H. Hypoglycemic effect of silychristin A from Silybum marianum fruit via protecting pancreatic islet β cells from oxidative damage and inhibiting α-glucosidase activity in vitro and in rats with type 1 diabetes. J. Funct. Foods 2017, 38, 168–179. [Google Scholar] [CrossRef]
- Sheehan, A.; Messer, A.; Papadaki, M.; Choudhry, A.; Křen, V.; Biedermann, D.; Blagg, B.; Kandelwahl, A.; Marston, S. Molecular defects in cardiac myofilament Ca2+- regulation leading to hypertrophic cardiomyopathy can be reversed by small molecules binding to troponin. Front. Physiol. Striated Muscle Physiol. 2018, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Pourová, J.; Applová, L.; Macáková, K.; Vopršalová, M.; Migkos, T.; Bentanachs, R.; Biedermann, D.; Petrásková, L.; Tvrdý, V.; Hrubša, M.; et al. The effect of silymarin flavonolignans and their sulfated conjugates on platelet aggregation and blood vessels ex vivo. Nutrients 2019, 11, 2286. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin B causes androgen receptor degradation in human prostate carcinoma cells via PI3K-Akt-Mdm2-mediated pathway. Oncogene 2008, 27, 3986–3998. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Identifying the differential effects of silymarin constituents on cell growth and cell cycle regulatory molecules in human prostate cancer cells. Int. J. Cancer 2008, 123, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Gangar, S.C.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin A induces apoptosis in human prostate cancer cells via targeting Akt, NF-kappaB, and androgen receptor signaling. Mol. Carcinog. 2010, 49, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Gangar, S.C.; Rajamanickam, S.; Raina, K.; Gu, M.; Agarwal, C.; Oberlies, N.H.; Agarwal, R. Angiopreventive efficacy of pure flavonolignans from milk thistle extract against prostate cancer: Targeting VEGF-VEGFR signaling. PLoS ONE 2012, 7, e34630. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.-S.; Shibano, M.; Nakagawa-Goto, K.; Tokuda, H.; Itokawa, H.; Morris-Natschke, S.L.; Lee, K.-H. Cancer preventive agents. 7. Antitumor-promoting effects of seven active flavonolignans from milk thistle (Silybum marianum) on Epstein-Barr virus activation. Pharm. Biol. 2007, 45, 735–738. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Luana, Q.; Liub, Y.; Lee, D.Y.-W.; Wang, Z. A comparison of the diastereoisomers, silybin A and silybin B, on the induction of apoptosis in K562 cells. Nat. Prod. Commun. 2011, 6, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Polyak, J.; Morishima, C.; Lohmann, V.; Pal, S.; Lee, D.Y.; Liu, Y.; Graf, T.N.; Oberlies, N.H. Identification of hepatoprotective flavonolignans from silymarin. Proc. Natl. Acad. Sci. USA 2010, 107, 5995–5999. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Belkacem, N.; Ahnou, L.; Barbotte, C.; Wychowski, C.; Pallier, R.; Brillet, R.-T.; Pohl, J.-M. Pawlotsky, Silibinin and related compounds are direct inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Gastroenterology 2010, 138, 1112–1122. [Google Scholar] [CrossRef]
- Olías-Molero, A.I.; Jiménez-Antón, M.D.; Biedermann, D.; Corral, M.J.; Alunda, J.M. In-vitro activity of silybin and related flavonolignans against Leishmania infantum and L. donovani. Molecules 2018, 23, 1560. [Google Scholar] [CrossRef]
- Chen, X.; Deng, X.; Han, X.; Liang, Y.; Meng, Z.; Liu, R.; Su, W.; Zhu, H.; Fu, T. Inhibition of lysozyme amyloid fibrillation by silybin diastereoisomers: The effects of stereochemistry. ACS Omega 2021, 6, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Filippopoulou, K.; Papaevgeniou, N.; Lefaki, M.; Paraskevopoulou, A.; Biedermann, D.; Křen, V.; Chondrogianni, N. 2,3-Dehydrosilybin A and B as a pro-longevity and anti-aggregation compound. Free Rad. Biol. Med. 2017, 103, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Differential effects of silibinin a on mitochondrial function in neuronal PC12 and HepG2 liver cells. Oxid. Med. Cell Longev. 2019, 1652609. [Google Scholar] [CrossRef] [PubMed]
