
Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1α in a Murine Model of NAFLD

 and
and 
Abstract
:1. Introduction
2. Results
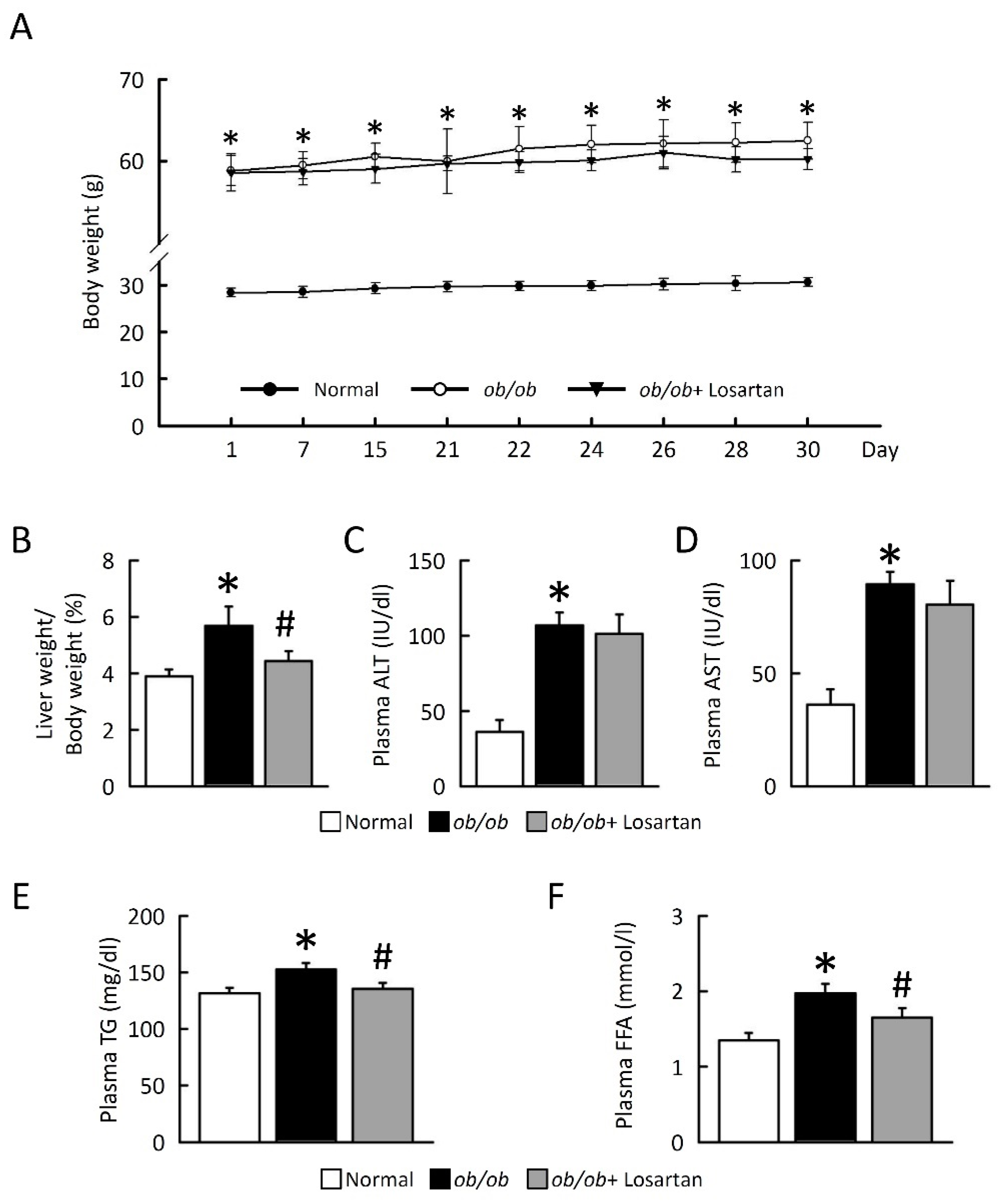
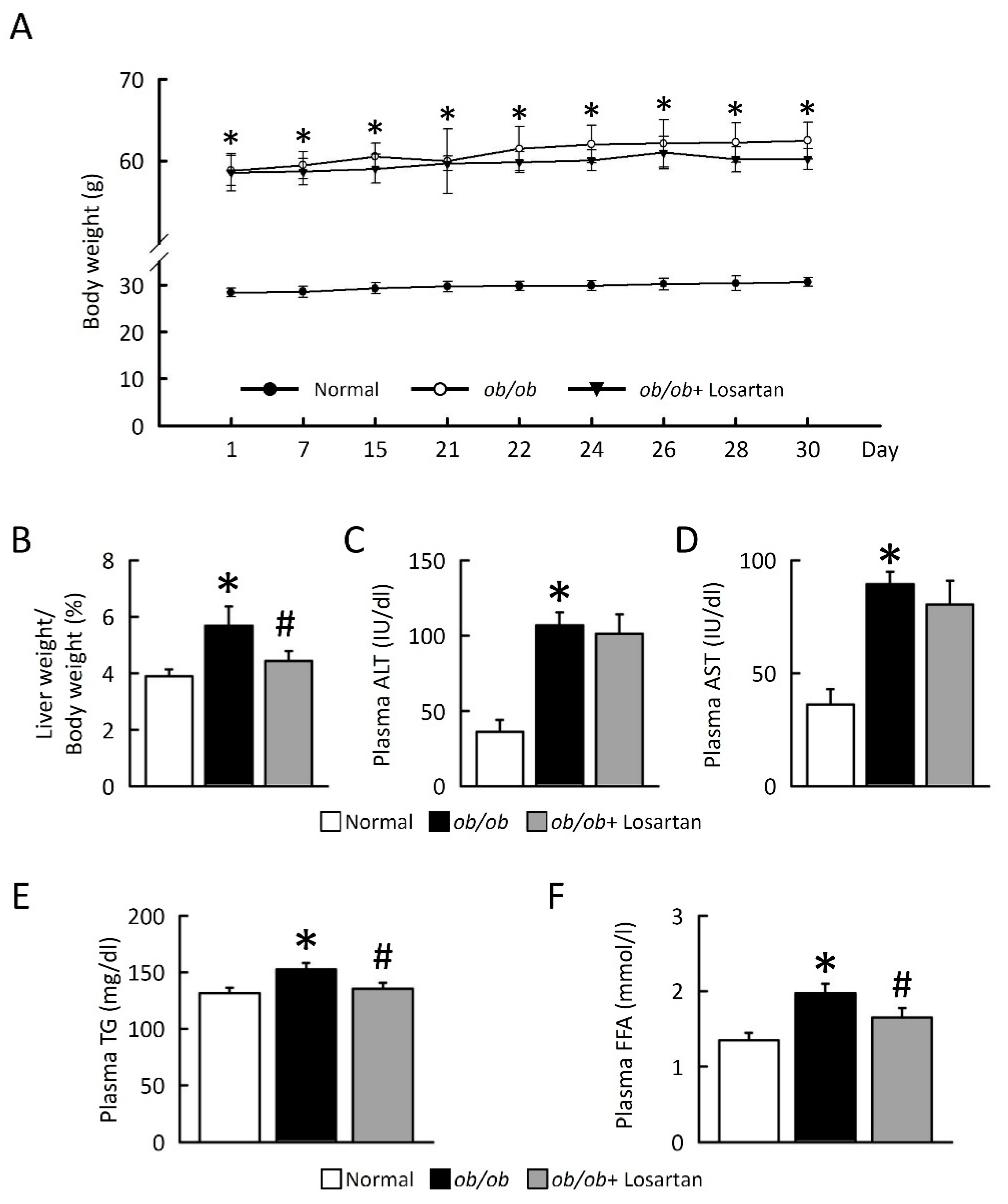
2.1. Losartan Prevents NAFLD Development
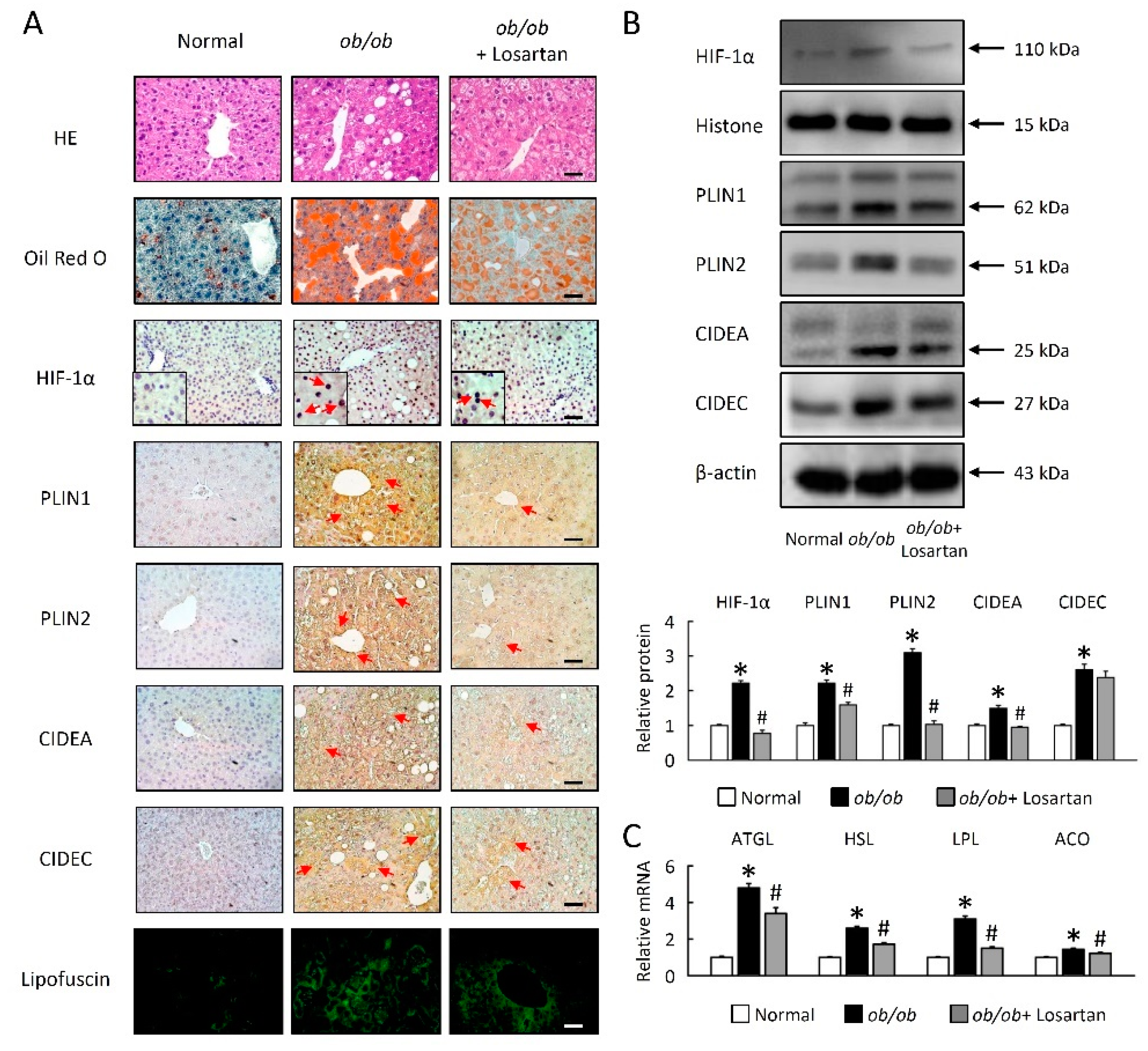
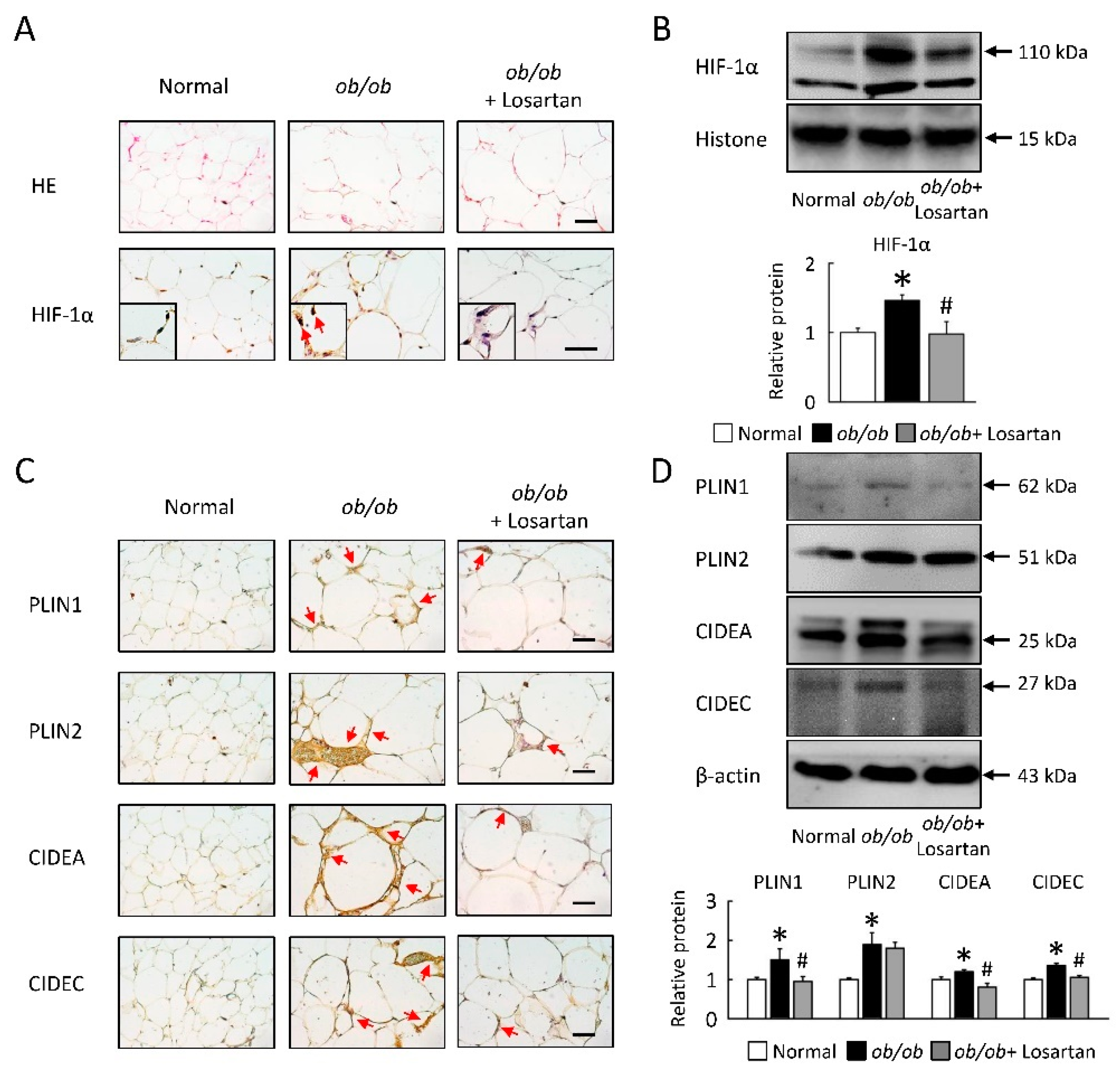
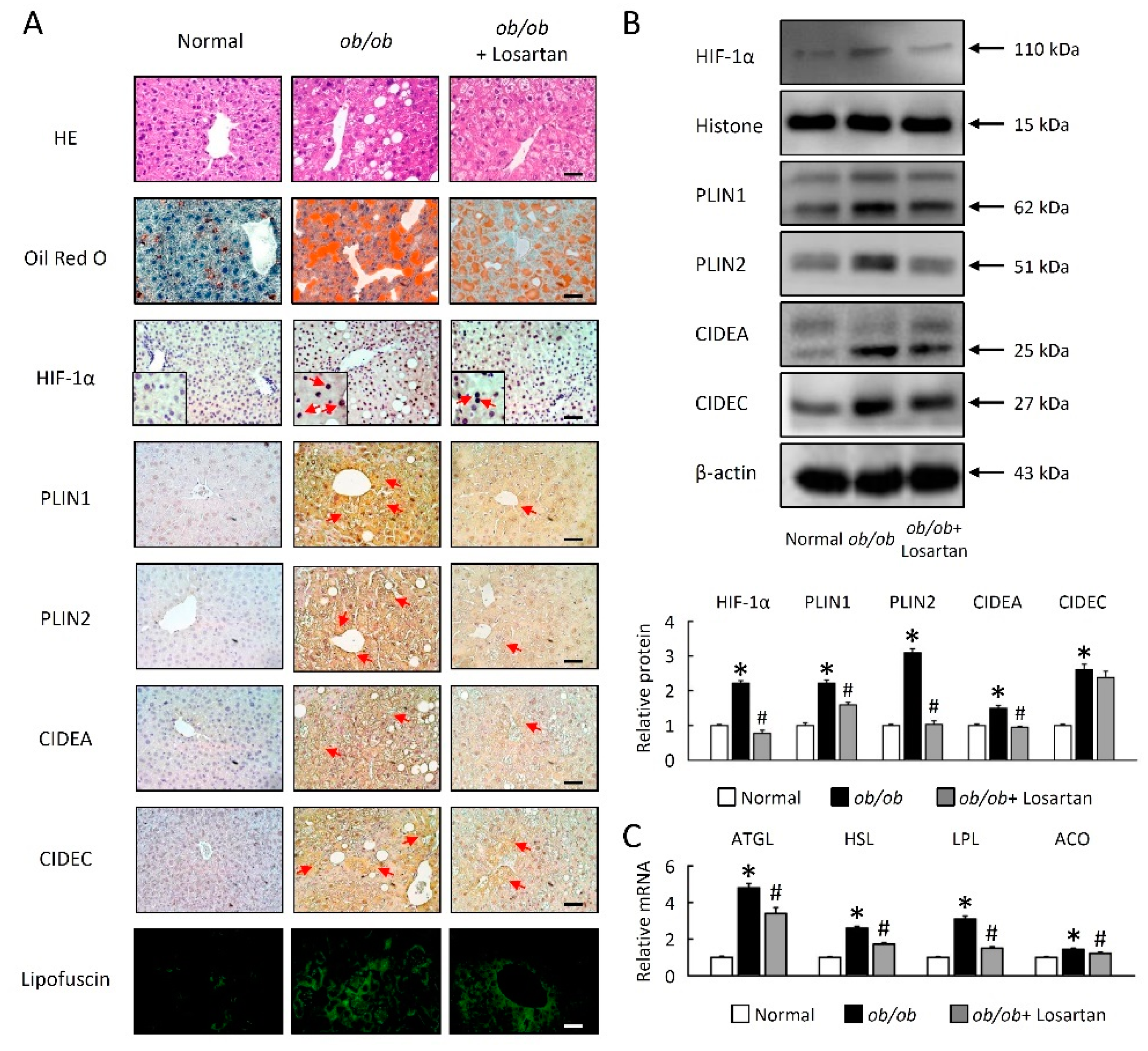
2.2. Losartan Attenuates Liver Steatosis and Hepatic HIF-1α Activation
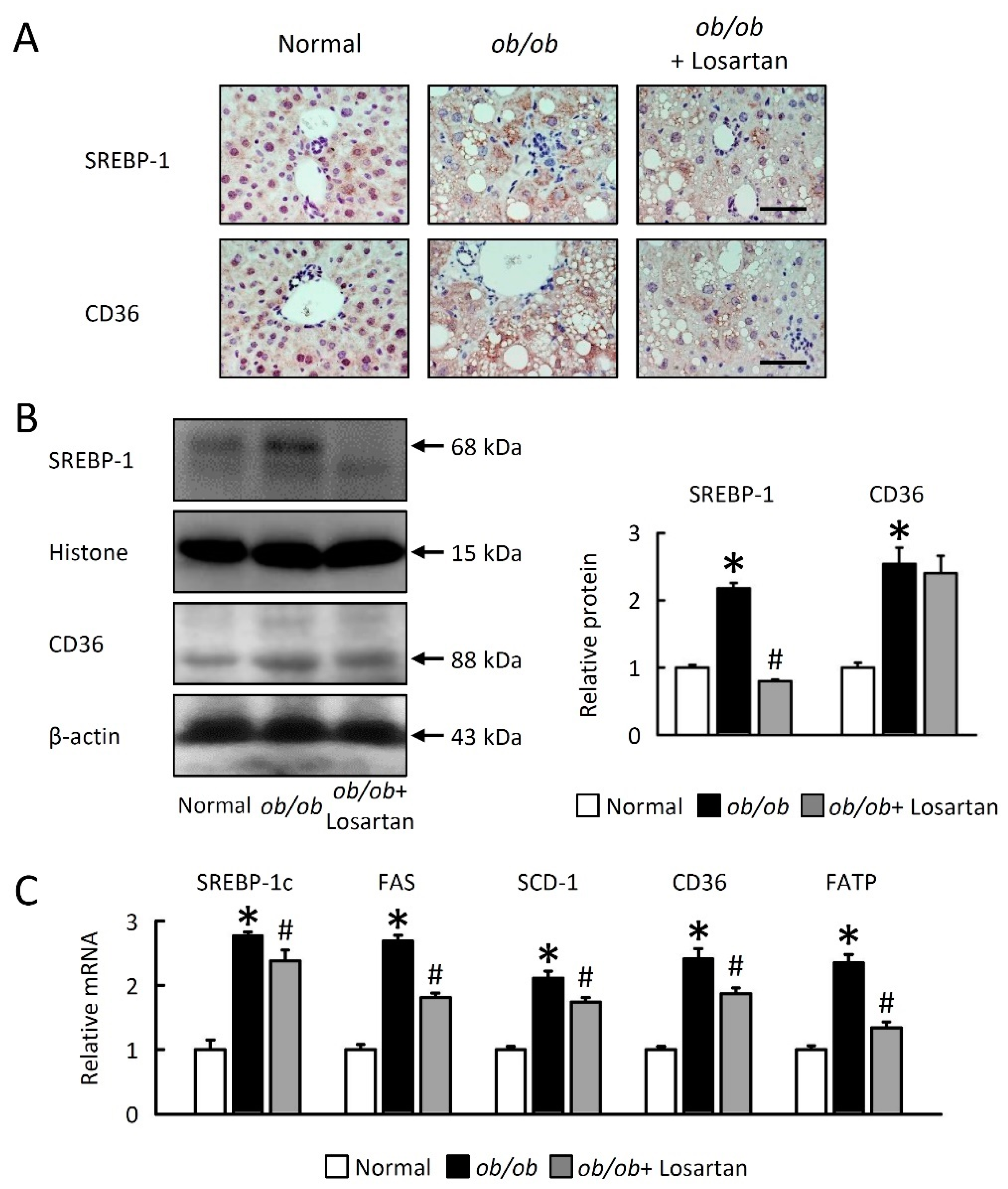
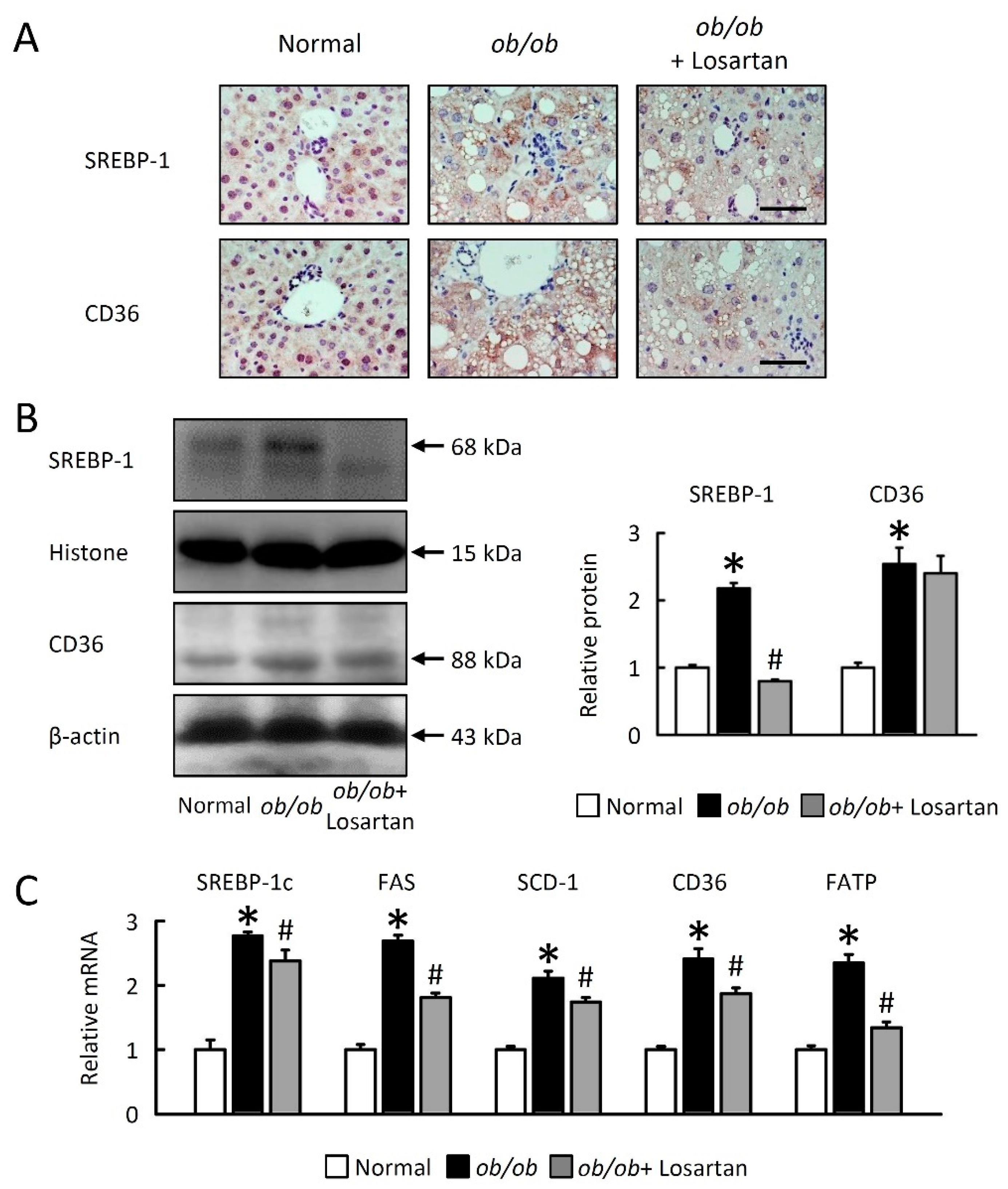
2.3. Losartan Reduces Hepatic Lipogenesis
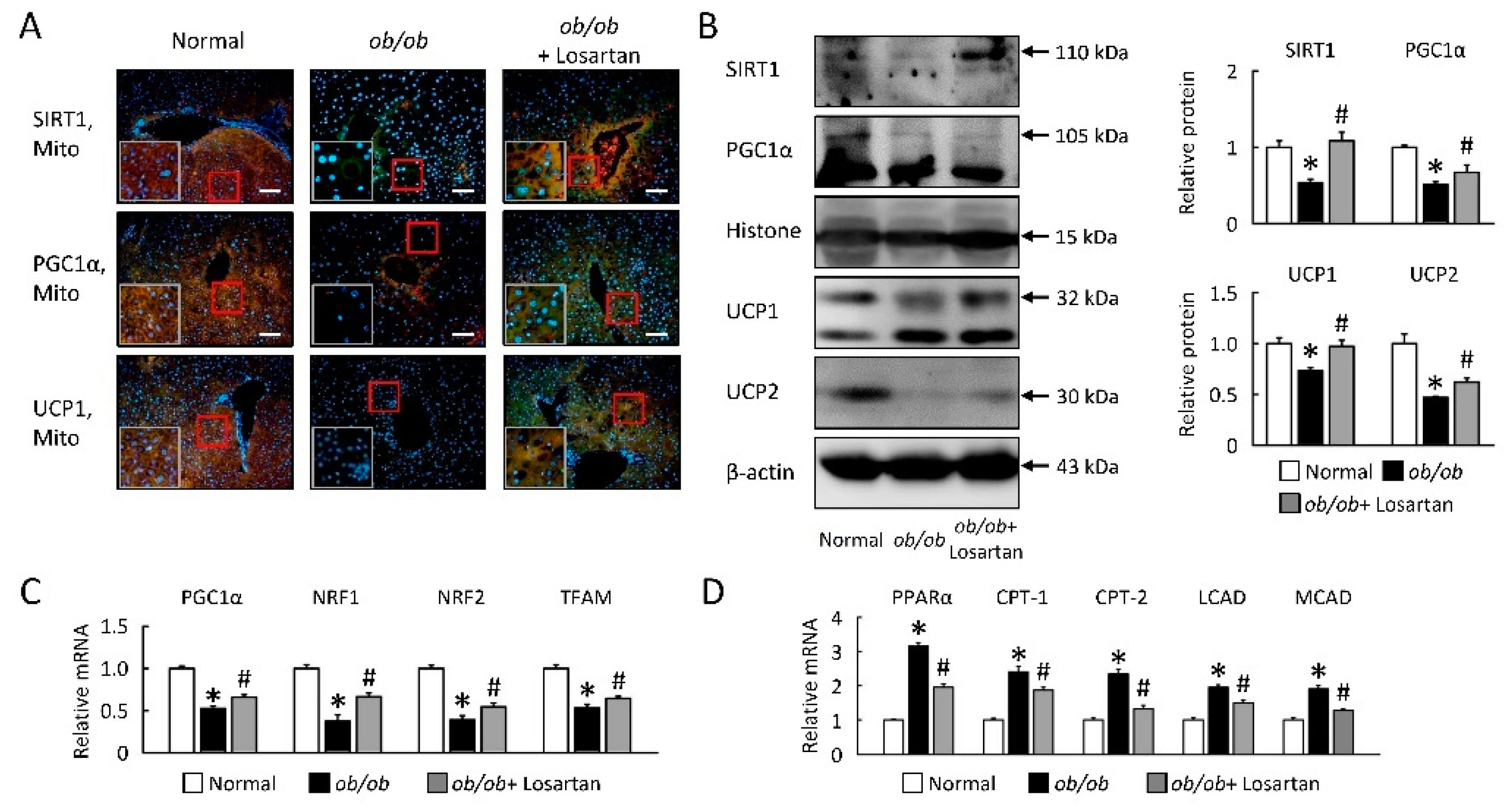
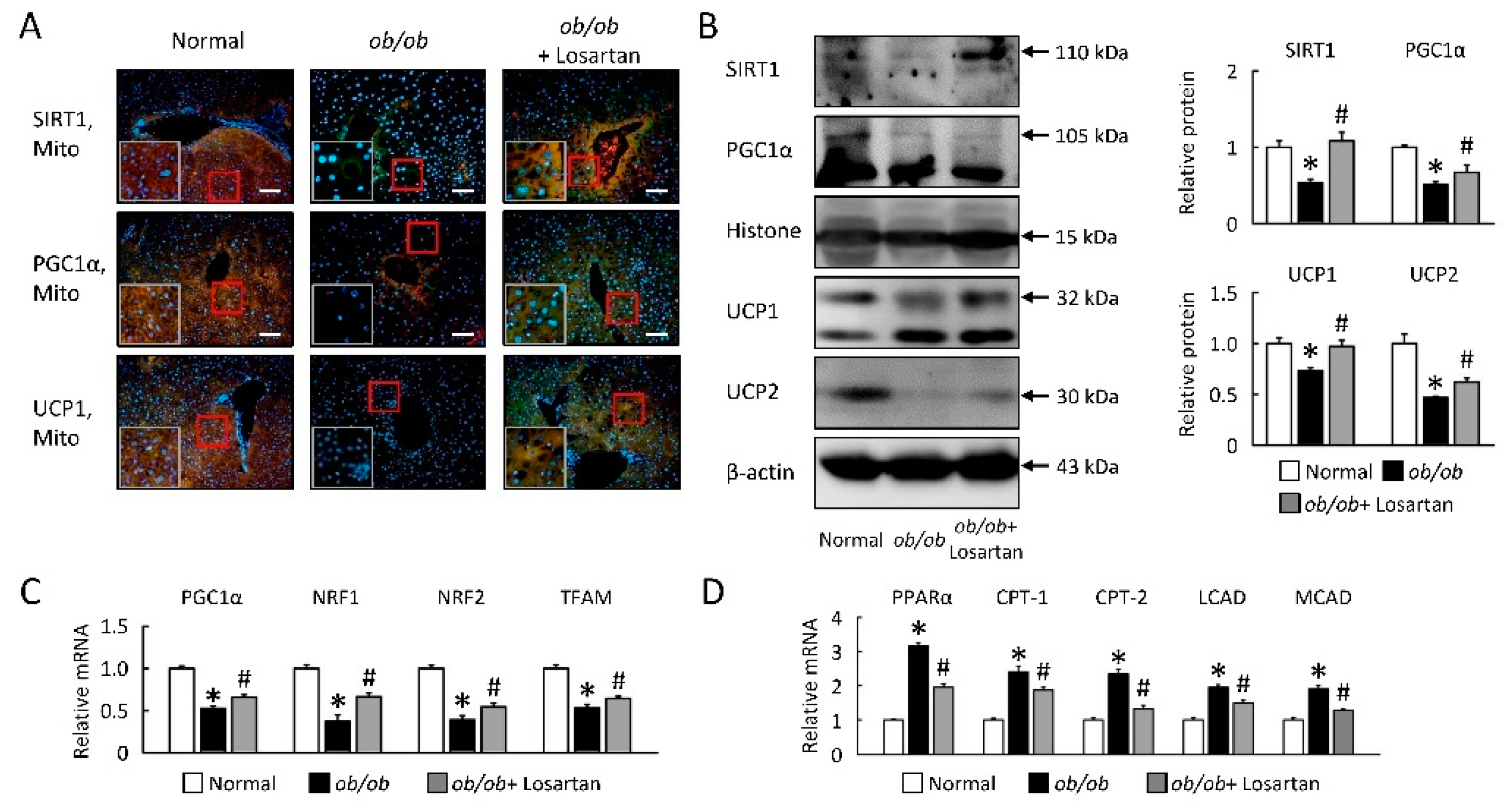
2.4. Losartan Improves Hepatic Mitochondrial Biogenesis and β-Oxidation
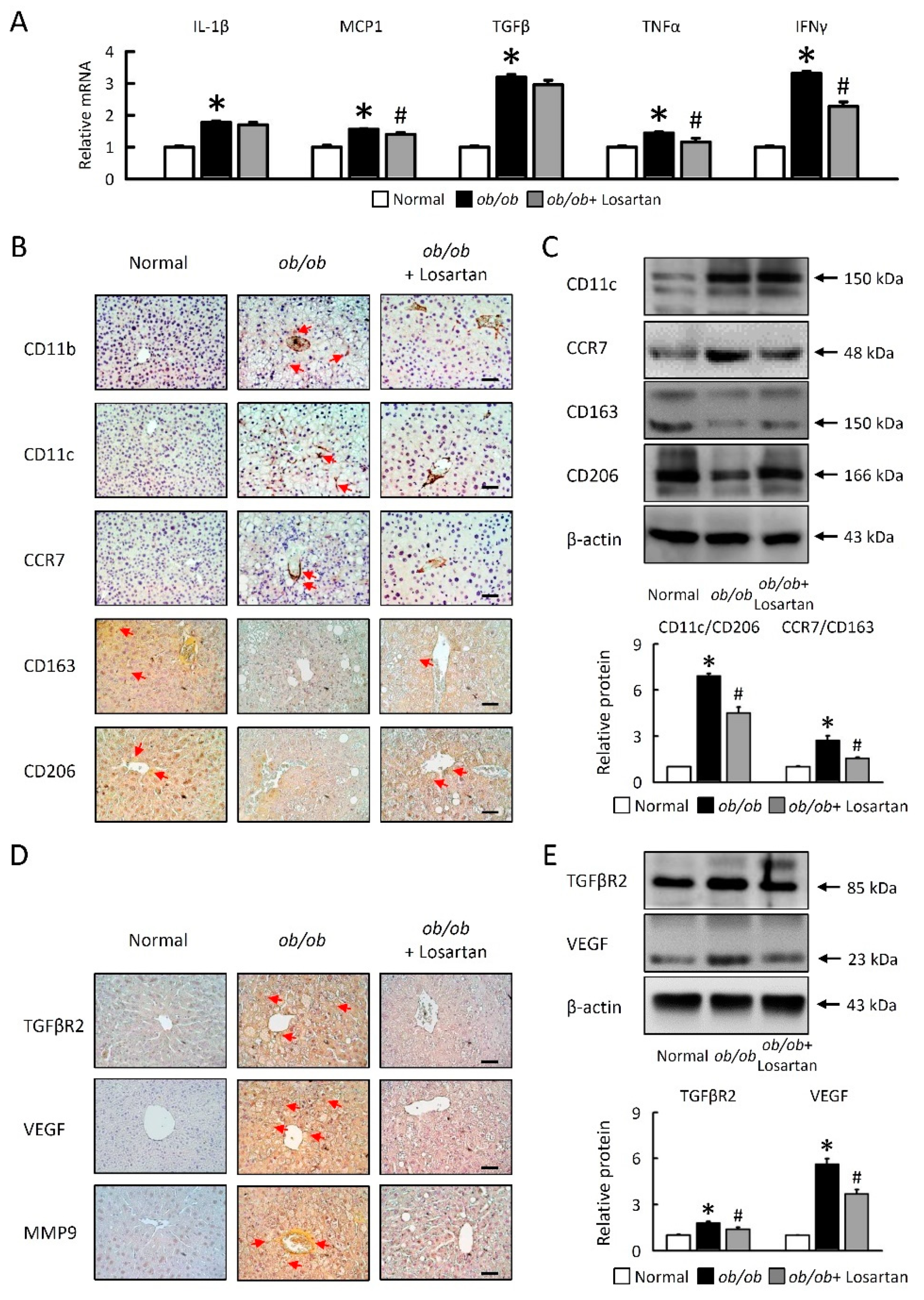
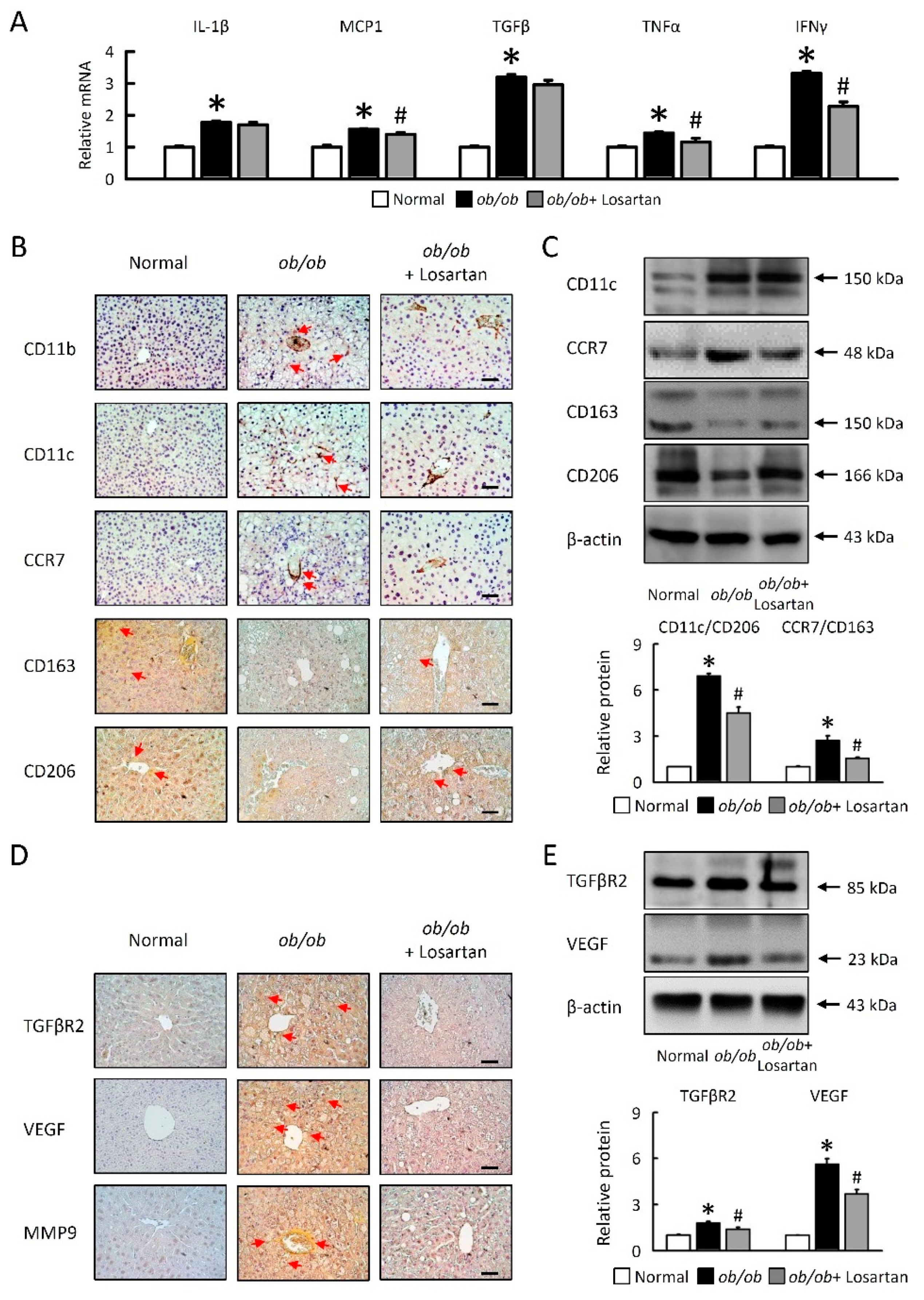
2.5. Losartan Attenuates Hepatic Inflammatory Cytokine mRNA Expression and Relate Hepatic Macrophage Polarization
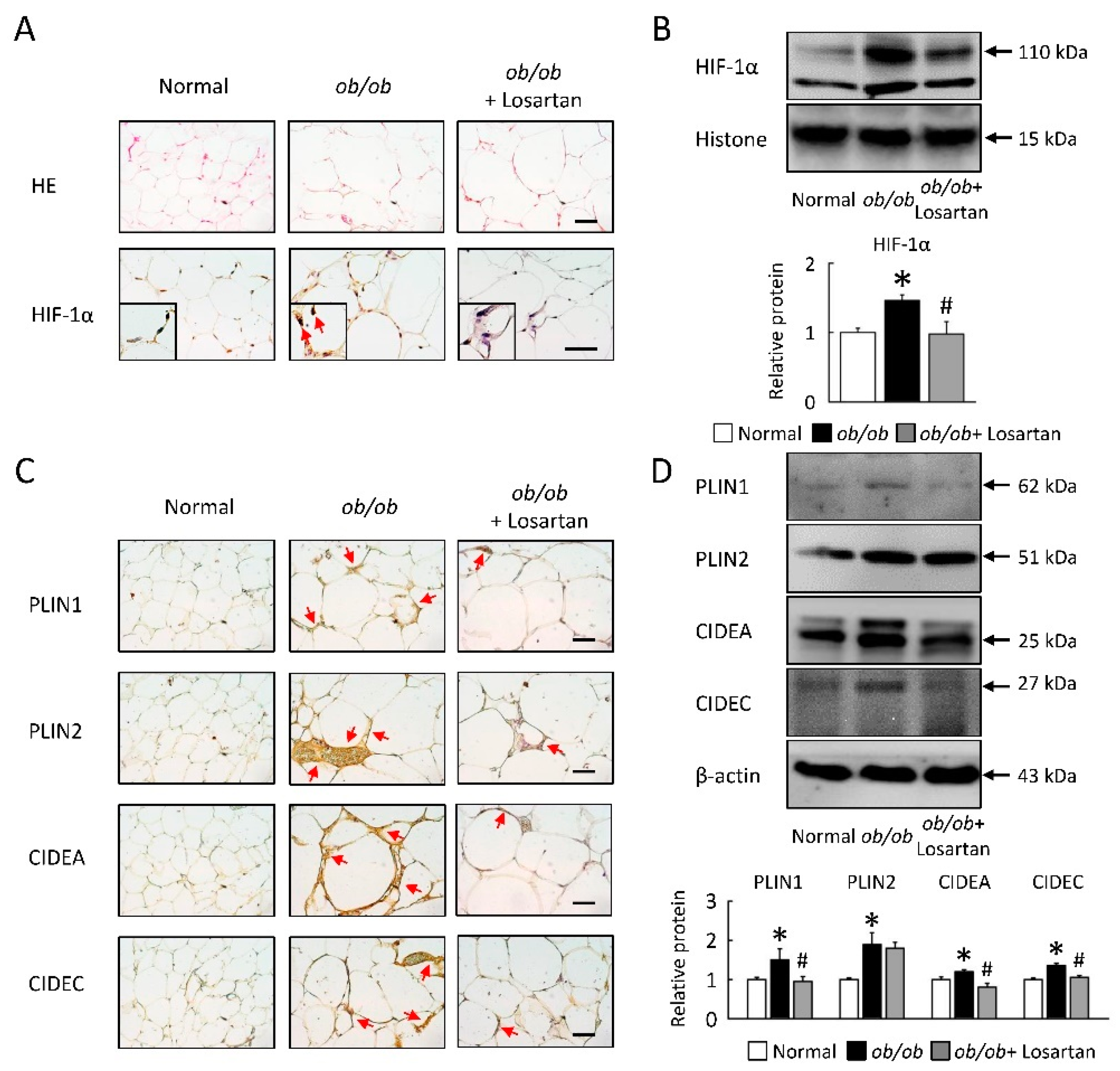
2.6. Losartan Attenuates Lipid Accumulate and HIF-1α Protein Level in EWAT
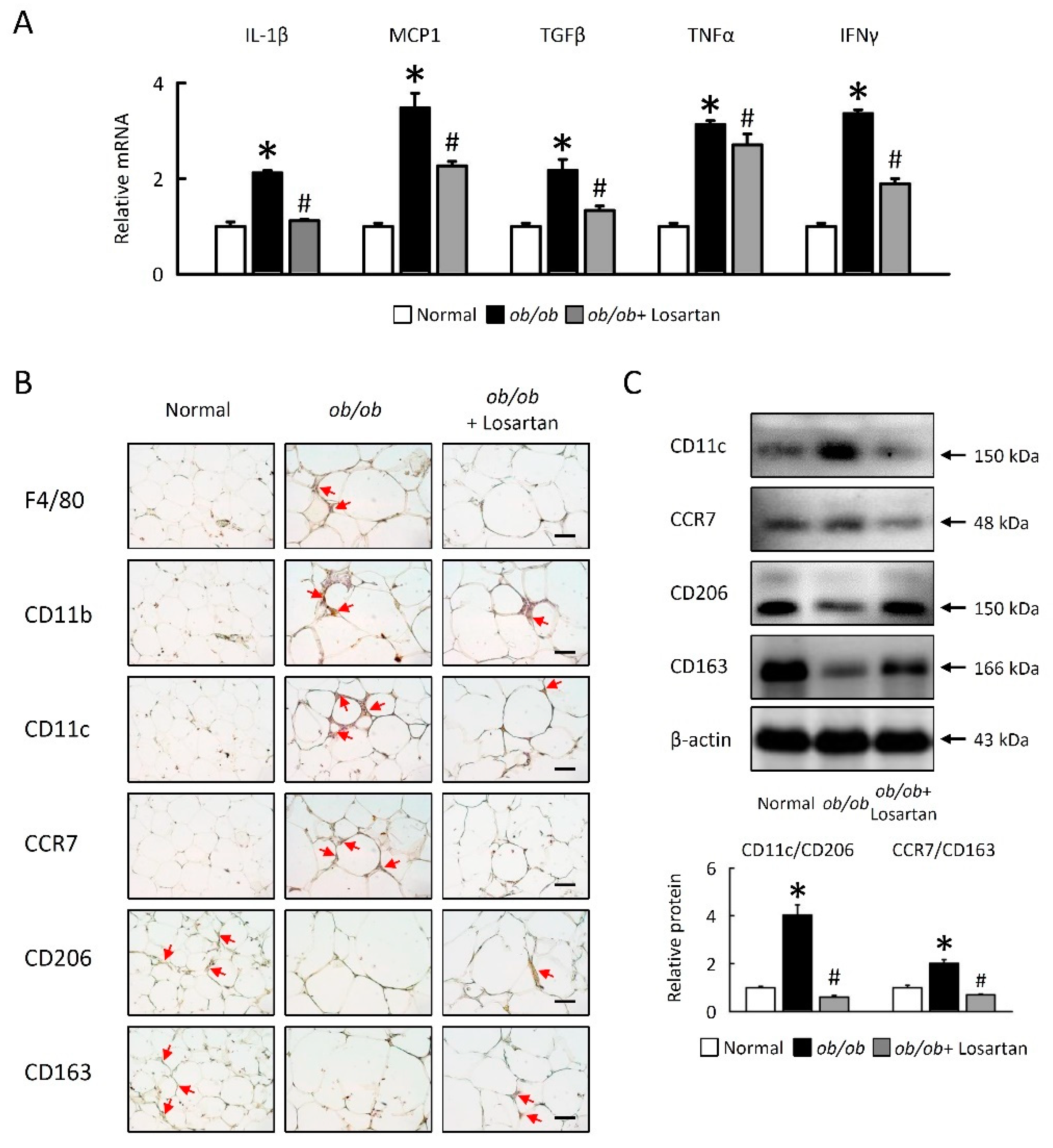
2.7. Losartan Attenuates Inflammation and Alters Macrophage Polarization in EWAT
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Sample Collection and Biochemical Analysis
4.3. Histological, Immunohistochemistry, and Immunofluorescence Analysis of Adipose Tissue and Liver
4.4. Oil Red O Staining
4.5. Auto-Fluorescence Detection of Lipofuscin
4.6. RNA Isolation and Quantitative Real-Time PCR
