
P62 Links the Autophagy Pathway and the Ubiquitin–Proteasome System in Endothelial Cells during Atherosclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
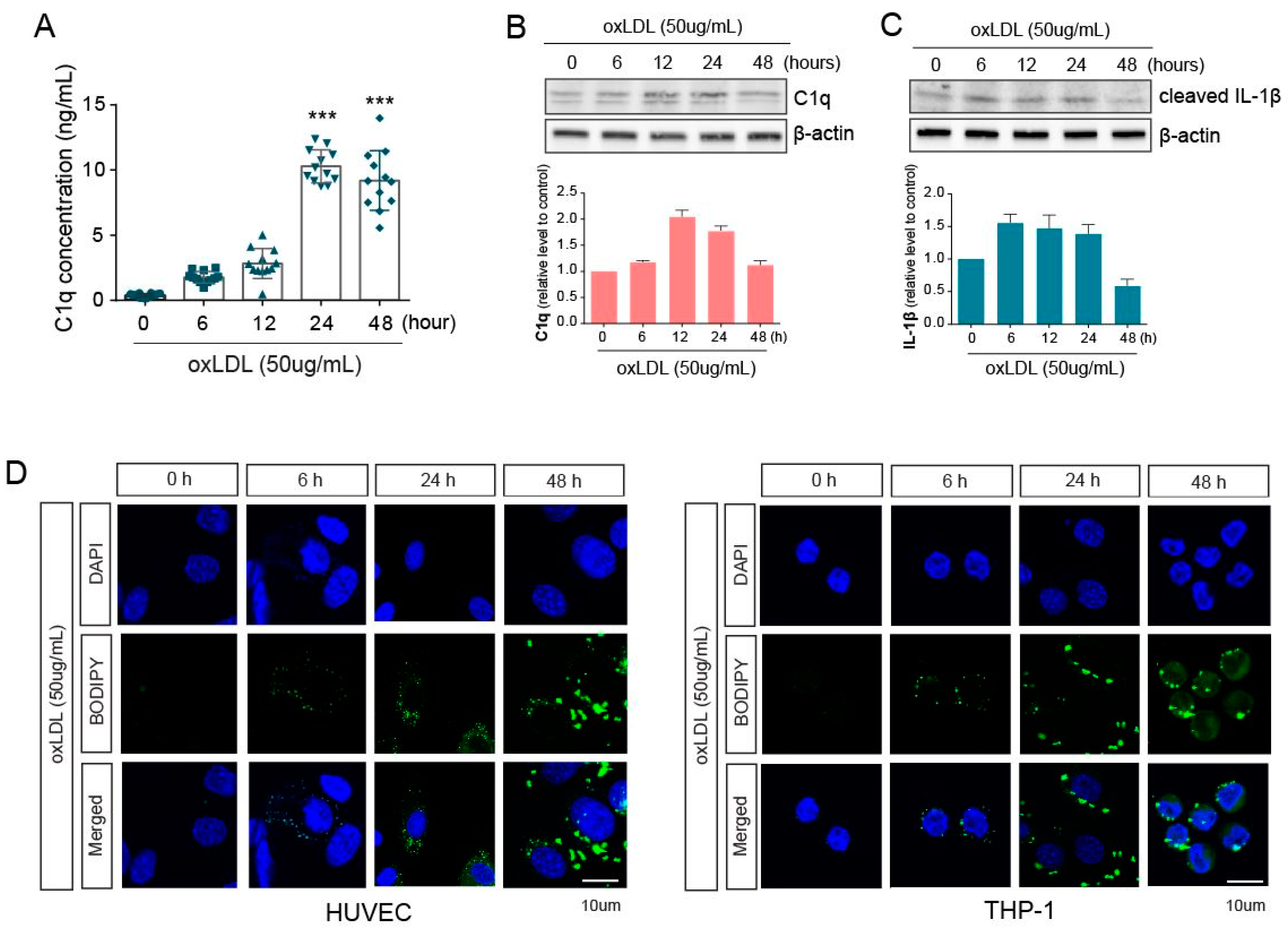
2.1. Innate Immune Responses and Inflammation Are Induced by oxLDL in Co-Cultured HUVEC and THP-1 Cells
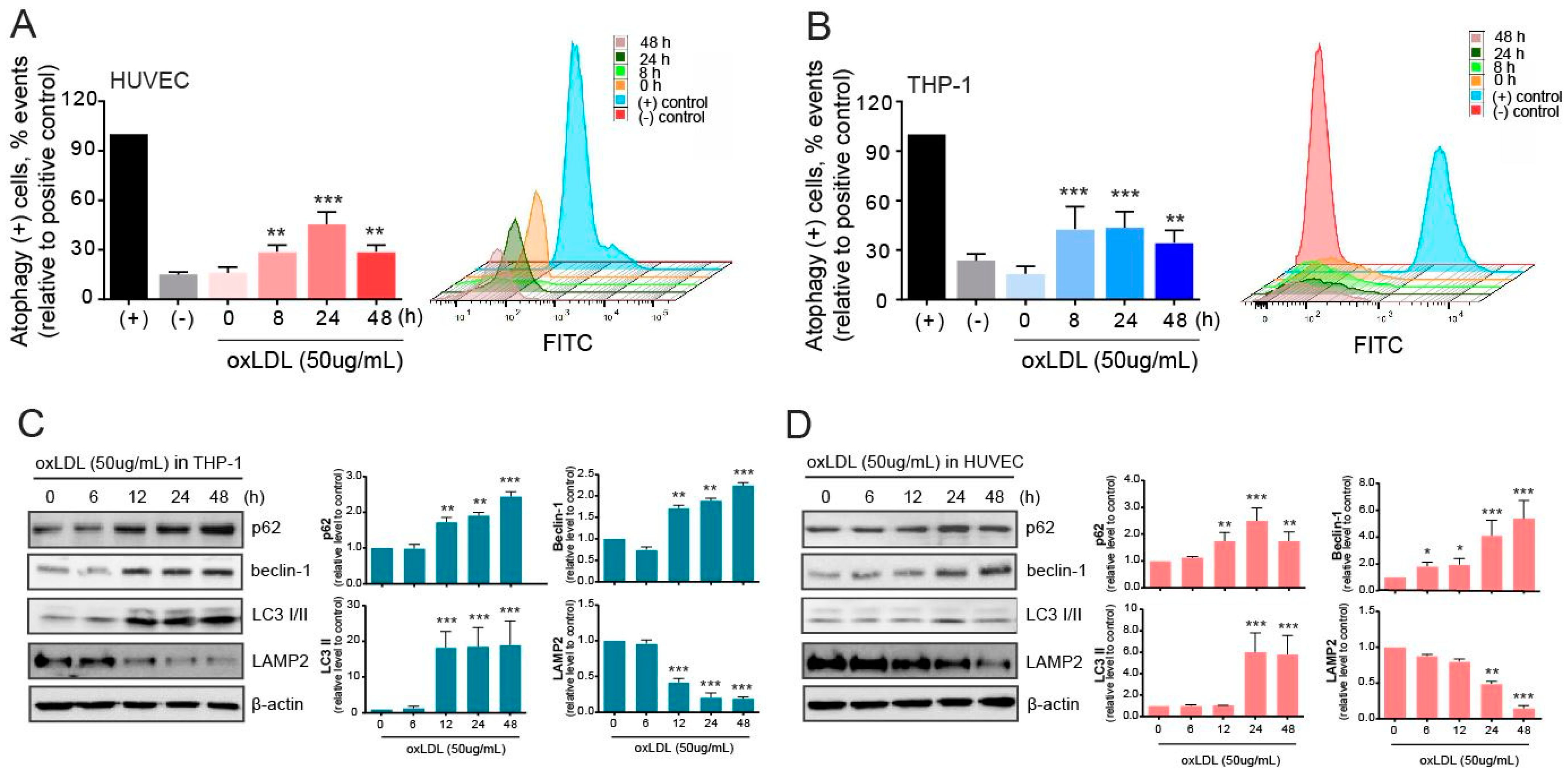
2.2. Autophagy Is Induced but Malfunctions in Atherosclerotic Conditions
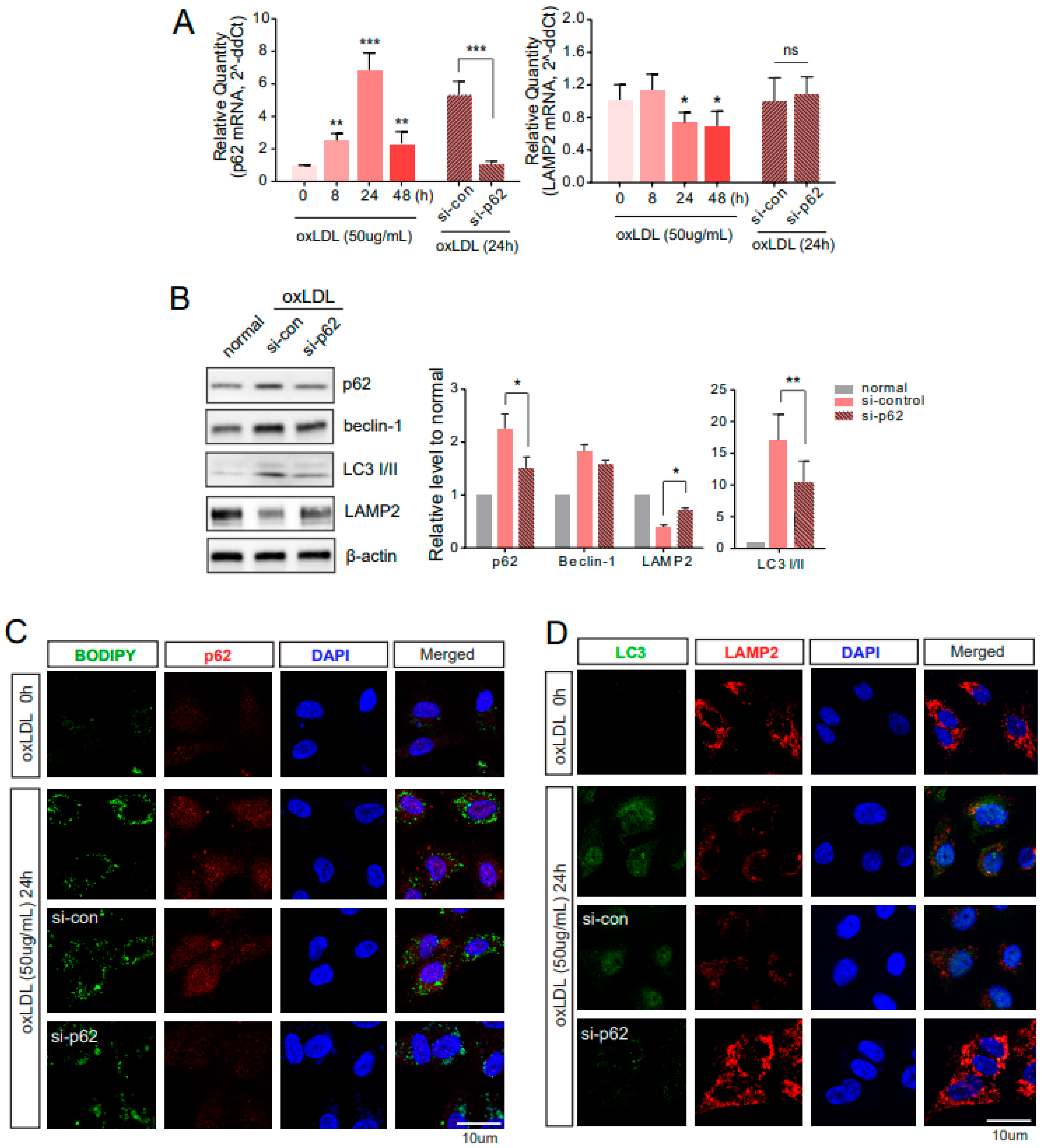
2.3. Downregulated p62 Induces Alternative Degrading Systems
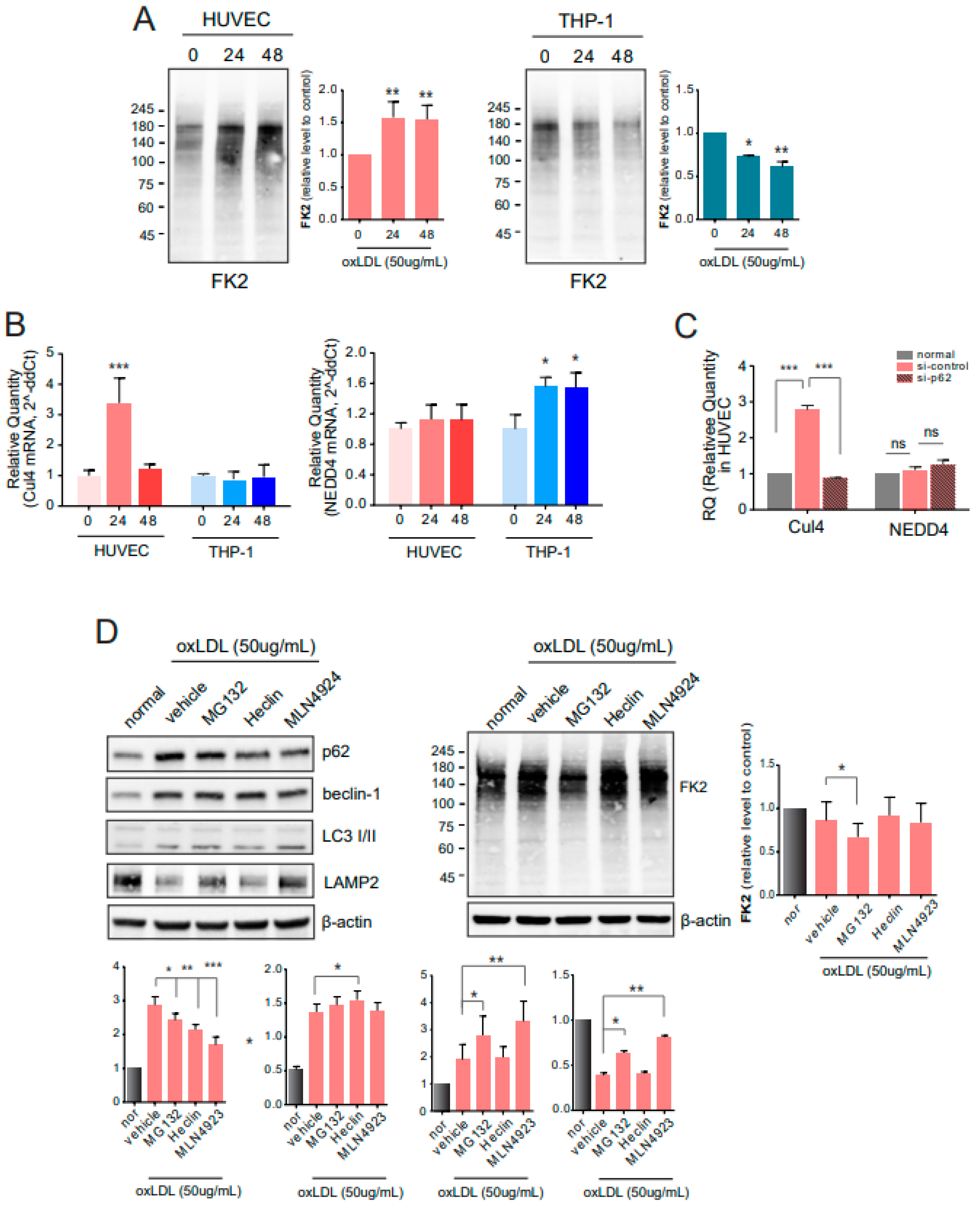
2.4. E3 Ligases Are Involved in Autophagy under Atherosclerotic Conditions
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drugs
4.2. RNA Silencing
4.3. Quantitative Real-Time PCR (qPCR) Analysis
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Autophagy Assay
4.6. BODIPY Staining
4.7. Immunocytochemistry
4.8. Western Blot Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Invest. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, C.C.; Gao, P.J. Role of Complement-Related Inflammation and Vascular Dysfunction in Hypertension. Hypertension 2019, 73, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Matsui, T. Anti-atherothrombogenic