
Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells

 ,
,  ,
,  ,
,
Abstract

1. Introduction
2. Results and Discussion
2.1. Biochemometric and LCMS Metabolomic Identification of Cytotoxic Metabolites of AP-1
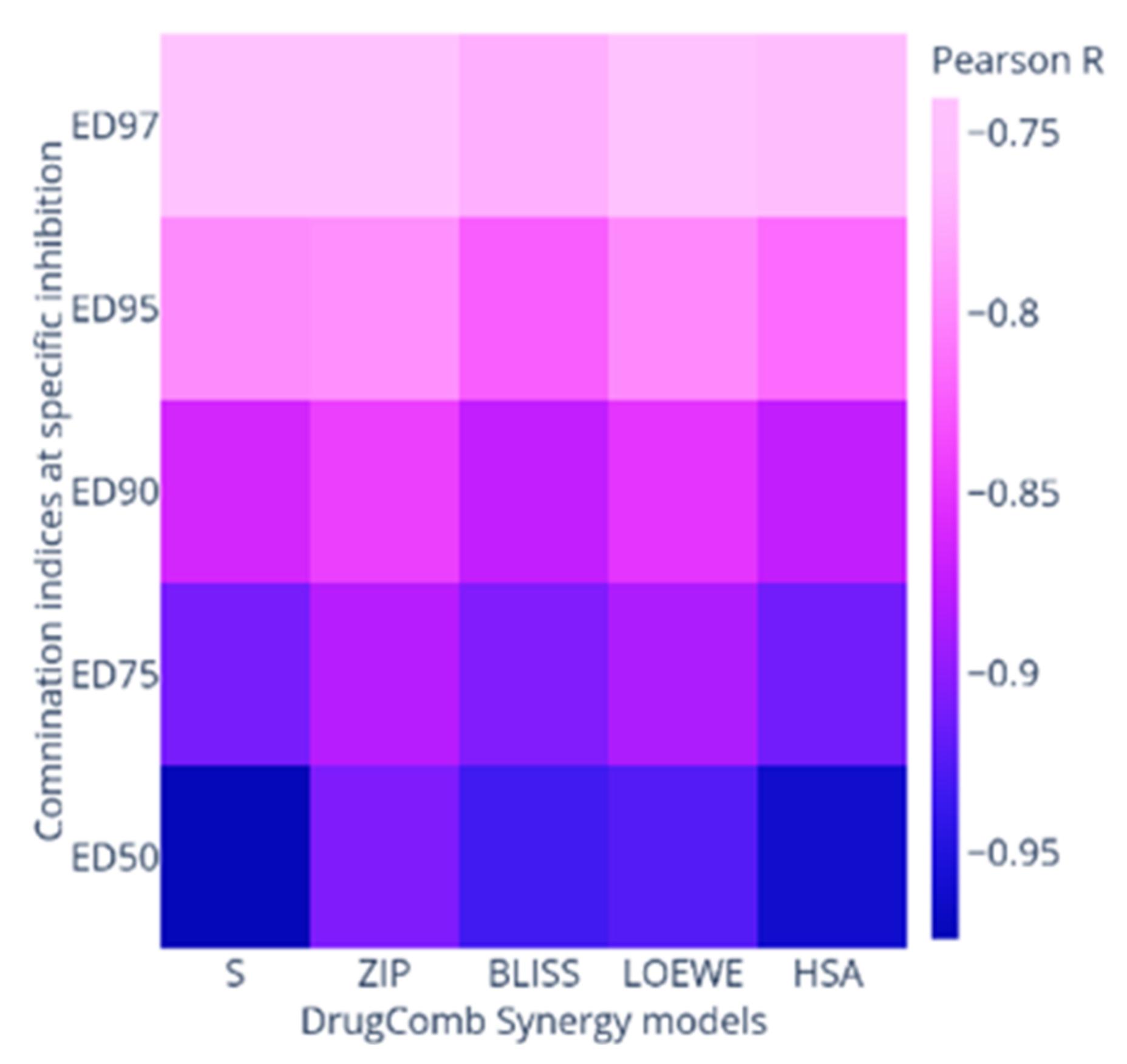
2.2. Synergy Quantification of AP-1 and DOX Combinations against the MCF7 Breast Adenocarcinoma Cells
2.3. Inhibition of Reactive Oxygen Species (ROS) Production in the MCF7 Cells after Mono and Combined Treatments with AP-1 and DOX
2.4. Flow Cytometric Analysis of Apoptosis in the MCF7 Cells Using Annexin V-CF Blue and 7AAD
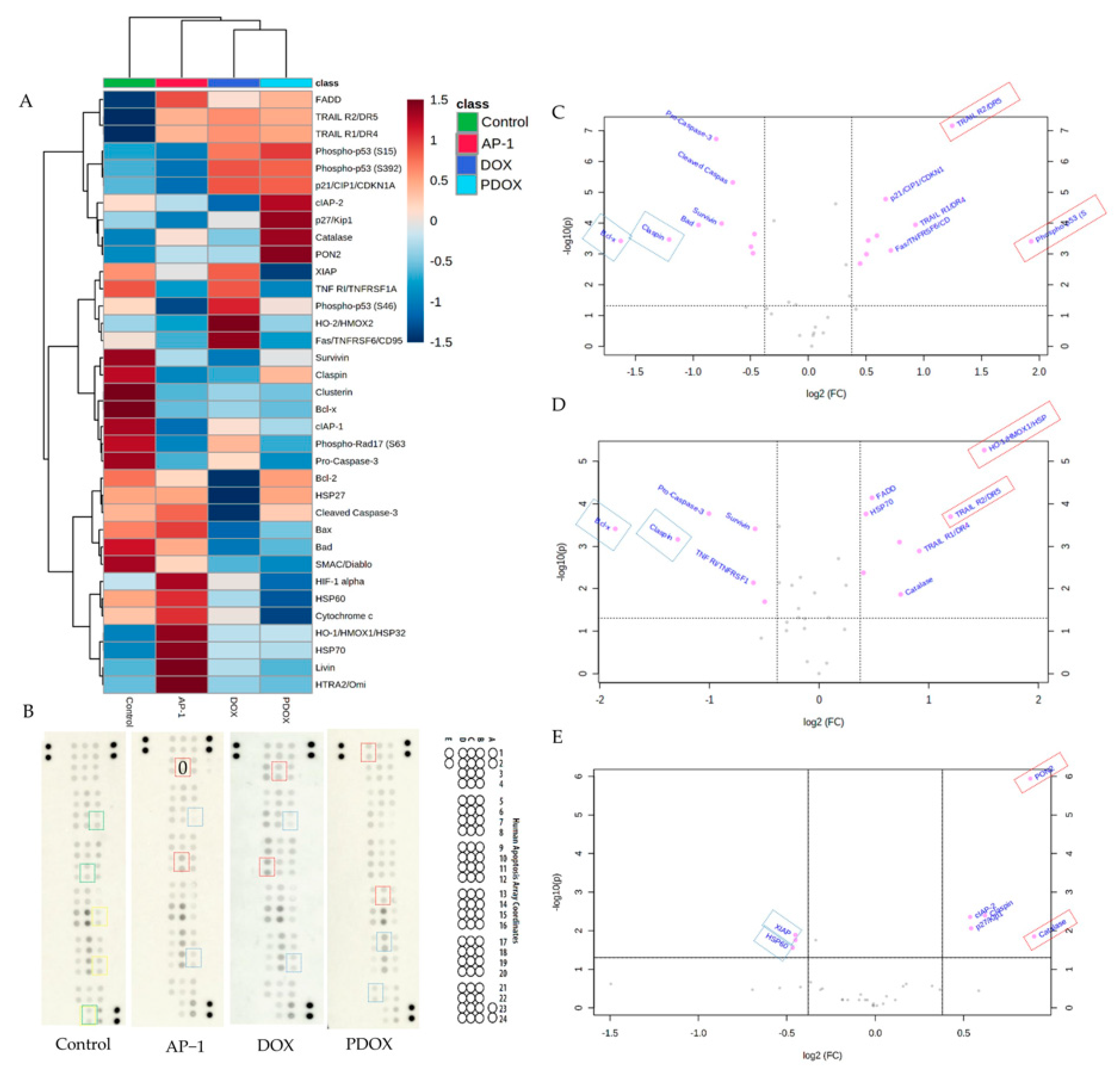
2.5. Apoptotic Proteome Profiler Array Analysis
2.5.1. Effects of AP-1 and DOX on Apoptotic Proteins of the MCF7 Cells
2.5.2. Effects of the Most Synergistic AP-1 and DOX Combination on Apoptotic Proteins of the MCF7 Cells
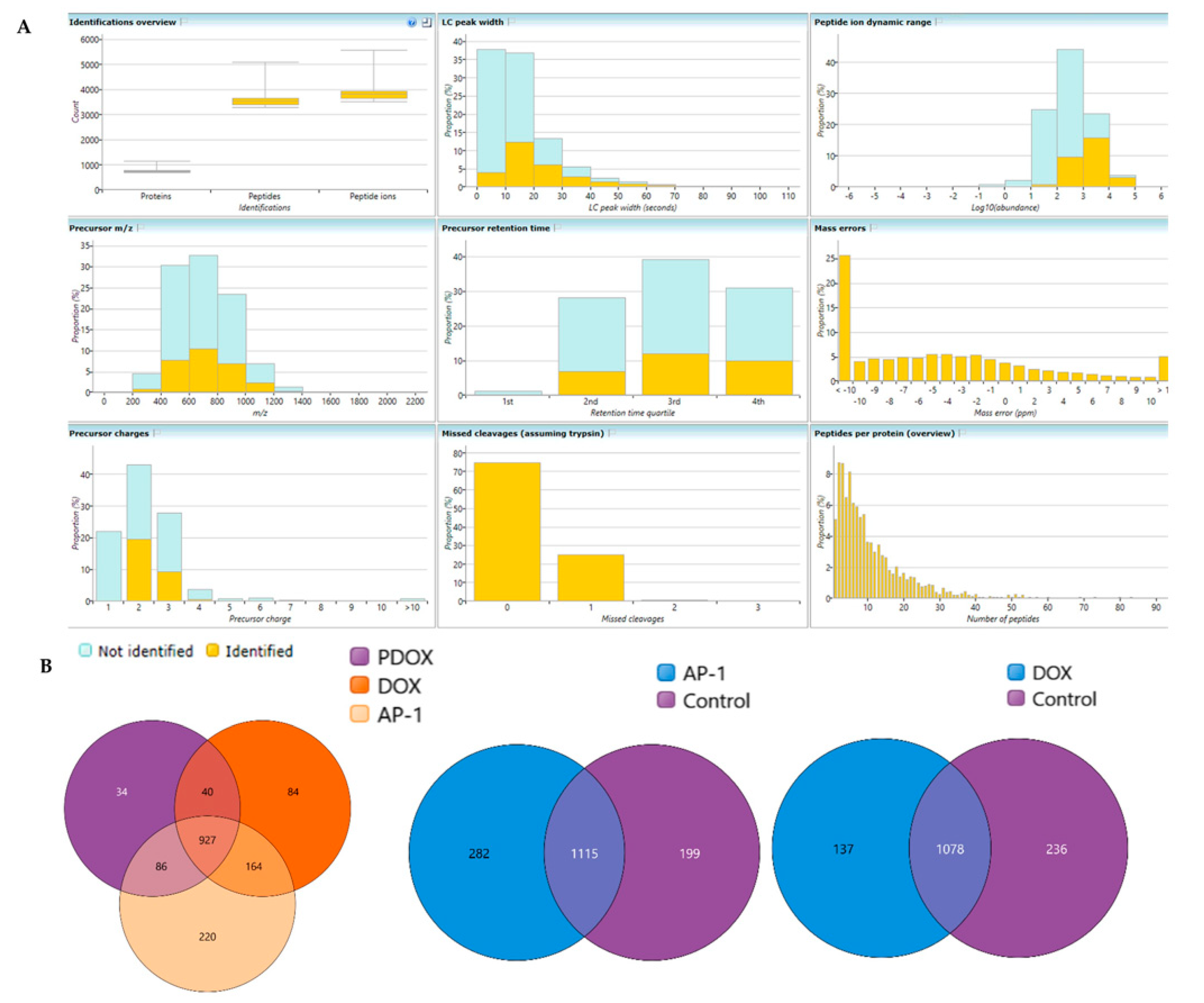
2.6. Bottom-Up Label-Free Quantification Proteomic Study of the MCF7 Cells after Treatment with AP-1, DOX and Their Synergistic Combination
3. Conclusions and Future Directions
4. Material and Methods
4.1. Chemicals and Preparation of Australian Propolis Extract
4.2. Cell Culture
4.3. Cell Viability Determination
4.4. Biochemometric and LCMS-Driven Metabolomic Identification of Anticancer Metabolites of AP-1 in the MCF7 Human Breast Adenocarcinoma Cells
4.5. Synergy Quantification of AP-1 and DOX Combinations against the MCF7 Human Breast Adenocarcinoma Cells
4.6. Reactive Oxygen Species (ROS) Assay
4.7. Flow Cytometric Analyses of Apoptosis in the MCF7 Human Breast Adenocarcinoma Cells Using Annexin V-CF Blue and 7-Aminoactinomycin D (7AAD)
4.8. Human Apoptosis Proteomic Array
4.8.1. Cell Culture, Treatment, and Protein Extraction
4.8.2. Protein Quantification
4.8.3. Apoptosis Proteome Array Analysis
4.9. Bottom-Up Label-Free Quantification Proteomic Study of the MCF7 Cell Lysates after Treatment with the Most Synergistic Combination
4.9.1. Cell Culture, Treatment and Protein Extraction
4.9.2. Peptides Preparation and Clean Up
4.9.3. Label-Free Bottom-Up Quantification via Nano-Ultra High-Performance Liquid Chromatography Coupled with Quadruple Time of Flight Mass Spectrometry (NanoUPLC-qTOF-MS)
4.9.4. Data Processing and Availability
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 7-AAD | 7-aminoactinomycin D |
| AC | Adriamycin/cyclophosphamide |
| AP-1 | Australian propolis sample 1 |
| Apaf-1 | Apoptotic protease-activating factor 1 |
| ARA | Adriamycin Resistance Associated long non-coding RNA |
| ATM | Ataxia-telangiectasia mutated |
| ATR | Ataxia-telangiectasia and Rad3-related protein |
| Bad | Bcl-2 associated agonist of cell death |
| Bax | BCL2 associated X |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-XL | B-cell lymphoma-extra large |
| CMF | cyclophosphamide/methotrextate/5-flurouricil |
| CAT | cyclophosphamide/adriamycin/taxanes |
| CI | Combination index |
| cIAP-1 | Cellular Inhibitor of Apoptosis Protein 1/Baculoviral IAP repeat-containing 2 |
| cIAP-2 | Baculoviral IAP repeat containing 3 |
| CSS | Combination sensitivity scores |
| DSBs | Double-strand breaks |
| DOX | Doxorubicin |
| EJC | Exon-junction complex |
| ER+ | Estrogen receptor positive |
| FADD | Fas-associated protein with death domain |
| Fas/TNFRSF6/CD95 | Fas receptor/tumour necrosis factor receptor superfamily member 6/cluster of differentiation 95 |
| HER-2 | Human epidermal growth factor receptor 2/Receptor tyrosine-protein kinase erbB-2 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HO-1/HMOX1/HSP32 | Heme oxygenase 1 |
| HO-2/HMOX2 | Heme oxygenase 2 |
| HSP27 | Heat shock protein 27 |
| HSP60 | Heat shock protein 60 chaperonins |
| HSP70 | Heat shock protein 70 |
| HTRA2/Omi | High-temperature requirement protein A |
| JNK | c-Jun N-terminal kinase |
| IAPs | Inhibitors of apoptosis |
| IGF-1 | Insulin-like growth factor-1 |
| NHEJ | Nonhomologous End-Joining |
| NMD | Nonsense-mediated decay |
| ROS | Reactive oxygen species |
| S | S synergy score |
| VIP | Variable Importance Projection score derived from PLS-Da model |
| TRAIL R1/DR4 | TNF-related apoptosis-inducing ligand receptor 1/Death receptor 4 |
| TRAIL R2/DR5 | TNF-related apoptosis-inducing ligand receptor 2/Death receptor 5 |
| PON2 | Serum paraoxonase/arylesterase 2 |
| p21/CIP1/CDKN1A | cyclin-dependent kinase inhibitor 1 |
| p27/Kip1 | Cyclin-dependent kinase inhibitor 1B |
| TNF RI/TNFRSF1A | Tumour necrosis factor receptor 1/tumour necrosis factor receptor superfamily member 1A |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Seddon, B.; Strauss, S.J.; Whelan, J.; Leahy, M.; Woll, P.J.; Cowie, F.; Rothermundt, C.; Wood, Z.; Benson, C.; Ali, N.; et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): A randomised controlled phase 3 trial. Lancet Oncol. 2017, 18, 1397–1410. [Google Scholar] [CrossRef]
- McMeekin, S.; Dizon, D.; Barter, J.; Scambia, G.; Manzyuk, L.; Lisyanskaya, A.; Oaknin, A.; Ringuette, S.; Mukhopadhyay, P.; Rosenberg, J.; et al. Phase III randomized trial of second-line ixabepilone versus paclitaxel or doxorubicin in women with advanced endometrial cancer. Gynecol. Oncol. 2015, 138, 18–23. [Google Scholar] [CrossRef]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Tap, W.D.; Wagner, A.J.; Schoffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs. Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.L.; Flynn, P.J.; Yothers, G.; Asmar, L.; Geyer, C.E., Jr.; Jacobs, S.A.; Robert, N.J.; Hopkins, J.O.; O’Shaughnessy, J.A.; Dang, C.T.; et al. Anthracyclines in Early Breast Cancer: The ABC Trials-USOR 06-090, NSABP B-46-I/USOR 07132, and NSABP B-49 (NRG Oncology). J. Clin. Oncol. 2017, 35, 2647–2655. [Google Scholar] [CrossRef]