4.7. Protein Isolation and Western Blot Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. Mol. Cell Proteom. 2018, 17, 2371–2386. [Google Scholar] [CrossRef] [Green Version]
- Lackner, C.; Tiniakos, D. Fibrosis and alcohol-related liver disease. J. Hepatol. 2019, 70, 294–304. [Google Scholar] [CrossRef]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Gross, D.A.; Silver, D.L. Cytosolic lipid droplets: From mechanisms of fat storage to disease. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 304–326. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Fujii, H.; Ikura, Y.; Arimoto, J.; Sugioka, K.; Iezzoni, J.C.; Park, S.H.; Naruko, T.; Itabe, H.; Kawada, N.; Caldwell, S.H.; et al. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J. Atheroscler. Thromb. 2009, 16, 893–901. [Google Scholar] [CrossRef] [Green Version]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, M.A.; Rennie, C.D.; Robertshaw, H.A.; Fedak-Tarnopolsky, S.N.; Devries, M.C.; Hamadeh, M.J. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1271–R1278. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Hoppins, S. The regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 2014, 29, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef] [Green Version]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.Y.; Wang, X.; Jiang, C. HIF1alpha-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, X.; Han, S.N.; Zhang, J.Y.; Dioletis, E.; Nemeth, B.T.; Pacher, P.; Feng, D.; Bataller, R.; Cabezas, J.; Starkel, P.; et al. Digoxin suppresses pyruvate kinase M2-promoted HIF-1alpha transactivation in steatohepatitis. Cell Metab. 