- Diukendjieva, A.; Zaharieva, M.M.; Mori, M.; Alov, P.; Tsakovska, I.; Pencheva, T.; Najdenski, H.; Křen, V.; Felici, C.; Bufalieri, F.; et al. Dual Smo/BRAF inhibition by flavonolignans from Silybum marianum. Antioxidants 2020, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Köck, K.; Xie, Y.; Hawke, R.L.; Oberlies, N.H.; Brouwer, K.L. Interaction of silymarin flavonolignans with organic anion-transporting polypeptides. Drug metabolism and disposition: The biological fate of chemicals. Drug Metab. Disp. 2013, 41, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Kubala, M.; Čechová, P.; Geletičová, J.; Biler, M.; Štenclová, T.; Trouillas, P.; Biedermann, D. Flavonolignans as a novel class of sodium pump inhibitors. Front. Physiol. 2016, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Johannes, J.; Jayarama-Naidu, R.; Meyer, F.; Wirth, E.K.; Schweizer, U.; Schomburg, L.; Köhrle, J.; Renko, K. Silychristin, a flavonolignan derived from the milk thistle is a potent inhibitor of the thyroid hormone transporter MCT8. Endocrinology 2016, 157, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Dobiasová, S.; Řehořová, K.; Kučerová, D.; Biedermann, D.; Káňová, K.; Petrásková, L.; Koucká, K.; Václavíková, R.; Valentová, K.; Ruml, T.; et al. Multidrug resistance modulation activity of silybin derivatives and their anti-inflammatory potential. Antioxidants 2020, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Viktorová, J.; Dobiášová, S.; Řehořová, K.; Biedermann, D.; Káňová, K.; Šeborová, K.; Václavíková, R.; Valentová, K.; Ruml, T.; Křen, V.; et al. Antioxidant, anti-inflammatory and multidrug resistance modulation activity of silychristin derivatives. Antioxidants 2019, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, J.Y.; Jenis, J.; Li, Z.P.; Ban, Y.J.; Baiseitova, A.; Park, K.H. Tyrosinase inhibitory study of flavonolignans from the seeds of Silybum marianum (Milk thistle). Bioorg. Med. Chem. 2019, 27, 2499–2507. [Google Scholar] [CrossRef]
- Brantley, S.J.; Oberlies, N.H.; Kroll, D.J.; Paine, M.F. Two flavonolignans from milk thistle (Silybum marianum) inhibit CYP2C9-mediated warfarin metabolism at clinically achievable concentrations. J. Pharmacol. Exp. Ther. 2010, 332, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Brantley, S.J.; Graf, T.N.; Oberlies, N.H.; Paine, M.F. A systematic approach to evaluate herb-drug interaction mechanisms: Investigation of milk thistle extracts and eight isolated constituents as CYP3A inhibitors. Drug Metab. Dispos. 2013, 41, 1662–1670. [Google Scholar] [CrossRef] [PubMed]
- Gufford, B.T.; Chen, G.; Lazarus, P.; Graf, T.N.; Oberlies, N.H.; Paine, M.F. Identification of diet-derived constituents as potent inhibitors of intestinal glucuronidation. Drug Metab. Disp. 2014, 42, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Šuk, J.; Jašprová, J.; Biedermann, D.; Petrásková, L.; Valentová, K.; Křen, V.; Muchová, L.; Vítek, L. Isolated silymarin flavonoids increase systemic and hepatic bilirubin concentrations and lower lipoperoxidation in mice. Oxid. Med. Cell. Longev. 2019, 2019, 6026902. [Google Scholar] [CrossRef] [PubMed]
- Seidlova-Wuttke, D.; Becker, T.; Christoffel, V.; Jarry, H.; Wuttke, W. Silymarin is a selective estrogen receptor β (ERβ) agonist and has estrogenic effects in the metaphysis of the femur but no or antiestrogenic effects in uterus of ovariectomized (ovx) rats. J. Steroid Biochem. Mol. Biol. 2003, 86, 179–188. [Google Scholar] [CrossRef]
- Škottová, N.; Krečman, V. Silymarin as a potential hypocholesterolaemic drug. Physiol. Res. 1998, 47, 1–7. [Google Scholar] [PubMed]