- Schneeweiss, A.; Mobus, V.; Tesch, H.; Hanusch, C.; Denkert, C.; Lubbe, K.; Huober, J.; Klare, P.; Kummel, S.; Untch, M.; et al. Intense dose-dense epirubicin, paclitaxel, cyclophosphamide versus weekly paclitaxel, liposomal doxorubicin (plus carboplatin in triple-negative breast cancer) for neoadjuvant treatment of high-risk early breast cancer (GeparOcto-GBG 84): A randomised phase III trial. Eur. J. Cancer 2019, 106, 181–192. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L., Jr.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-lnduced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.L.; Renu, K.; Valsala Gopalakrishnan, A. New molecular and biochemical insights of doxorubicin-induced hepatotoxicity. Life Sci. 2020, 250, 117599. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Gopalakrishnan, A.V. Deciphering the molecular mechanism during doxorubicin-mediated oxidative stress, apoptosis through Nrf2 and PGC-1α in a rat testicular milieu. Reprod. Biol. 2019, 19, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Sruthy, K.; Parthiban, S.; Sugunapriyadharshini, S.; George, A.; Tirupathi Pichiah, P.B.; Suman, S.; Abilash, V.; Arunachalam, S. Elevated lipolysis in adipose tissue by doxorubicin via PPARα activation associated with hepatic steatosis and insulin resistance. Eur. J. Pharmacol. 2019, 843, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy–An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, K.J.; Kim, J.H.; Kwak, J.H.; Song, H.; Cho, J.Y.; Hwang, D.Y.; Kim, K.S.; Jung, Y.S. Comparision of doxorubicin-induced cardiotoxicity in the ICR mice of different sources. Lab. Anim. Res. 2017, 33, 165–170. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Thomas, S.A. Chemotherapy agents that cause cardiotoxicity. US Pharm. 2017, 42, HS24. [Google Scholar]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updates 2020, 49, 100671. [Google Scholar] [CrossRef]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anticancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Xiao, Q.; Wang, X.; Zhu, J.; Li, J.; Liang, X.; Peng, Y.; Wu, C.; Lu, R.; Pan, Y.; et al. A Biomimetic Drug Delivery System by Integrating Grapefruit Extracellular Vesicles and Doxorubicin-Loaded Heparin-Based Nanoparticles for Glioma Therapy. Nano Lett. 2021, 21, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Ruzycka-Ayoush, M.; Kowalik, P.; Kowalczyk, A.; Bujak, P.; Nowicka, A.M.; Wojewodzka, M.; Kruszewski, M.; Grudzinski, I.P. Quantum dots as targeted doxorubicin drug delivery nanosystems. Cancer Nanotechnol. 2021, 12, 1–27. [Google Scholar]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Update 2016, 29, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E. Curcumin Combination Chemotherapy: The Implication and Efficacy in Cancer. Molecules 2019, 24, 2527. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Arabi, L.; Alibolandi, M. Doxorubicin-loaded composite nanogels for cancer treatment. J. Control. Release 2020, 328, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Makwana, V.; Karanjia, J.; Haselhorst, T.; Anoopkumar-Dukie, S.; Rudrawar, S. Liposomal doxorubicin as targeted delivery platform: Current trends in surface functionalization. Int. J. Pharm. 2021, 593, 120117. [Google Scholar] [CrossRef]
- Borisev, I.; Mrdanovic, J.; Petrovic, D.; Seke, M.; Jovic, D.; Srdenovic, B.; Latinovic, N.; Djordjevic, A. Nanoformulations of doxorubicin: How far have we come and where do we go from here? Nanotechnology 2018, 29, 332002. [Google Scholar] [CrossRef]
- Yarmohammadi, F.; Rezaee, R.; Karimi, G. Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/ARE signaling pathway. Phytother. Res. PTR 2021, 35, 1163–1175. [Google Scholar] [CrossRef]
- Alsherbiny, M.A.; Abd-Elsalam, W.H.; El Badawy, S.A.; Taher, E.; Fares, M.; Torres, A.; Chang, D.; Li, C.G. Ameliorative and protective effects of ginger and its main constituents against natural, chemical and radiation-induced toxicities: A comprehensive review. Food Chem. Toxicol. 2019, 123, 72–97. [Google Scholar] [CrossRef]
- Alsherbiny, M.A.; Li, C.G. Medicinal cannabis—Potential drug interactions. Medicines 2019, 6, 3. [Google Scholar] [CrossRef]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e13. [Google Scholar] [CrossRef]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.J.; Hegreness, M.J.; Aiden, A.P.; Kishony, R. Drug interactions and the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.T.; Wooten, D.J.; Lopez, C.F.; Quaranta, V. Charting the Fragmented Landscape of Drug Synergy. Trends Pharm. Sci. 2020, 41, 266–280. [Google Scholar] [CrossRef]
- Vlot, A.H.; Aniceto, N.; Menden, M.P.; Ulrich-Merzenich, G.; Bender, A. Applying drug synergy metrics to oncology combination screening data: Agreements, disagreements and pitfalls. Drug Discov. Today 2019, 24, 2286–2298. [Google Scholar] [CrossRef]
- Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef]
- Berenbaum, M.C. What is synergy? Pharmacol. Rev. 1989, 41, 93–141. [Google Scholar]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly1. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- O’Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Gilvary, C.; Dry, J.R.; Elemento, O. Multi-Task learning predicts drug combination synergy in cells and in the clinic. bioRxiv 2019, 576017. [Google Scholar] [CrossRef]
- Hosseini, A.; Hosseinzadeh, H. Antidotal or protective effects of Curcuma longa (turmeric) and its active ingredient, curcumin, against natural and chemical toxicities: A review. Biomed. Pharmacother. 2018, 99, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Tavakkoli, A.; Ahmadi, A.; Razavi, B.M.; Hosseinzadeh, H. Black Seed (Nigella sativa) and its Constituent Thymoquinone as an Antidote or a Protective Agent Against Natural or Chemical Toxicities. Iran. J. Pharm. Res. 2017, 16, 2–23. [Google Scholar] [PubMed]
- Fanoudi, S.; Alavi, M.S.; Karimi, G.; Hosseinzadeh, H. Milk thistle (Silybum marianum) as an antidote or a protective agent against natural or chemical toxicities: A review. Drug Chem. Toxicol. 2020, 43, 240–254. [Google Scholar] [CrossRef]
- Dorri, M.; Hashemitabar, S.; Hosseinzadeh, H. Cinnamon (Cinnamomum zeylanicum) as an antidote or a protective agent against natural or chemical toxicities: A review. Drug Chem. Toxicol. 2018, 41, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, N.; Mehri, S.; Hosseinzadeh, H. Berberis vulgaris and its constituent berberine as antidotes and protective agents against natural or chemical toxicities. Iran. J. Basic Med. Sci. 2017, 20, 538–551. [Google Scholar] [CrossRef]
- Rameshrad, M.; Razavi, B.M.; Hosseinzadeh, H. Protective effects of green tea and its main constituents against natural and chemical toxins: A comprehensive review. Food Chem. Toxicol. 2017, 100, 115–137. [Google Scholar] [CrossRef]