2018, 27, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Rosselli, M.S.; Burgueño, A.L.; Carabelli, J.; Schuman, M.; Carlos, J.; Pirola, C.J.; Sookoian, S. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis 2009, 206, 119–126. [Google Scholar] [CrossRef]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Shirai, Y.; Aihara, Y.; Namisaki, T.; Yoshii, J.; Yanase, K.; et al. Combination treatment of angiotensin II type I receptor blocker and new oral iron chelator attenuates progression of nonalcoholic steatohepatitis in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1094–G1104. [Google Scholar] [CrossRef] [Green Version]
- Hirata, T.; Tomita, K.; Kawai, T.; Yokoyama, H.; Shimada, A.; Kikuchi, M.; Hirose, H.; Ebinuma, H.; Irie, J.; Ojiro, K.; et al. Effect of telmisartan or losartan for treatment of nonalcoholic fatty liver disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int. J. Endocrinol. 2013, 2013, 587140. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Lin, H.C.; Huang, Y.T.; Lee, T.Y.; Hou, M.C.; Lee, F.Y.; Liu, R.S.; Chang, F.Y.; Lee, S.D. Effect of 1-week losartan administration on bile duct-ligated cirrhotic rats with portal hypertension. J. Hepatol. 2002, 36, 600–606. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Lee, P.C.; Huang, Y.T.; Lee, W.P.; Kuo, Y.J.; Lee, K.C.; Hsieh, Y.C.; Lee, T.Y.; Lin, H.C. Involvement of the HIF-1alpha and Wnt/beta-catenin pathways in the protective effects of losartan on fatty liver graft with ischaemia/reperfusion injury. Clin. Sci. 2014, 126, 163–174. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Q.; Yang, J.; Ma, Y.; Ding, G. Angiotensin II induces cholesterol accumulation and injury in podocytes. Sci. Rep. 2017, 7, 10672. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Sugioka, T.; Tsukada, K.; Aizawa, M.; Takizawa, M.; Shimizu, K.; Morimoto, M.; Suematsu, M.; Goda, N. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, improves hepatic microcirculatory patency and oxygen availability in a high-fat-diet-induced fatty liver in mice. Adv. Exp. Med. Biol. 2010, 662, 77–82. [Google Scholar]
- Suzuki, T.; Shinjo, S.; Arai, T.; Kanai, M.; Goda, N. Hypoxia and fatty liver. World J. Gastroenterol. 2014, 20, 15087–15097. [Google Scholar] [CrossRef]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Qingdu Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Safran, M.; Buckley, M.R.M.; Ebert, B.L.; Glickman, J.; Bosenberg, M.; Regan, M.; Kaelin, W.G., Jr. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation In Vivo. EMBO J. 2006, 25, 4650–4662. [Google Scholar] [CrossRef]