- MacDonald-Ramos, K.; Michán, L.; Martínez-Ibarraa, A.; Cerbón, M. Silymarin is an ally against insulin resistance: A review. Ann. Hepatol. 2021, 23, 100255. [Google Scholar] [CrossRef]
- Agarwal, R.; Agarwal, C.; Ichikawa, H.; Singh, R.P.; Aggarwal, B.B. Anticancer potential of silymarin: From bench to bed side. Anticancer Res. 2006, 26, 4457–4498. [Google Scholar] [PubMed]
- Deep, G.; Raina, K.; Singh, R.P.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilibinin inhibits advanced human prostate cancer growth in athymic nude mice: Comparison with silymarin and silibinin. Int. J. Cancer 2008, 123, 2750–2758. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Ohta, T.; Igura, K.; Hara, Y.; Kaji, K. Tea catechins inhibit angiogenesis in vitro, measured by human endothelial cell growth, migration and tube formation, through inhibition of VEGF receptor binding. Cancer Lett. 2002, 180, 139–144. [Google Scholar] [CrossRef]
- Vue, B.; Zhang, S.; Zhang, X.; Parisis, K.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. Silibinin derivatives as anti-prostate cancer agents: Synthesis and cell-based evaluations. Eur. J. Med. Chem. 2016, 109, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Vue, B.; Chen, Q.H. The potential of flavonolignans in prostate cancer management. Curr. Med. Chem. 2016, 23, 3925–3950. [Google Scholar] [CrossRef]
- Liu, C.-H.; Jassey, A.; Hsu, H.-Y.; Lin, L.-T. Antiviral activities of silymarin and derivatives. Molecules 2019, 24, 1552. [Google Scholar] [CrossRef] [PubMed]
- Polyak, J.; Ferenci, P.; Pawlotsky, J.-M. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology 2013, 57, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Martin-Castillo, B.; Buxó, M.; Brunet, J.; Encinar, J.A.; Menendez, J.A. Silibinin and SARS-CoV-2: Dual targeting of host cytokine storm and virus replication machinery for clinical management of COVID-19 patients. J. Clin. Med. 2020, 9, 1770. [Google Scholar] [CrossRef]
- Srivastava, R.; Tripathi, S.; Unni, S.; Hussain, A.; Haque, S.; Dasgupta, N.; Singh, V.; Mishra, B.N. Silybin B and cianidanol inhibit Mpro and spike protein of SARS-CoV-2: Evidence from in silico molecular docking studies. Curr. Pharm. Design 2021, 27, in press. [Google Scholar] [CrossRef]
- Faixová, D.; Hrčková, G.; Kubašková, T.M.; Mudroňová, D. Antiparasitic effects of selected isoflavones on flatworms. Helminthologia 2021, 58, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.A.; Agarwal, R.; Glodé, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Investig. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L.; Bellarosa, C.; Tiribelli, C. Induction of mild hyperbilirubinemia: Hype or real therapeutic opportunity? Clin. Pharm. Ther. 2019, 106, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.; Viktorová, J.; Řehořová, K.; Biedermann, D.; Turková, L.; Macek, T.; Křen, V.; Valentová, K. Defying multidrug resistance! Modulation of related transporters by flavonoids and flavonolignans. J. Agric. Food Chem. 2020, 68, 1763–1779. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Effect | MODEL USED | Active Substance | Reference |
|---|---|---|---|
| Estrogen receptor activation | ER- and AhR-mediated luciferase activity in cell lines T47D.Luc and H4IIE.Gud.Luc | Silybin B | [117] |
| PPARγ activation | PPARγ luciferase reporter transactivation nuclear receptor activation in HEK-293 cells | Isosilybin A | [118] |
| Cholesterol efflux transporter ABCA1 expression induction | ABCA1 protein expression in THP-1 macrophages | Silybin B a Isosilybin A Silychristin Isosilychristin | [119] |
| (Nrf2-HO-1/SOD2 pathway activation; Decrease in apoptosis and upregulation of GLP-1 | Intestinal murine enteroendocrine L-cell line GLUTag apoptotic activity and production of GLP-1 | Silychristin A | [120] |