- Bhuyan, D.J.; Alsherbiny, M.A.; Low, M.N.; Zhou, X.; Kaur, K.; Li, G.; Li, C.G. Broad-Spectrum pharmacological activity of Australian propolis and metabolomic-driven identification of marker metabolites of propolis samples from three continents. Food Funct. 2021, 12, 2498–2519. [Google Scholar] [CrossRef]
- Kocot, J.; Kielczykowska, M.; Luchowska-Kocot, D.; Kurzepa, J.; Musik, I. Antioxidant Potential of Propolis, Bee Pollen, and Royal Jelly: Possible Medical Application. Oxidative Med. Cell. Longev. 2018, 2018, 7074209. [Google Scholar] [CrossRef]
- Chiu, H.F.; Han, Y.C.; Shen, Y.C.; Golovinskaia, O.; Venkatakrishnan, K.; Wang, C.K. Chemopreventive and Chemotherapeutic Effect of Propolis and Its Constituents: A Mini-review. J. Cancer Prev. 2020, 25, 70–78. [Google Scholar] [CrossRef]
- Patel, S. Emerging Adjuvant Therapy for Cancer: Propolis and its Constituents. J. Diet. Suppl. 2016, 13, 245–268. [Google Scholar] [CrossRef]
- Abu-Mellal, A.; Koolaji, N.; Duke, R.K.; Tran, V.H.; Duke, C.C. Prenylated cinnamate and stilbenes from Kangaroo Island propolis and their antioxidant activity. Phytochemistry 2012, 77, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Duke, C.C.; Tran, V.H.; Duke, R.K.; Abu-Mellal, A.; Plunkett, G.T.; King, D.I.; Hamid, K.; Wilson, K.L.; Barrett, R.L.; Bruhl, J.J. A sedge plant as the source of Kangaroo Island propolis rich in prenylated p-coumarate ester and stilbenes. Phytochemistry 2017, 134, 87–97. [Google Scholar] [CrossRef] [PubMed]
- El-Bassuony, A.; AbouZid, S. A new prenylated flavanoid with antibacterial activity from propolis collected in Egypt. Nat. Prod. Commun. 2010, 5, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Omar, R.M.; Igoli, J.; Gray, A.I.; Ebiloma, G.U.; Clements, C.; Fearnley, J.; Ebel, R.A.; Zhang, T.; De Koning, H.P.; Watson, D.G. Chemical characterisation of Nigerian red propolis and its biological activity against Trypanosoma Brucei. Phytochem. Anal. 2016, 27, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Trusheva, B.; Popova, M.; Koendhori, E.B.; Tsvetkova, I.; Naydenski, C.; Bankova, V. Indonesian propolis: Chemical composition, biological activity and botanical origin. Nat. Prod. Res. 2011, 25, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, S.; Goto, H.; Hamasaka, T.; Fukumoto, S.; Fujimoto, T.; Nakayama, T. A new prenylated flavonoid from propolis collected in Okinawa, Japan. Biosci. Biotechnol. Biochem. 2004, 68, 260–262. [Google Scholar] [CrossRef]
- Chen, C.N.; Wu, C.L.; Lin, J.K. Propolin C from propolis induces apoptosis through activating caspases, Bid and cytochrome c release in human melanoma cells. Biochem. Pharmacol. 2004, 67, 53–66. [Google Scholar] [CrossRef]
- Mukaide, K.; Honda, S.; Vongsak, B.; Kumazawa, S. Prenylflavonoids from propolis collected in Chiang Mai, Thailand. Phytochem. Lett. 2021, 43, 88–93. [Google Scholar] [CrossRef]
- Raghukumar, R.; Vali, L.; Watson, D.; Fearnley, J.; Seidel, V. Antimethicillin-resistant Staphylococcus aureus (MRSA) activity of ‘pacific propolis’ and isolated prenylflavanones. Phytother. Res. 2010, 24, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Inui, S.; Hosoya, T.; Shimamura, Y.; Masuda, S.; Ogawa, T.; Kobayashi, H.; Shirafuji, K.; Moli, R.T.; Kozone, I.; Shin-Ya, K. Solophenols B–D and Solomonin: New Prenylated Polyphenols Isolated from Propolis Collected from The Solomon Islands and Their Antibacterial Activity. J. Agric. Food Chem. 2012, 60, 11765–11770. [Google Scholar] [CrossRef] [PubMed]
- Arvouet-Grand, A.; Lejeune, B.; Bastide, P.; Pourrat, A.; Privat, A.M.; Legret, P. Propolis extract. I. Acute toxicity and determination of acute primary cutaneous irritation index. J. Pharm. Belg. 1993, 48, 165–170. [Google Scholar]
- Schmidt, J.; Buchmann, S.J.H. Other Products of the Hive. The Hive and the Honey Bee; Dadant and Sons: Hamilton, IL, USA, 1992; pp. 927–987. [Google Scholar]
- Burdock, G.J.F. Review of the biological properties and toxicity of bee propolis (propolis). J. Food Chem. Toxicol. 1998, 36, 347–363. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Zeraik, M.L.; Pauli, I.; Dutra, L.A.; Cruz, R.S.; Valli, M.; Paracatu, L.C.; de Faria, C.; Ximenes, V.F.; Regasini, L.O.; Andricopulo, A.D.; et al. Identification of a Prenyl Chalcone as a Competitive Lipoxygenase Inhibitor: Screening, Biochemical Evaluation and Molecular Modeling Studies. Molecules 2021, 26, 2205. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.L.; Stevens, J.F.; Ivanov, V.; McCall, M.; Frei, B.; Deinzer, M.L.; Buhler, D.R. Antioxidant and prooxidant actions of prenylated and nonprenylated chalcones and flavanones in vitro. J. Agric. Food Chem. 2000, 48, 3876–3884. [Google Scholar] [CrossRef]
- Venturelli, S.; Burkard, M.; Biendl, M.; Lauer, U.M.; Frank, J.; Busch, C. Prenylated chalcones and flavonoids for the prevention and treatment of cancer. Nutrition 2016, 32, 1171–1178. [Google Scholar] [CrossRef]
- Miranda, C.L.; Aponso, G.L.M.; Stevens, J.F.; Deinzer, M.L.; Buhler, D.R. Prenylated chalcones and flavanones as inducers of quinone reductase in mouse Hepa 1c1c7 cells. Cancer Lett. 2000, 149, 21–29. [Google Scholar] [CrossRef]
- Go, M.L.; Wu, X.; Liu, X.L. Chalcones: An update on cytotoxic and chemoprotective properties. Curr. Med. Chem. 2005, 12, 481–499. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Pon, A.; Karu, N.; Zheng, J.; Li, C.; Arndt, D.; Gautam, M.; Allen, F.; Wishart, D.S. CFM-ID 3.0: Significantly Improved ESI-MS/MS Prediction and Compound Identification. Metabolites 2019, 9, 72. [Google Scholar] [CrossRef]
- Chou, T.-C. The combination index (CI < 1) as the definition of synergism and of synergy claims. Synergy 2018, 7, 49–50. [Google Scholar]