- Wang, R.; Kong, X.; Cui, A.; Liu, X.; Xiang, R.; Yang, Y.; Guan, Y.; Fang, F.; Chang, Y. Sterol-regulatory-element-binding protein 1c mediates the effect of insulin on the expression of Cidea in mouse hepatocytes. Biochem. J. 2010, 430, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Toh, S.Y.; Gong, J.; Du, G.; Li, J.Z.; Yang, S.; Ye, J.; Yao, H.; Zhang, Y.; Xue, B.; Li, Q.; et al. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS ONE 2008, 3, e2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Kusakabe, T.; Noguchi, T.; Takiguchi, S.; Suzuki, T.; Yamano, S.; Gonzalez, F.J. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008, 7, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Yon Toh, S.; Chen, Z.; Guo, K.; Ng, C.P.; Ponniah, S.; Lin, S.C.; Hong, W.; Li, P. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat. Genet. 2003, 35, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding pro-tein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Graus-Nunes, F.; Santos, F.O.; Marinho, T.S.; Miranda, C.S.; Barbosa-da-Silva, S.; Souza-Mello, V. Beneficial effects of losartan or telmisartan on the local hepatic renin-angiotensin system to counter obesity in an experimental model. World J. Hepatol. 2019, 11, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Jia, Y.; Yang, G.; Zhang, X.; Boddu, P.C.; Petersen, B.; Narsingam, S.; Zhu, Y.J.; Thimmapaya, B.; Kanwar, Y.S.; et al. PPARalpha-deficient ob/ob obese mice become more obese and manifest severe hepatic steatosis due to decreased fatty acid oxidation. Am. J. Pathol. 