| α-Glucosidase inhibition; improvement of oral sucrose tolerance | α-Glucosidase from Saccharomyces cerevisiae | Silychristin A | [121] |
| Lowering fasting blood glucose and stimulation of insulin secretion by improving the viability of pancreatic β cells | Streptozotocin-induced diabetes mellitus type 1 in rats | Silychristin A | [121] |
| Inhibition of respective phosphatases | Protein tyrosine phosphatase 1B (human recombinant); T-cell protein tyrosine phosphatase (human recombinant); Vaccinia H1-related phosphatase (human recombinant) | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin A Silychristin B Isosilychristin A Dehydrosilychristin A Silydianin | [39] |
| Ability to re-couple troponin in thin myofilaments; Restoration of inherited cardiomyopathy | Difference between motility in phosphorylated and unphosphorylated forms of thin filaments reconstituted from rabbit skeletal or mouse cardiac muscle α-actin with TPM1 E180G HCM mutation. | Silybin B a Silybin A 2,3-Dehydrosilybin B 2,3-Dehydrosilybin A Silychristin Silydianin | [122] |
| Vasodilatory activity | Rat aortic ring contraction/dilatation | Silybin A a Silybin B 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B 2,3-Dehydrosilychristin Respective sulfated metabolites | [123] |
| Antiproliferative activities against LNCaP, DU145, and PC3 human prostate carcinoma cell lines; Suppresion of DNA topoisomerase IIa gene promoter in DU145 cells | LNCaP (CRL-1740, androgen-dependent line from lymph node metastasis of PA), DU145 (HTB-81, androgen-independent line derived from central nervous system metastasis of PA), and PC-3 (CRL-1435, androgen-independent line derived from bone metastasis of PA); topoisomerase IIA promoter-reporter transfection and reporter activity assay in DU145 cells. | Silybin A Silybin B Isosilybin A Isosilybin B a | [34] |
| Growth inhibition and cell death and significant G1 arrest and apoptosis; Decrease in the levels of both cyclins (D1, D3, E, and A) and Cdk2, Cdk4, and cell division cycle 25A, increase in p21, p27, and p53 concentrations | Human prostate carcinoma cell lines LNCaP and 22Rv1. | Isosilybin A Isosilybin B a | [35] |
| Decrease in AR and PSA; Increase in phosphorylation of Akt (Ser-473 and Thr-308) and Mdm2 (Ser-166) | LNCaP cells with single AR mutation (T877A), LAPC4 cells with wild-type AR, and androgen-independent 22Rv1 cells with mutated AR (H874Y) | Isosilybin B | [124] |
| Inhibition of colony formation of cell lines tested; Induction of cell cycle arrest (in PC3 line) | HPC PC3 and LNCaP cells | Silybin A Silybin B Isosilybin A Isosilybin B a | [125] |
| Activation of apoptotic pathways in PA cells via targeting the Akt-NF-kB-AR pathway | HPC 22Rv1, LNCaP, and LAPC4 cells | Isosilybin A | [126] |
| Competitive anti-angiogenetic effects;Down-regulation of prostate tumor angiogenesis biomarkers—CD31 and nestin; Target VEGF-induced signaling cascade; Down-regulation of the cyclin A, D1, D3, and E levels; Down-regulation of the level of Cdk4 and Cdk6, but not of Cdk2 | Athymic (nu/nu) nude mice with DU145 cell line xenograft | Silybin A Silybin B Isosilybin A Isosilybin B | [127] |
| Antiproliferative/cytotoxic activity | HPC DU-145, PC3, and LNCaP cells | Silybin A Silybin B Isosilybin A Isosilybin B Isosilybin C Isosilybin D | [53] |
| Cell line growth inhibition in 72 h assay | HPC DU-145, PC-3, and LNCaP cells and a HH cell line Huh7.5.1 | Silybin B Respective mono-, di-, tri-, and tetra-O-methylated derivatives | [97] |
| Cell line growth inhibition | HH Huh7.5.1 cell line | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin Silydianin All respective 7-O-methyl derivatives | [96] |
| Inhibition of Epstein-Barr virus early antigen activation | 12-O-Tetradecanoylphorbol-13-acetate-induced Epstein-Barr virus early antigen activation in Raji cells | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin A Silychristin B Silydianin | [128] |