- Doroshow, J.H.; Simon, R.M. On the Design of Combination Cancer Therapy. Cell 2017, 171, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Malyutina, A.; Majumder, M.M.; Wang, W.; Pessia, A.; Heckman, C.A.; Tang, J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput. Biol. 2019, 15, e1006752. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Sarmiento-Salinas, F.L.; Delgado-Magallon, A.; Montes-Alvarado, J.B.; Ramirez-Ramirez, D.; Flores-Alonso, J.C.; Cortes-Hernandez, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Kurbacher, C.M.; Wagner, U.; Kolster, B.; Andreotti, P.E.; Krebs, D.; Bruckner, H.W. Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 1996, 103, 183–189. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Dong, K.; Zhao, Z.Z.; Kang, J.; Lin, L.R.; Chen, W.T.; Liu, J.X.; Wu, X.L.; Lu, T.L. Cinnamaldehyde and Doxorubicin Co-Loaded Graphene Oxide Wrapped Mesoporous Silica Nanoparticles for Enhanced MCF-7 Cell Apoptosis. Int. J. Nanomed. 2020, 15, 10285–10304. [Google Scholar] [CrossRef]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms: Intermediacy of H2O2-and p53-dependent pathways. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Punganuru, S.R.; Srivenugopal, K.S. Piperlongumine exerts cytotoxic effects against cancer cells with mutant p53 proteins at least in part by restoring the biological functions of the tumor suppressor. Int. J. Oncol. 2016, 48, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-N.; Kang, K.-J. The effect of blueberry on ROS accumulation and cell death in human normal breast epithelial (MCF10A) and breast cancer (MCF7) cells. Korean J. Food Nutr. 2008, 21, 416–424. [Google Scholar]
- Kumar, R.; Saneja, A.; Panda, A.K. An Annexin V-FITC—Propidium Iodide-Based Method for Detecting Apoptosis in a Non-Small Cell Lung Cancer Cell Line. In Lung Cancer: Methods and Protocols; Santiago-Cardona, P.G., Ed.; Springer: New York, NY, USA, 2021; pp. 213–223. [Google Scholar]
- Wadkins, R.M.; Jovin, T.M. Actinomycin D and 7-aminoactinomycin D binding to single-stranded DNA. Biochemistry 1991, 30, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Schmit, T.; Klomp, M.; Khan, M.N. An Overview of Flow Cytometry: Its Principles and Applications in Allergic Disease Research. Methods Mol. Biol. 2021, 2223, 169–182. [Google Scholar] [CrossRef]
- Golbidi, S.; Botta, A.; Gottfred, S.; Nusrat, A.; Laher, I.; Ghosh, S. Glutathione administration reduces mitochondrial damage and shifts cell death from necrosis to apoptosis in ageing diabetic mice hearts during exercise. Br. J. Pharmacol. 2014, 171, 5345–5360. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Czeczuga-Semeniuk, E.; Wołczyński, S.; Dabrowska, M.; Dziecioł, J.; Anchim, T. The effect of doxorubicin and retinoids on proliferation, necrosis and apoptosis in MCF-7 breast cancer cells. Folia Histochem. Cytobiol. 2004, 42, 221–227. [Google Scholar]
- Sugimoto, K.; Tamayose, K.; Sasaki, M.; Hayashi, K.; Oshimi, K. Low-dose doxorubicin-induced necrosis in Jurkat cells and its acceleration and conversion to apoptosis by antioxidants. Br. J. Haematol. 2002, 118, 229–238. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Renner, S.; Xu, F.; Fleishman, A.; Taylor, J.; Weisz, J.; Vescio, R.; Rettig, M.; Berenson, J.; Krajewski, S.; et al. BCL-X expression in multiple myeloma: Possible indicator of chemoresistance. Cancer Res. 1998, 58, 256–262. [Google Scholar] [PubMed]
- Jaattela, M.; Benedict, M.; Tewari, M.; Shayman, J.A.; Dixit, V.M. Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene 1995, 10, 2297–2305. [Google Scholar] [PubMed]
- Lima, R.T.; Martins, L.M.; Guimaraes, J.E.; Sambade, C.; Vasconcelos, M.H. Specific downregulation of bcl-2 and xIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene Ther. 2004, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Akar, U.; Chaves-Reyez, A.; Barria, M.; Tari, A.; Sanguino, A.; Kondo, Y.; Kondo, S.; Arun, B.; Lopez-Berestein, G.; Ozpolat, B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.L.; Pandey, V.; Zhu, T.; Ma, L.; Basappa; Lobie, P.E. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 2018, 415, 177–186. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef]
- Chini, C.C.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef]
- Bianco, J.N.; Bergoglio, V.; Lin, Y.L.; Pillaire, M.J.; Schmitz, A.L.; Gilhodes, J.; Lusque, A.; Mazieres, J.; Lacroix-Triki, M.; Roumeliotis, T.I.; et al. Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun. 2019, 10, 910. [Google Scholar] [CrossRef]
- Shojaei, F.; Yazdani-Nafchi, F.; Banitalebi-Dehkordi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M. Trace of survivin in cancer. Eur. J. Cancer Prev. 2019, 28, 365–372. [Google Scholar] [CrossRef]
- Nestal de Moraes, G.; Vasconcelos, F.C.; Delbue, D.; Mognol, G.P.; Sternberg, C.; Viola, J.P.; Maia, R.C. Doxorubicin induces cell death in breast cancer cells regardless of Survivin and XIAP expression levels. Eur. J. Cell Biol. 2013, 92, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Long, L.M.; Li, L.; Marciniak, R.A.; Kazhdan, I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer 2008, 8, 129. [Google Scholar] [CrossRef]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef]
- Jonsson, G.; Paulie, S.; Grandien, A. cIAP-2 block apoptotic events in bladder cancer cells. Anticancer Res. 2003, 23, 3311–3316. [Google Scholar]
- Shao, F.; Wang, L.; Chu, X. Lonidamine induces apoptosis via endoplasmic reticulum stress response and down-regulating cIAP expression in human breast carcinoma MCF-7 cells. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 883–887. [Google Scholar]
- Park, S.H.; Ham, S.; Kwon, T.H.; Kim, M.S.; Lee, D.H.; Kang, J.W.; Oh, S.R.; Yoon, D.Y. Luteolin induces cell cycle arrest and apoptosis through extrinsic and intrinsic signaling pathways in MCF-7 breast cancer cells. J. Environ. Pathol. Toxicol. Oncol. 2014, 33, 219–231. [Google Scholar] [CrossRef]
- Verma, A.K.; Ahmad, I.; Yadav, P.; Rahmani, A.H.; Khan, B.; Alsahli, M.A.; Joshi, P.C.; Ahmad, H.; Ali Beg, M.M. Expression and Correlation of Cell-Free cIAP-1 and cIAP-2 mRNA in Breast Cancer Patients: A Study from India. J. Oncol. 2020, 2020, 3634825. [Google Scholar] [CrossRef] [PubMed]
- Pluta, P.; Jeziorski, A.; Cebula-Obrzut, A.P.; Wierzbowska, A.; Piekarski, J.; Smolewski, P. Expression of IAP family proteins and its clinical importance in breast cancer patients. Neoplasma 2015, 62, 666–673. [Google Scholar] [CrossRef]
- Yang, X.H.; Sladek, T.L.; Liu, X.; Butler, B.R.; Froelich, C.J.; Thor, A.D. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001, 61, 348–354. [Google Scholar] [PubMed]
