
Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (NETs) Differ in Obesity and Systemic Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
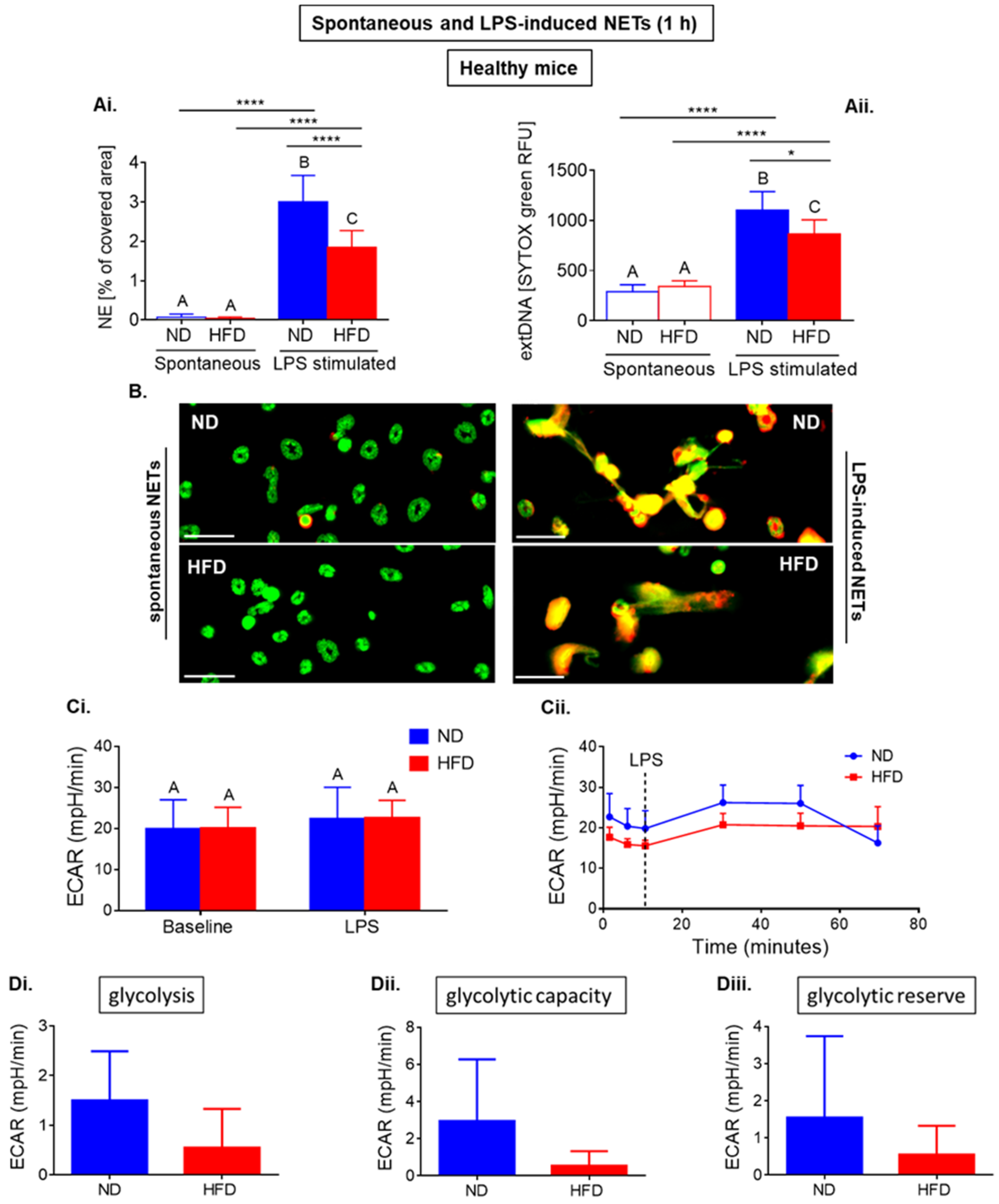