| Inhibition of cell growth and colony formation of tested cell lines—trypan blue dye exclusion assay | Human bladder cancer HTB9, colon cancer HCT116, and HPC PC3 cells | Silybin A Silybin B 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B 7-O-Methylsilybin A 7-O-Methylsilybin B 7-O-Galloylsilybin A 7-O-Galloylsilybin B | [86] |
| Cell growth inhibition and apoptosis induction | Chronic myeloid leukemia K562 cells | Silybin A Silybin B | [129] |
| HUVEC viability inhibition; Inhibition of HUVEC proliferation; Inhibition of endothelial cell differentiation (tube formation) and migration | HUVEC | Silybin A Silybin B 7-O-Galloylsilybin A 7-O-Galloylsilybin B | [99] |
| Inhibition of HCVcc infection, NS5B polymerase activity, HCVcc induced oxidative stress, TNF-α-induced NF-κBtranscription, and TCR-mediated induction of T-cell proliferation | HCV infected Huh7.5.1 HH cells by virus strain JFH-1 | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin Isosilychristin Silydianin Taxifolin | [130] |
| HCV RNA-dependent RNA polymerase inhibition and NS3/4A protease inhibition; Inhibition of JFH1 replication in Huh7 cells | Recombinant HCV RdRp (NS5BΔ21) RNA-dependent RNA polymerase, HCV NS3/4A protease, and HCV JFH1 infected Huh7 cells | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin Silydianin Plus all the above flavonolignans in the water-soluble form of bishemisuccinate di-Na salts | [131] |
| Inhibition of multiplication of promastigotes in vitro and ex vivo on intracellular amastigotes of Leishmania infantum (L. chagasi) and L. donovani | Intracellular amastigotes of L. infantum and L. donovani, causative agents of human and canine visceral leishmaniasis. L. infantum Li UCM9 (M/CAN/ES/2001/UCM9) and Li BCN150 (M/CAN/ES/96/BCN150 zymodeme MON-1) and L. donovani (MHOM/SD/43/124). | Silybin A Silybin B Isosilybin A Silychristin A Silydianin 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B 2,3-Dehydrosilychristin A 2,3-Dehydroisosilybin A 2,3-Dehydrosilydianin | [132] |
| Lifespan extension of C. elegans; Inhibition of FGT-1 in C. elegans; antiaggregation activity ofAβ in C. elegans GMC101 strain overproducing Aβ and protection against toxicity of Aβ in human neuroblastoma SH-SY5Y cells | Caenorhabditis elegans N2, DR26, CL2179, CL2331, and GMC101 strains; human dopaminergic neuroblastoma cell line SH-SY5Y | Silybin A Silybin B Isosilybin A Silychristin A Silydianin 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B | [133] |
| Antiaggregation activity of Aβ in C. elegans CL4176 strain overproducing Aβ; inhibition of Aβ40 fiber formation by ThT assay | Aβ40 fiber formation kinetics measured by ThT assay; transgenic Caenorhabditis elegans CL4176 strain expressing human Aβ42 in the body-wall muscle | Silybin A Silybin B 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B | [101] |
| Inhibition of fibrillation of Aβ42 monomers | Human dopaminergic neuroblastoma cell line SH-SY5Y challenged Aβ42; Aβ40 fiber formation kinetics measured by ThT assay | Silybin A Silybin B Silybin A-23-phosphotrehaloside Silybin B phosphotrehaloside | [102] |
| Inhibition of HEWL by fibril-induced cytotoxicity in SH-SY5Y cells | HEWL guanidine-induced fibrillation process assay (ThT); neuroblastoma cell line SH-SY5Y treated with HEWL fibrils | Silybin A Silybin B | [134] |
| Attenuating of nitrosative stress in both PC12 and HepG2 cells; increase in mitochondrial membrane potential and ATP levels | PC12 and human hepatoblastoma HepG2 cell lines | Silybin A | [135] |
| Inhibition of BRAF V600E kinase activity | HEK293T cells transfected with human Myc-DDK-tagged SMO; BRAF V600E mutant kinase assay | Silybin A Silybin B 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B | [136] |