- Yeruva, L.; Elegbede, J.A.; Carper, S.W. Methyl jasmonate decreases membrane fluidity and induces apoptosis through tumor necrosis factor receptor 1 in breast cancer cells. Anticancer Drugs 2008, 19, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Yu, J.H.; Wu, J.N.; Tashiro, S.; Onodera, S.; Minami, M.; Ikejima, T. P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharm. Sin. 2007, 28, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.P.; Lu, Y.Y.; Lv, J.C.; Ou, H.J. [Effect of ursolic acid on caspase-3 and PARP expression of human MCF-7 cells]. Zhongguo Zhong Yao Za Zhi 2006, 31, 141–144. [Google Scholar]
- Yang, H.L.; Chen, C.S.; Chang, W.H.; Lu, F.J.; Lai, Y.C.; Chen, C.C.; Hseu, T.H.; Kuo, C.T.; Hseu, Y.C. Growth inhibition and induction of apoptosis in MCF-7 breast cancer cells by Antrodia camphorata. Cancer Lett. 2006, 231, 215–227. [Google Scholar] [CrossRef]
- Chen, J.S.; Konopleva, M.; Andreeff, M.; Multani, A.S.; Pathak, S.; Mehta, K. Drug-resistant breast carcinoma (MCF-7) cells are paradoxically sensitive to apoptosis. J. Cell. Physiol. 2004, 200, 223–234. [Google Scholar] [CrossRef]
- Wang, S.; He, M.; Li, L.; Liang, Z.; Zou, Z.; Tao, A. Cell-in-cell death is not restricted by caspase-3 deficiency in MCF-7 cells. J. Breast Cancer 2016, 19, 231. [Google Scholar] [CrossRef]
- Janicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res. Treat. 2009, 117, 219–221. [Google Scholar] [CrossRef]
- Mackay, M.; Perez-Lopez, A.M.; Bradley, M.; Lilienkampf, A. Eliminating caspase-7 and cathepsin B cross-reactivity on fluorogenic caspase-3 substrates. Mol. Biosyst. 2016, 12, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, D.J.; Vuong, Q.V.; Bond, D.R.; Chalmers, A.C.; Bowyer, M.C.; Scarlett, C.J. Eucalyptus microcorys leaf extract derived HPLC-fraction reduces the viability of MIA PaCa-2 cells by inducing apoptosis and arresting cell cycle. Biomed. Pharmacother. 2018, 105, 449–460. [Google Scholar] [CrossRef]
- Zhang, H.; Kim, J.K.; Edwards, C.A.; Xu, Z.; Taichman, R.; Wang, C.Y. Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Djeu, J.Y.; Wei, S. Clusterin and Chemoresistance. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2009; Volume 105, pp. 77–92. [Google Scholar]
- Redondo, M.; Tellez, T.; Roldan, M.J.; Serrano, A.; Garcia-Aranda, M.; Gleave, M.E.; Hortas, M.L.; Morell, M. Anticlusterin treatment of breast cancer cells increases the sensitivities of chemotherapy and tamoxifen and counteracts the inhibitory action of dexamethasone on chemotherapy-induced cytotoxicity. Breast Cancer Res. Treat. 2007, 9, R86. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Capoulade, C.; Moyret-Lalle, C.; Amor-Gueret, M.; Feunteun, J.; Larsen, A.K.; Paillerets, B.B.; Chouaib, S. Resistance of MCF7 human breast carcinoma cells to TNF-induced cell death is associated with loss of p53 function. Oncogene 1997, 15, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Whibley, C.; Pharoah, P.D.; Hollstein, M. p53 polymorphisms: Cancer implications. Nat. Rev. Cancer 2009, 9, 95–107. [Google Scholar] [CrossRef]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed]
- Xuan, H.; Li, Z.; Yan, H.; Sang, Q.; Wang, K.; He, Q.; Wang, Y.; Hu, F. Antitumor Activity of Chinese Propolis in Human Breast Cancer MCF-7 and MDA-MB-231 Cells. Evid. Based Complement. Altern. Med. 2014, 2014, 280120. [Google Scholar] [CrossRef]
- Misir, S.; Aliyazicioglu, Y.; Demir, S.; Turan, I.; Hepokur, C. Effect of Turkish Propolis on miRNA Expression, Cell Cycle, and Apoptosis in Human Breast Cancer (MCF-7) Cells. Nutr. Cancer 2020, 72, 133–145. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar] [CrossRef]
- Kong, W.; Jiang, X.; Mercer, W.E. Downregulation of Wip-1 phosphatase expression in MCF-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of Bax. Cancer Biol. Ther. 2009, 8, 555–563. [Google Scholar] [CrossRef]
- Millour, J.; de Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef]
- Mohammad, N.; Singh, S.V.; Malvi, P.; Chaube, B.; Athavale, D.; Vanuopadath, M.; Nair, S.S.; Nair, B.; Bhat, M.K. Strategy to enhance efficacy of doxorubicin in solid tumor cells by methyl-β-cyclodextrin: Involvement of p53 and Fas receptor ligand complex. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Lee, Y.K.; Thomas, S.N.; Yang, A.J.; Ann, D.K. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J. Biol. Chem. 2007, 282, 1595–1606. [Google Scholar] [CrossRef]
- Liu, W.H.; Chang, L.S. Fas/FasL-dependent and -independent activation of caspase-8 in doxorubicin-treated human breast cancer MCF-7 cells: ADAM10 down-regulation activates Fas/FasL signaling pathway. Int. J. Biochem. Cell Biol. 2011, 43, 1708–1719. [Google Scholar] [CrossRef]
- Li, S.; Zhou, Y.; Dong, Y.; Ip, C. Doxorubicin and selenium cooperatively induce fas signaling in the absence of Fas/Fas ligand interaction. Anticancer Res. 2007, 27, 3075–3082. [Google Scholar]
- Wang, T.T.Y.; Jeng, J. Coordinated regulation of two TRAIL-R2/KILLER/DR5 mRNA isoforms by DNA damaging agents, serum and 17β-estradiol in human breast cancer cells. Breast Cancer Res. Treat. 2000, 61, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.D.; Huang, Y.; Mao, S.H.; Chen, S.C.; Qiu, M.; Ji, L.L.; Yi, C. Synergistic antitumor effect of TRAIL and adriamycin on the human breast cancer cell line MCF-7. Braz. J. Med. Biol. Res. 2009, 42, 854–862. [Google Scholar] [CrossRef][Green Version]
- Malin, D.; Chen, F.; Schiller, C.; Koblinski, J.; Cryns, V.L. Enhanced metastasis suppression by targeting TRAIL receptor 2 in a murine model of triple-negative breast cancer. Clin. Cancer Res. 2011, 17, 5005–5015. [Google Scholar] [CrossRef]
- Pileczki, V.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. TNF-α gene knockout in triple negative breast cancer cell line induces apoptosis. Int. J. Mol. Sci. 2013, 14, 411–420. [Google Scholar] [CrossRef]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.V.; Hushmandi, K.; Ashrafizadeh, M.; et al. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef]
- Chun, K.S.; Kim, D.H.; Surh, Y.J. Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance. Cells 2021, 10, 758. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, S.A.; Jung, E.K.; Lee, K.H.; Lee, J.K.; Kang, H.C.; Joo, Y.E.; Lim, S.C.; Yoon, T.M. Inhibitor of apoptosis protein Livin promotes tumor progression and chemoradioresistance in human anaplastic thyroid cancer. Oncol. Rep. 2021, 45, 1. [Google Scholar] [CrossRef] [PubMed]