2015, 185, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.O.Y.; Soro-Arnaiz, I.; Aragones, J. Age-dependent obesity and mitochondrial dysfunction. Adipocyte 2017, 6, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARgamma-coactivator 1alpha (PGC1alpha) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Pessayre, D.; Mansouri, A.; Fromenty, B. Nonalcoholic steatosis and steatohepatitis. V. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G193–G199. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Vascotto, C.; Tiribelli, C. Alterations in the redox state and liver damage: Hints from the EASL Basic School of Hepatology. J. Hepatol. 2013, 58, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; de Carvalho Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-specific hypoxia-inducible factor-1alpha contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Tarling, E.J.; McMillen, T.S.; Tang, C.; LeBoeuf, R.C. ABCG1 regulates mouse adipose tissue macrophage cholesterol levels and ratio of M1 to M2 cells in obesity and caloric restriction. J. Lipid Res. 2015, 56, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.N.; Valentin, J.E.; Stewart-Akers, A.M.; McCabe, G.P.; Badylak, S.F. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 2009, 30, 1482–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romijn, H.J.; van Uum, J.F.; Breedijk, I.; Emmering, J.; Radu, I.; Pool, C.W. Double immunolabeling of neuropeptides in the human hypothalamus as analyzed by confocal laser scanning fluorescence microscopy. J. Histochem. Cytochem. 1999, 47, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| ATGL | 5′ aacaccagcatccagttcaa 3′ | 5′ ggttcagtaggccattcctc 3′ |