| Inhibition of estradiol-17β-glucuronide and rosuvastatin uptake by the cells | Human embryonic kidney cell lines expressing drug transporters—HEK293-Mock, HEK293-OATP1B3, and HEK293-OATP1B1 | Silybin A Silybin B Isosilibinin A Isosilybin B Silychristin Silydianin | [137] |
| Inhibition of Na+/K+-ATPase | Ouabain-sensitive Na+/K+-ATPase from porcine kidney outer medulla | Silybin AB Silychristin A 2,3-Dehydrosilychristin A Silydianin 2,3-Dehydrosilydianin | [138] |
| Inhibition of 3,3′,5-triiodothyronine (T3) transport by MCT8 | Thyroid hormone transmembrane transporters in MCT8 -overexpressing MDCK1-cells; primary murine astrocytes expressing endogenous Mct8 but not MCT10-overexpressing MDCK1-cells | Silychristin Silydianin Isosilibinin A Isosilybin B Silybin AB | [139] |
| Dose-dependent inhibition of P-gp pump; sensitization of doxorubicin-resistant ovarian carcinoma; downregulating the MDR gene expression; inhibition of acetylcholinesterase | P-gp-Glo assay system; human ovarian carcinoma cell line resistant to doxorubicin (HOC/ADR, A2780/ADR); macrophages (RAW 264.7); acetylcholinesterase activity | Silybin A Silybin B 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B Quercetin | [140] |
| Inhibition of P-glycoprotein and/or its expression; Sensitization of doxorubicin-resistant ovarian carcinoma; anti-inflammatory activity; Inhibition of ABC transporter expression | P-gp-Glo assay system; human ovarian carcinoma cell line resistant to doxorubicin (HOC/ADR, A2780/ADR); macrophages (RAW 264.7); ORAC assay | Silybin A Silychristin A Isosilychristin 2,3-Dehydrosilychristin A Anhydrosilychristin | [141] |
| Tyrosinase inhibition | Mushroom tyrosinase with l-tyrosine or l-DOPA as substrates | Silybin AB Isosilybin A Isosilybin B Silychristin A Silychristin B Silydianin 2,3-Dihydrosilychristin | [142] |
| Inhibition of (S)-warfarin 7-hydroxylation with CytP450 isoenzyme CYP2C9 | Human liver microsomes; recombinant CYP2C9 | Silybin A Silybin B Isosilybin A Isosilybin B | [143] |
| Inhibition of midazolam 1’-hydroxylation with CYP3A | Human liver microsomes; human intestinal microsomes; recombinant human rCYP3A4 with midazolam as a substrate. | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin Isosilychristin Silydianin Taxifolin | [144] |
| Inhibition of intestinal glucuronidation | Human liver microsomes; Human intestinal microsomes; HEK293-cells overexpressing UGT1A, UGT1A8, and UGT1A10 with 4-methylumbelliferone as a substrate | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin Isosilychristin Silydianin | [145] |
| Increase in intracellular bilirubin concentration in HepG2 cells; downregulation of UGT1A1 mRNA expression in mice (liver), the elevation of serum bilirubin concentrations, decrease in lipoperoxidation in liver | Human hepatoblastoma HepG2 cells; C57BL/6 mice intraperitoneal or oral administration of tested compounds | Silybin A Silybin B Isosilybin A Isosilybin B Silychristin A Silydianin 2,3-Dehydrosilybin A 2,3-Dehydrosilybin B 2,3-Dehydrosilychristin 2,3-Dehydrosilydianin | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Křen, V. Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners. Int. J. Mol. Sci. 2021, 22, 7885. https://doi.org/10.3390/ijms22157885
Křen V. Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners. International Journal of Molecular Sciences. 2021; 22(15):7885. https://doi.org/10.3390/ijms22157885
Chicago/Turabian StyleKřen, Vladimír. 2021. "Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners" International Journal of Molecular Sciences 22, no. 15: 7885. https://doi.org/10.3390/ijms22157885
APA StyleKřen, V. (2021). Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners. International Journal of Molecular Sciences, 22(15), 7885. https://doi.org/10.3390/ijms22157885