2.1. Neutrophils of Healthy ND and HFD Mice Depend on Glycolysis in Basal Conditions with Higher Tendency to Glycolytic Functions in Lean Mice
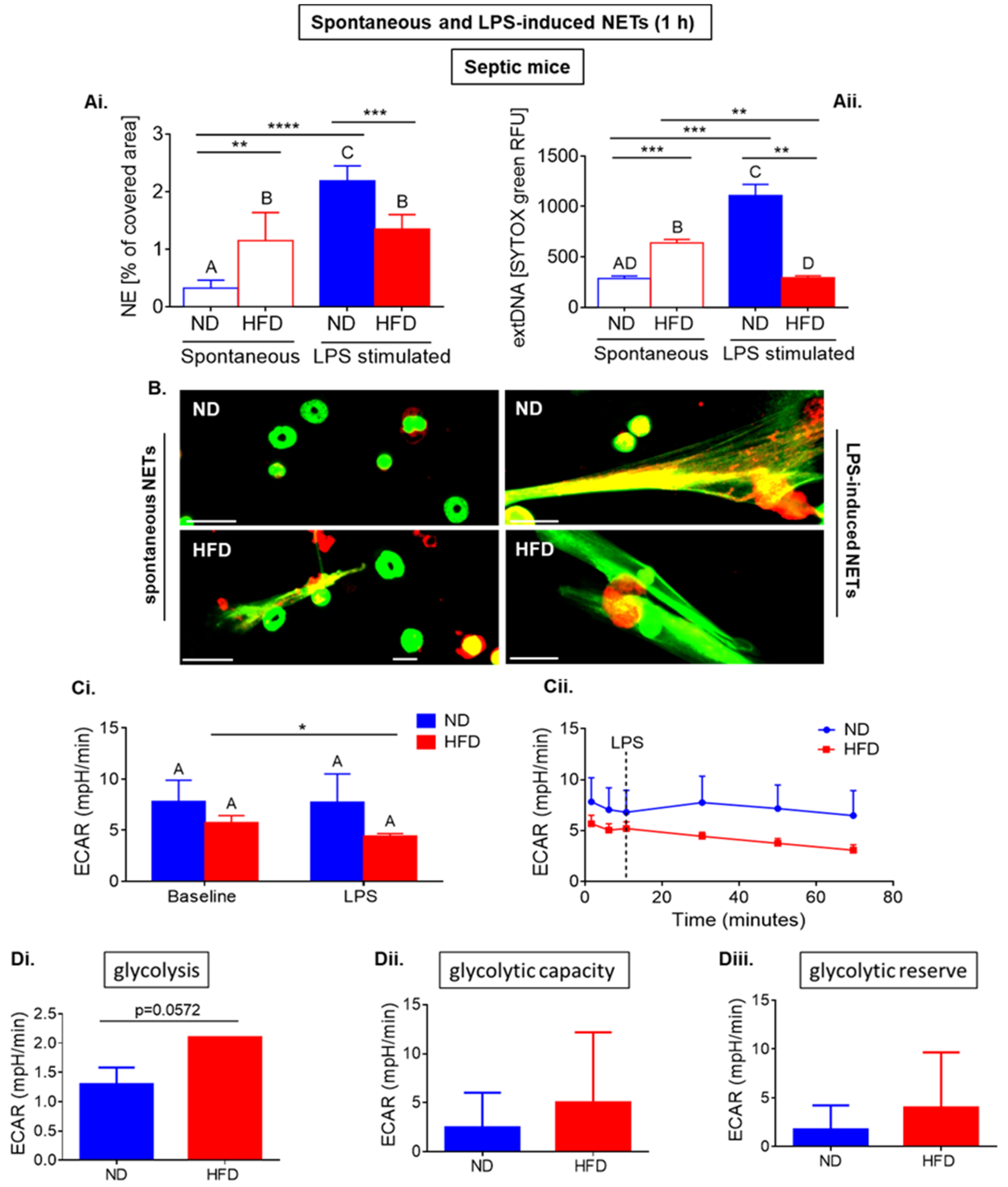
2.2. Neutrophils of Septic Obese Mice Are