| HSL | 5′ agacaccagccaacggatac 3′ | 5′ catcaccctcgaagaagagca 3′ |
| LPL | 5′ actcatctccgccatgcc 3′ | 5′ ccagctttctcctagcaagg 3′ |
| ACO | 5′ atgaatcccgatctgcgcaaggagc 3′ | 5′ aaaggcatgtaacccgtagcactcc 3′ |
| SREBP-1c | 5′ actgtcttggttgttgatgagctggagcat 3′ | 5′ atcggcgcggaagctgtcggggtagcgtc 3′ |
| FAS | 5′ tgtcattggcctcctcaaaaagggcgtcca 3′ | 5′ tcaccactgtgggctctgcagagaagcgag 3′ |
| SCD-1 | 5′ ccggagaccccttagatcga 3′ | 5′ tagcctgtaaaagatttctgcaaacc 3′ |
| FATP | 5′ gcttcaacagccgtatcctc 3′ | 5′ tcttcttgttggtggcactg 3′ |
| CD36 | 5′ gcaaaacgactgcaggtcaac 3′ | 5′ tggtcccagtctcatttagcca 3′ |
| CPT-1 | 5′ ggacagagactgtgcgttcct 3′ | 5′ gcgatatccaacagtgcttga 3′ |
| CPT-2 | 5′ caaggccctggctgatgatgtg 3′ | 5′ agtctctgtccgcccctctcg 3′ |
| LCAD | 5′ tcaacagcagttacttgg 3′ | 5′ gacaatatctgagtggag 3′ |
| MCAD | 5′ ggggaggatgacggagcagc 3′ | 5′ cgggtactttaggatctggg 3′ |
| PGC1α | 5′ gactcagtgtcaccaccgaaa-3′ | 5′ tgaacgagagcgcatcctt 3′ |
| TFAM | 5′ ggaatgtggagcgtgctaaaa 3′ | 5′-tgctggaaaaacacttcggaata 3′ |
| UCP1 | 5′ cctgcctctctcggaaacaa 3′ | 5′-tgtaggctgcccaatgaaca 3′ |
| UCP2 | 5′ gcctctggaaagggacttctc 3′ | 5′ accagctcagcacagttgaca 3′ |
| NRF1 | 5′ cgcagcacctttggagaa 3′ | 5′-cccgacctgtggaatacttg-3′ |
| NRF2 | 5′ atggatttgattgacatcctt 3′ | 5′ catgtttttctttgtatctgg 3′ |