- Moradi-Marjaneh, R.; Paseban, M.; Moradi Marjaneh, M. Hsp70 inhibitors: Implications for the treatment of colorectal cancer. IUBMB Life 2019, 71, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.A.; Dix, D.J.; Collins, B.W.; Luft, C.; Allen, J.W. Expression of inducible Hsp70 enhances the proliferation of MCF-7 breast cancer cells and protects against the cytotoxic effects of hyperthermia. Cell Stress Chaperones 2001, 6, 316–325. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Dhamad, A.E.; Zhou, Z.; Zhou, J.; Du, Y. Systematic Proteomic Identification of the Heat Shock Proteins (Hsp) that Interact with Estrogen Receptor Alpha (ERalpha) and Biochemical Characterization of the ERalpha-Hsp70 Interaction. PLoS ONE 2016, 11, e0160312. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase down-regulation in cancer cells exposed to arsenic trioxide is involved in their increased sensitivity to a pro-oxidant treatment. Cancer Cell Int. 2018, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- De Moreno de LeBlanc, A.; LeBlanc, J.G.; Perdigon, G.; Miyoshi, A.; Langella, P.; Azevedo, V.; Sesma, F. Oral administration of a catalase-producing Lactococcus lactis can prevent a chemically induced colon cancer in mice. J. Med. Microbiol. 2008, 57, 100–105. [Google Scholar] [CrossRef]
- Goh, J.; Enns, L.; Fatemie, S.; Hopkins, H.; Morton, J.; Pettan-Brewer, C.; Ladiges, W. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011, 11, 191. [Google Scholar] [CrossRef]
- Baker, A.M.; Oberley, L.W.; Cohen, M.B. Expression of antioxidant enzymes in human prostatic adenocarcinoma. Prostate 1997, 32, 229–233. [Google Scholar] [CrossRef]
- Bostwick, D.G.; Alexander, E.E.; Singh, R.; Shan, A.; Qian, J.; Santella, R.M.; Oberley, L.W.; Yan, T.; Zhong, W.; Jiang, X.; et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer 2000, 89, 123–134. [Google Scholar] [CrossRef]
- Ho, J.C.M.; Zheng, S.; Comhair, S.A.A.; Farver, C.; Erzurum, S.C. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res. 2001, 61, 8578–8585. [Google Scholar]
- Coursin, D.B.; Cihla, H.P.; Sempf, J.; Oberley, T.D.; Oberley, L.W. An immunohistochemical analysis of antioxidant and glutathione S-transferase enzyme levels in normal and neoplastic human lung. Histol. Histopathol. 1996, 11, 851–860. [Google Scholar]
- Cullen, J.J.; Mitros, F.A.; Oberley, L.W. Expression of antioxidant enzymes in diseases of the human pancreas: Another link between chronic pancreatitis and pancreatic cancer. Pancreas 2003, 26, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Guner, G.; Islekel, H.; Oto, O.; Hazan, E.; Acikel, U. Evaluation of some antioxidant enzymes in lung carcinoma tissue. Cancer Lett. 1996, 103, 233–239. [Google Scholar] [CrossRef]
- Kwei, K.A.; Finch, J.S.; Thompson, E.J.; Bowden, G.T. Transcriptional repression of catalase in mouse skin tumor progression. Neoplasia 2004, 6, 440–448. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lauer, C.; Volkl, A.; Riedl, S.; Fahimi, H.D.; Beier, K. Impairment of peroxisomal biogenesis in human colon carcinoma. Carcinogenesis 1999, 20, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.L.; Westman, N.G.; Lundgren, E.; Roos, G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982, 42, 1955–1961. [Google Scholar]
- Oberley, T.D.; Sempf, J.M.; Oberley, L.W. Immunogold analysis of antioxidant enzymes in common renal cancers. Histol. Histopathol. 1996, 11, 153–160. [Google Scholar]
- Subapriya, R.; Kumaraguruparan, R.; Ramachandran, C.R.; Nagini, S. Oxidant-antioxidant status in patients with oral squamous cell carcinomas at different intraoral sites. Clin. Biochem. 2002, 35, 489–493. [Google Scholar] [CrossRef]
- Yoo, D.G.; Song, Y.J.; Cho, E.J.; Lee, S.K.; Park, J.B.; Yu, J.H.; Lim, S.P.; Kim, J.M.; Jeon, B.H. Alteration of APE1/ref-1 expression in non-small cell lung cancer: The implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer 2008, 60, 277–284. [Google Scholar] [CrossRef]
- Glorieux, C.; Auquier, J.; Dejeans, N.; Sid, B.; Demoulin, J.B.; Bertrand, L.; Verrax, J.; Calderon, P.B. Catalase expression in MCF-7 breast cancer cells is mainly controlled by PI3K/Akt/mTor signaling pathway. Biochem. Pharmacol. 2014, 89, 217–223. [Google Scholar] [CrossRef]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Matsuyoshi, S.; Shimada, K.; Nakamura, M.; Ishida, E.; Konishi, N. FADD phosphorylation is critical for cell cycle regulation in breast cancer cells. Br. J. Cancer 2006, 94, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Takahashi-Niki, K.; Akagi, T.; Hashikawa, T.; Takahashi, R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004, 11, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Walle, L.V.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef]
- Li, W.; Srinivasula, S.M.; Chai, J.; Li, P.; Wu, J.W.; Zhang, Z.; Alnemri, E.S.; Shi, Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Mol. Biol. 2002, 9, 436–441. [Google Scholar] [CrossRef]
- Nestal de Moraes, G.; Delbue, D.; Silva, K.L.; Robaina, M.C.; Khongkow, P.; Gomes, A.R.; Zona, S.; Crocamo, S.; Mencalha, A.L.; Magalhaes, L.M.; et al. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell Signal. 2015, 27, 2496–2505. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yeh, C.T. Functional Compartmentalization of HSP60-Survivin Interaction between Mitochondria and Cytosol in Cancer Cells. Cells 2019, 9, 23. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Su, F.; Grijalva, V.; Yalamanchi, M.; Yalamanchi, A.; Gao, F.; Trost, H.; Nwokedi, J.; Farias-Eisner, G.; Farias-Eisner, R.; et al. Paraoxonase 2 overexpression inhibits tumor development in a mouse model of ovarian cancer. Cell Death Dis. 2018, 9, 392. [Google Scholar] [CrossRef]
- Muoio, M.G.; Talia, M.; Lappano, R.; Sims, A.H.; Vella, V.; Cirillo, F.; Manzella, L.; Giuliano, M.; Maggiolini, M.; Belfiore, A.; et al. Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers 2021, 13, 621. [Google Scholar] [CrossRef] [PubMed]