properties of PEDF. Curr. Mol. Med. 2010, 10, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Ramos, C.; Machado-Oliveira, G.; Vieira, O.V. Lysosome (Dys)function in Atherosclerosis-A Big Weight on the Shoulders of a Small Organelle. Front. Cell Dev. Biol. 2021, 9, 658995. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxidative Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef] [PubMed]
- Stoneman, V.; Braganza, D.; Figg, N.; Mercer, J.; Lang, R.; Goddard, M.; Bennett, M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 2007, 100, 884–893. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, L.; Blanchard, S.; Rama, S.; Beier, V.; Kaufmann, A.; Wollert, T. TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nat. Commun 2020, 11, 2993. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Razani, B. Self-eating in the plaque: What macrophage autophagy reveals about atherosclerosis. Trends Endocrinol. Metab. 2014, 25, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaClair, K.D.; Donde, A.; Ling, J.P.; Jeong, Y.H.; Chhabra, R.; Martin, L.J.; Wong, P.C. Depletion of TDP-43 decreases fibril and plaque beta-amyloid and exacerbates neurodegeneration in an Alzheimer’s mouse model. Acta Neuropathol. 2016, 132, 859–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.S.; Cheng, W.C.; Chen, C.Y.; Wu, M.C.; Wang, Y.C.; Tseng, Y.H.; Chuang, T.J.; Shen, C.J. Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yoshikawa, Y.; Kobayashi, T.; Mimuro, H.; Fukumatsu, M.; Kiga, K.; Piao, Z.; Ashida, H.; Yoshida, M.; Kakuta, S.; et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell. Host Microbe 2011, 9, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Terpstra, E.J. Ubiquitin receptors and protein quality control. J. Mol. Cell Cardiol. 2013, 55, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Li, S.; Zhao, Y.; Ma, X.; Zhang, K.; He, X.; Wang, Z. Interaction domains of p62: A bridge between p62 and selective autophagy. DNA Cell Biol. 2013, 32, 220–227. [Google Scholar] [CrossRef]
- Shin, J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharm. Res. 1998, 21, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Figueiredo-Pereira, M.E. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: Association with sequestosome 1/p62. J. Biol. Chem. 2011, 286, 22426–22440. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; La Spada, A.R. Autophagy in polyglutamine disease: Imposing order on disorder or contributing to the chaos? Mol. Cell Neurosci. 2015, 66, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A.; Keir, M.; et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; De Meyer, G.R. Autophagy in atherosclerosis: A cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009, 104, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.; Choi, S.H. ASK1 Mediates Apoptosis and Autophagy during oxLDL-CD36 Signaling in Senescent Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2840437. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.F.; Kurdi, A.; Timmermans, J.P.; De Meyer, G.R.Y.; Martinet, W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis 2016, 251, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Du, Y.; Zhao, Z.; Shi, L.; Sun, L.; Huang, G.; Ye, B.; Li, C.; Dai, Z.; et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J. 2013, 32, 2685–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Dan, K.; Goto, F.; Tshuchihashi, N.; Nomura, Y.; Fujioka, M.; Kanzaki, S.; Ogawa, K. The autophagy pathway maintained signaling crosstalk with the Keap1-Nrf2 system through p62 in auditory cells under oxidative stress. Cell Signal. 2015, 27, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Jung, H.Y.; Park, K.Y.; Kim, S.K. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology 2014, 53, 1043–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Lee, W.; Cho, K. P62 Links the Autophagy Pathway and the Ubiquitin–Proteasome System in Endothelial Cells during Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 7791. https://doi.org/10.3390/ijms22157791
Kim S, Lee W, Cho K. P62 Links the Autophagy Pathway and the Ubiquitin–Proteasome System in Endothelial Cells during Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(15):7791. https://doi.org/10.3390/ijms22157791
Chicago/Turabian StyleKim, SeJeong, WoongJin Lee, and KyoungJoo Cho. 2021. "P62 Links the Autophagy Pathway and the Ubiquitin–Proteasome System in Endothelial Cells during Atherosclerosis" International Journal of Molecular Sciences 22, no. 15: 7791. https://doi.org/10.3390/ijms22157791
APA StyleKim, S., Lee, W., & Cho, K. (2021). P62 Links the Autophagy Pathway and the Ubiquitin–Proteasome System in Endothelial Cells during Atherosclerosis. International Journal of Molecular Sciences, 22(15), 7791. https://doi.org/10.3390/ijms22157791