| IL-1β | 5′ aacctgctggtgtgtgacgttc 3′ | 5′ cagcacgaggcttttttgttgt 3′ |
| MCP1 | 5′ aggtccctgtcatgcttctg 3′ | 5′ tctggacccattccttcttg 3′ |
| TGFβ | 5′ tatagcaacaattcctggcg 3′ | 5′ tgctgtcacaggagcagtg 3′ |
| TNFα | 5′ ttgacctcagcgctgagttg 3′ | 5′ cctgtagcccacgtcgtagc 3′ |
| IFNγ | 5′ cctcaaacttggcaatactc 3′ | 5′ agcaacaacataagcgtcat 3′ |
| GAPDH | 5′ tcaccaccatggagaaggc 3′ | 5′ gctaagcagttggtggtgca 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-H.; Liu, H.-M.; Chang, Z.-Y.; Huang, T.-H.; Lee, T.-Y. Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1α in a Murine Model of NAFLD. Int. J. Mol. Sci. 2021, 22, 7841. https://doi.org/10.3390/ijms22157841
Wang C-H, Liu H-M, Chang Z-Y, Huang T-H, Lee T-Y. Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1α in a Murine Model of NAFLD. International Journal of Molecular Sciences. 2021; 22(15):7841. https://doi.org/10.3390/ijms22157841
Chicago/Turabian StyleWang, Cheng-Hui, Hsuan-Miao Liu, Zi-Yu Chang, Tse-Hung Huang, and Tzung-Yan Lee. 2021. "Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1α in a Murine Model of NAFLD" International Journal of Molecular Sciences 22, no. 15: 7841. https://doi.org/10.3390/ijms22157841
APA StyleWang, C.-H., Liu, H.-M., Chang, Z.-Y., Huang, T.-H., & Lee, T.-Y. (2021). Losartan Prevents Hepatic Steatosis and Macrophage Polarization by Inhibiting HIF-1α in a Murine Model of NAFLD. International Journal of Molecular Sciences, 22(15), 7841. https://doi.org/10.3390/ijms22157841