- Biello, F.; Platini, F.; D’Avanzo, F.; Cattrini, C.; Mennitto, A.; Genestroni, S.; Martini, V.; Marzullo, P.; Aimaretti, G.; Gennari, A. Insulin/IGF Axis in Breast Cancer: Clinical Evidence and Translational Insights. Biomolecules 2021, 11, 125. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.F.; Corthay, A.; Koutsilieris, M. Aiming for the Insulin-like Growth Factor-1 system in breast cancer therapeutics. Cancer Treat. Rev. 2018, 63, 79–95. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The role of the insulin-like growth factor-1 system in breast cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F. The genetics of breast and ovarian cancer. Br. J. Cancer 1995, 72, 805–812. [Google Scholar] [CrossRef]
- Wooster, R.; Weber, B.L. Breast and ovarian cancer. N. Engl. J. Med. 2003, 348, 2339–2347. [Google Scholar] [CrossRef]
- Simard, J.; Tonin, P.; Durocher, F.; Morgan, K.; Rommens, J.; Gingras, S.; Samson, C.; Leblanc, J.F.; Belanger, C.; Dion, F.; et al. Common origins of BRCA1 mutations in Canadian breast and ovarian cancer families. Nat. Genet. 1994, 8, 392–398. [Google Scholar] [CrossRef]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Kharbanda, S.; Pandey, P.; Schofield, L.; Israels, S.; Roncinske, R.; Yoshida, K.; Bharti, A.; Yuan, Z.M.; Saxena, S.; Weichselbaum, R.; et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6939–6942. [Google Scholar] [CrossRef]
- Garcia-Heredia, J.M.; Diaz-Quintana, A.; Salzano, M.; Orzaez, M.; Perez-Paya, E.; Teixeira, M.; De la Rosa, M.A.; Diaz-Moreno, I. Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti-apoptotic switch. J. Biol. Inorg. Chem. 2011, 16, 1155–1168. [Google Scholar] [CrossRef]
- Polyak, K.; Lee, M.H.; Erdjument-Bromage, H.; Koff, A.; Roberts, J.M.; Tempst, P.; Massague, J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78, 59–66. [Google Scholar] [CrossRef]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Chu, I.; Sun, J.; Arnaout, A.; Kahn, H.; Hanna, W.; Narod, S.; Sun, P.; Tan, C.K.; Hengst, L.; Slingerland, J. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 2007, 128, 281–294. [Google Scholar] [CrossRef]
- Chappuis, P.O.; Kapusta, L.; Begin, L.R.; Wong, N.; Brunet, J.S.; Narod, S.A.; Slingerland, J.; Foulkes, W.D. Germline BRCA1/2 mutations and p27(Kip1) protein levels independently predict outcome after breast cancer. J. Clin. Oncol. 2000, 18, 4045–4052. [Google Scholar] [CrossRef]
- Wilson, C.A.; Ramos, L.; Villasenor, M.R.; Anders, K.H.; Press, M.F.; Clarke, K.; Karlan, B.; Chen, J.J.; Scully, R.; Livingston, D.; et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat. Genet. 1999, 21, 236–240. [Google Scholar] [CrossRef]
- Holt, J.T.; Thompson, M.E.; Szabo, C.; Robinson-Benion, C.; Arteaga, C.L.; King, M.C.; Jensen, R.A. Growth retardation and tumour inhibition by BRCA1. Nat. Genet. 1996, 12, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.R.; Roskelley, C.D. Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. Treat. 2003, 5, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, F.V.; Esteller, M. Mutator pathways unleashed by epigenetic silencing in human cancer. Mutagenesis 2007, 22, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Young, D.; Cerosaletti, K.M.; Desai-Mehta, A.; Spring, K.; Kozlov, S.; Lavin, M.F.; Gatti, R.A.; Concannon, P.; Khanna, K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 2000, 25, 115–119. [Google Scholar] [CrossRef]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Carpenter, P.B.; Elledge, S.J. 53BP1, a mediator of the DNA damage checkpoint. Science 2002, 298, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.A.; Stern, D.F. NFBD1/MDC1, 53BP1 and BRCA1 have both redundant and unique roles in the ATM pathway. Cell Cycle 2008, 7, 3584–3594. [Google Scholar] [CrossRef][Green Version]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef]
- Jiang, M.; Huang, O.; Xie, Z.; Wu, S.; Zhang, X.; Shen, A.; Liu, H.; Chen, X.; Wu, J.; Lou, Y.; et al. A novel long non-coding RNA-ARA: Adriamycin resistance-associated. Biochem. Pharmacol. 2014, 87, 254–283. [Google Scholar] [CrossRef]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef]
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000, 103, 1121–1131. [Google Scholar] [CrossRef]
- Gehring, N.H.; Kunz, J.B.; Neu-Yilik, G.; Breit, S.; Viegas, M.H.; Hentze, M.W.; Kulozik, A.E. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 2005, 20, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mamoor, S. UPF2 is a differentially expressed gene in brain metastatic human breast cancer. OSFPREPRINTS 2021. [Google Scholar] [CrossRef]
- Xu, C.; Chen, H.; Wang, X.; Gao, J.; Che, Y.; Li, Y.; Ding, F.; Luo, A.; Zhang, S.; Liu, Z. S100A14, a member of the EF-hand calcium-binding proteins, is overexpressed in breast cancer and acts as a modulator of HER2 signaling. J. Biol. Chem. 2014, 289, 827–837. [Google Scholar] [CrossRef]
- Tanaka, M.; Ichikawa-Tomikawa, N.; Shishito, N.; Nishiura, K.; Miura, T.; Hozumi, A.; Chiba, H.; Yoshida, S.; Ohtake, T.; Sugino, T. Co-expression of S100A14 and S100A16 correlates with a poor prognosis in human breast cancer and promotes cancer cell invasion. BMC Cancer 2015, 15, 53. [Google Scholar] [CrossRef]
- Ravipati, A.S.; Zhang, L.; Koyyalamudi, S.R.; Jeong, S.C.; Reddy, N.; Bartlett, J.; Smith, P.T.; Shanmugam, K.; Munch, G.; Wu, M.J.; et al. Antioxidant and anti-inflammatory activities of selected Chinese medicinal plants and their relation with antioxidant content. BMC Complement. Altern. Med. 2012, 12, 173. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M., Jr.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H]thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Farag, O.M.; Abd-Elsalam, R.M.; Ogaly, H.A.; Ali, S.E.; El Badawy, S.A.; Alsherbiny, M.A.; Li, C.G.; Ahmed, K.A. Metabolomic Profiling and Neuroprotective Effects of Purslane Seeds Extract Against Acrylamide Toxicity in Rat’s Brain. Neurochem. Res. 2021, 46, 819–842. [Google Scholar] [CrossRef] [PubMed]
- Azouz, A.A.; Ali, S.E.; Abd-Elsalam, R.M.; Emam, S.R.; Galal, M.K.; Elmosalamy, S.H.; Alsherbiny, M.A.; Hassan, B.B.; Li, C.G.; El Badawy, S.A. Modulation of steroidogenesis by Actaea racemosa and vitamin C combination, in letrozole induced polycystic ovarian syndrome rat model: Promising activity without the risk of hepatic adverse effect. Chin. Med. 2021, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Chou, T.; Martin, N. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values; ComboSyn: Paramus, NJ, USA, 2005. [Google Scholar]
- Chou, T.-C.; Martin, N. The mass-action law-based new computer software, CompuSyn, for automated simulation of synergism and antagonism in drug combination studies. Exp. Mol. Ther. 2007, 67, 637. [Google Scholar]
- Zagidullin, B.; Aldahdooh, J.; Zheng, S.; Wang, W.; Wang, Y.; Saad, J.; Malyutina, A.; Jafari, M.; Tanoli, Z.; Pessia, A.; et al. DrugComb: An integrative cancer drug combination data portal. Nucleic Acids Res. 2019, 47, W43–W51. [Google Scholar] [CrossRef] [PubMed]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, gkw937. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g: Profiler—A web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Cavill, R.; Ebbels, T.M.; Herwig, R.; Keun, H.C. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics 2011, 27, 2917–2918. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combo ID | Highest Dose (µg mL−1) | IC50 ± SD | Ratio (w/w) | CI Values at: | CSS | S | ZIP | BLISS | LOEWE | HSA | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AP-1 | DOX | (µg mL−1) | ED50 | ED75 | ED90 | ED95 | ED97 | ||||||||
| Propolis | 62.95 ± 9.28 | ||||||||||||||
| Doxorubicin | 0.24 ± 0.03 | ||||||||||||||
| PDOX19 | 20 | 0.52 | 62.95 ± 9.28 | 100:2.61 | 1.67 | 1.3 | 1.03 | 0.88 | 0.79 | 75.38 | 44.86 | −10.43 | −12.09 | −5.89 | −2.22 |
| PDOX28 | 40 | 0.46 | 11.25 ± 4.59 | 100:1.16 | 2.5 | 2.23 | 2.05 | 1.95 | 1.9 | 72.4 | 41.07 | −14.33 | −16.63 | −10.69 | −6.43 |
| PDOX37 | 60 | 0.41 | 28.10 ± 4.32 | 100:0.68 | 2.93 | 2.75 | 2.69 | 2.7 | 2.72 | 73.35 | 40.57 | −15.11 | −19.6 | −13.43 | −8.88 |
| PDOX46 | 80 | 0.35 | 40.55 ± 4.22 | 100:0.43 | 1.96 | 2.72 | 2.75 | 3.2 | 3.59 | 77.25 | 43.88 | −13.51 | −16.12 | −9.71 | −5.03 |
| PDOX55 | 100 | 0.29 | 49.03 ± 16.99 | 100:0.29 | 0.77 | 0.57 | 0.44 | 0.38 | 0.35 | 84.41 | 51.85 | −6.69 | −8.52 | −2.82 | 1.78 |
| PDOX64 | 120 | 0.23 | 36.93 ± 15.24 | 100:0.19 | 0.75 | 0.64 | 0.58 | 0.55 | 0.54 | 84.81 | 51.87 | −8.05 | −9.62 | −3.52 | 1.55 |
| PDOX73 | 140 | 0.17 | 41.88 ± 18.87 | 100:0.12 | 0.98 | 0.88 | 0.83 | 0.82 | 0.82 | 82.35 | 49.75 | −9.64 | −11.25 | −4.93 | −0.21 |
| PDOX82 | 160 | 0.12 | 54.97 ± 7.04 | 100:0.07 | 0.98 | 0.82 | 0.71 | 0.66 | 0.63 | 83.27 | 51.94 | −6.66 | −8.04 | −1.59 | 2.49 |
| PDOX91 | 180 | 0.06 | 58.48 ± 3.10 | 100:0.03 | 1.24 | 1.34 | 1.4 | 1.47 | 1.64 | 80.54 | 49.78 | −6.05 | −7.95 | −1.31 | 1.98 |
| CI to DC | 80.74 | 55.69 | −9.05 | −10.59 | −6.46 | −2.17 | |||||||||
| Checkerboard | 65.39 | 32.65 | 4.5 | 4.49 | 0.96 | 4.28 | |||||||||
| Selected Dose * | 0.11 (94% inhibition) # | ||||||||||||||
| ➢ CI to DC | 22.55 | 16.65 | 9.95 | 40.08 | |||||||||||
| ➢ Checkerboard design | −1.27 | 0.25 | 4.52 | 10.05 | |||||||||||
| Uniprot Accession | Gene | Fold Change | Description |
|---|---|---|---|
| Upregulated Proteins | |||
| H0YMP8 | ATP8B4 | 15.05 | Phospholipid-transporting ATPase |
| A0A0A0MQS9;Q16363 | LAMA4 | 4.60 | Laminin subunit alpha-4 |
| Q9HAU5 | UPF2 | 3.22 | Regulator of nonsense transcripts 2 |
| Q9Y216 | SEC23A | 2.53 | Myotubularin-related protein 7 |
| P38398 | BRCA1 | 1.77 | Breast cancer type 1 susceptibility protein |
| E9PDF1 | SH3TC2 | 1.74 | SH3 domain and tetratricopeptide repeats 2 |
| Downregulated Proteins | |||
| Q9HCY8 | S100A14 | 18.96 | Protein S100-A14 |
| H3BUX2 | CYB5B | 5.09 | Cytochrome b5 type B |
| Q8N5U6 | RNF10 | 3.73 | RING finger protein 10 |
| H7C2B5 | RIF1 | 3.58 | Telomere-associated protein RIF1 |
| Q8N9V7 | TOPAZ1 | 2.86 | Protein TOPAZ1 |
| Q14676 | MDC1 | 2.63 | Mediator of DNA damage checkpoint protein 1 |
| Q12873 | CHD3 | 2.59 | Chromodomain-helicase-DNA-binding protein 3 |
| Q9NSB2 | KRT84 | 2.49 | Keratin_ type II cuticular Hb4 |
| A0A087WWY9; Q8TB24 | RIN3 | 2.35 | Ras and Rab interactor 3 |
| P62913 | RPL11 | 2.32 | 60S ribosomal protein L11 |
| D6RCP5 | ARHGAP24 | 2.08 | Rho GTPase-activating protein 24 |
| Q9NYL9 | TMOD3 | 1.88 | Tropomodulin-3 |
| Q9Y2H9 | MAST1 | 1.83 | Microtubule-associated serine/threonine-protein kinase 1 |
| Q6ZRV2 | FAM83H | 1.82 | Protein FAM83H |
| P14854 | COX6B1 | 1.77 | Cytochrome c oxidase subunit 6B1 |
| Platform. | Process/Pathway | p-Value | FDR | Present Entities |
|---|---|---|---|---|
| Reactome | TP53 Regulates Transcription of DNA Repair Genes | 1.78 × 10−8 | 3.29 × 10−06 | MDC1; BRCA1 |
| Transcriptional Regulation by TP53 | 2.72 × 10−06 | 2.50 × 10−04 | MDC1; BRCA1; CHD3; COX6B1 | |
| NHEJ | 1.84 × 10−04 | 1.12 × 10−02 | MDC1, RIF1, BRCA1 | |
| G2/M DNA damage checkpoint | 1.07 × 10−02 | 1.07 × 10−01 | MDC1; BRCA1 | |
| Cell Cycle Checkpoints | 1.64 × 10−02 | 1.47 × 10−01 | MDC1; MAST1; BRCA1 | |
| Nonsense-Mediated Decay (NMD) | 2.38 × 10−02 | 1.67 × 10−01 | UPF2; RPL11 | |
| STRING | DSBs repair via NHEJ | NA | 0.047 | MDC1, RIF1, BRCA1 |
| BRCT, breast cancer carboxy-terminal domain | NA | 0.022 | MDC1, BRCA1 | |
| g:Profiler | NHEJ | 1.30 × 10−2 | NA | MDC1, RIF1, BRCA1 |
| DNA IR-DSBs and cellular response via ATM | 7.00 × 10−3 | NA | MDC1, RIF1, BRCA1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsherbiny, M.A.; Bhuyan, D.J.; Radwan, I.; Chang, D.; Li, C.-G. Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells. Int. J. Mol. Sci. 2021, 22, 7840. https://doi.org/10.3390/ijms22157840
Alsherbiny MA, Bhuyan DJ, Radwan I, Chang D, Li C-G. Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells. International Journal of Molecular Sciences. 2021; 22(15):7840. https://doi.org/10.3390/ijms22157840
Chicago/Turabian StyleAlsherbiny, Muhammad A., Deep J. Bhuyan, Ibrahim Radwan, Dennis Chang, and Chun-Guang Li. 2021. "Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells" International Journal of Molecular Sciences 22, no. 15: 7840. https://doi.org/10.3390/ijms22157840
APA StyleAlsherbiny, M. A., Bhuyan, D. J., Radwan, I., Chang, D., & Li, C.-G. (2021). Metabolomic Identification of Anticancer Metabolites of Australian Propolis and Proteomic Elucidation of Its Synergistic Mechanisms with Doxorubicin in the MCF7 Cells. International Journal of Molecular Sciences, 22(15), 7840. https://doi.org/10.3390/ijms22157840