Highly Glycolytic but Exhibit “Exhausted Phenotype” after Ex Vivo LPS Stimulation
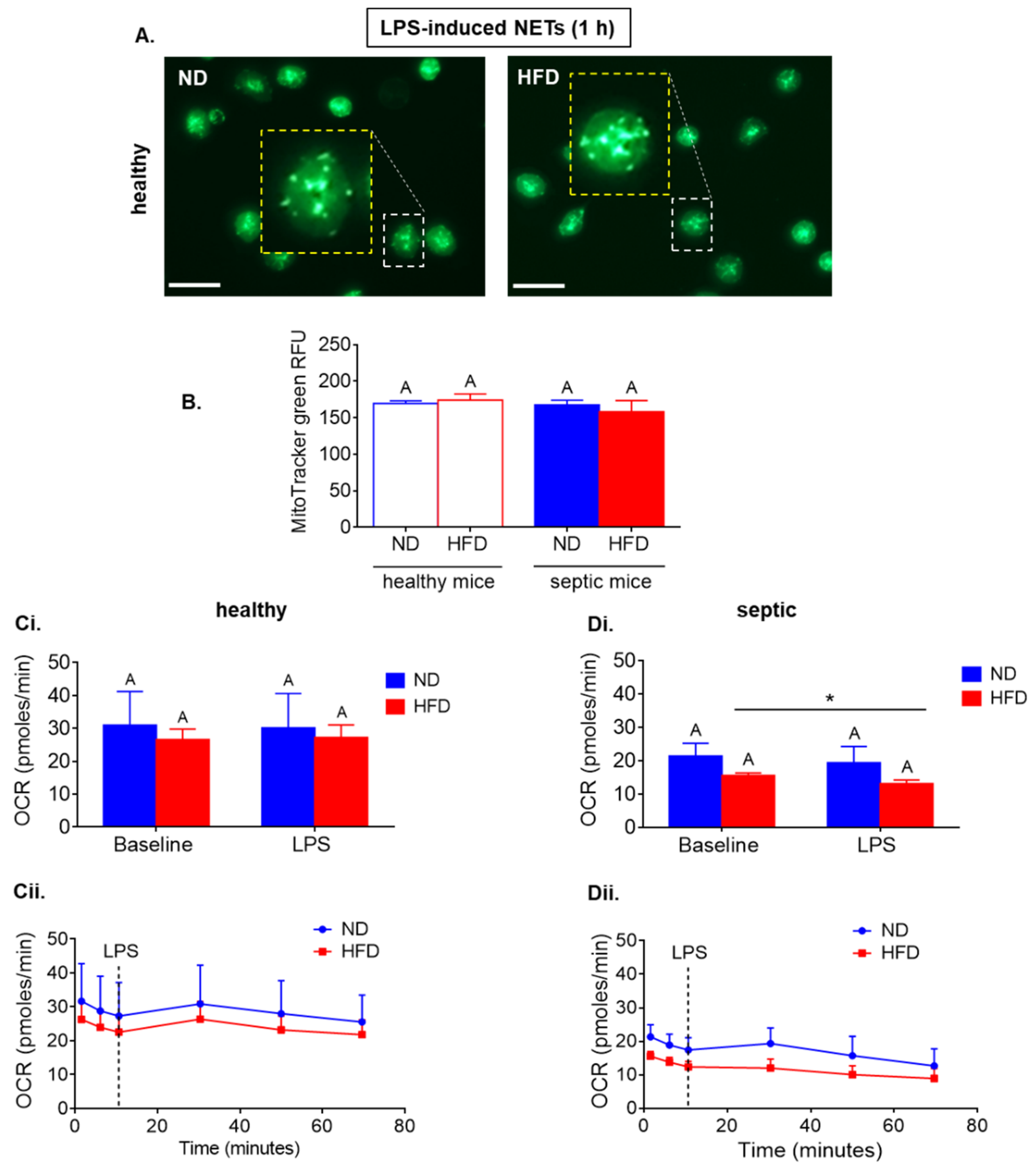
2.3. Neutrophils of Septic, but Not Healthy, Obese Mice Display Reduced Respiration
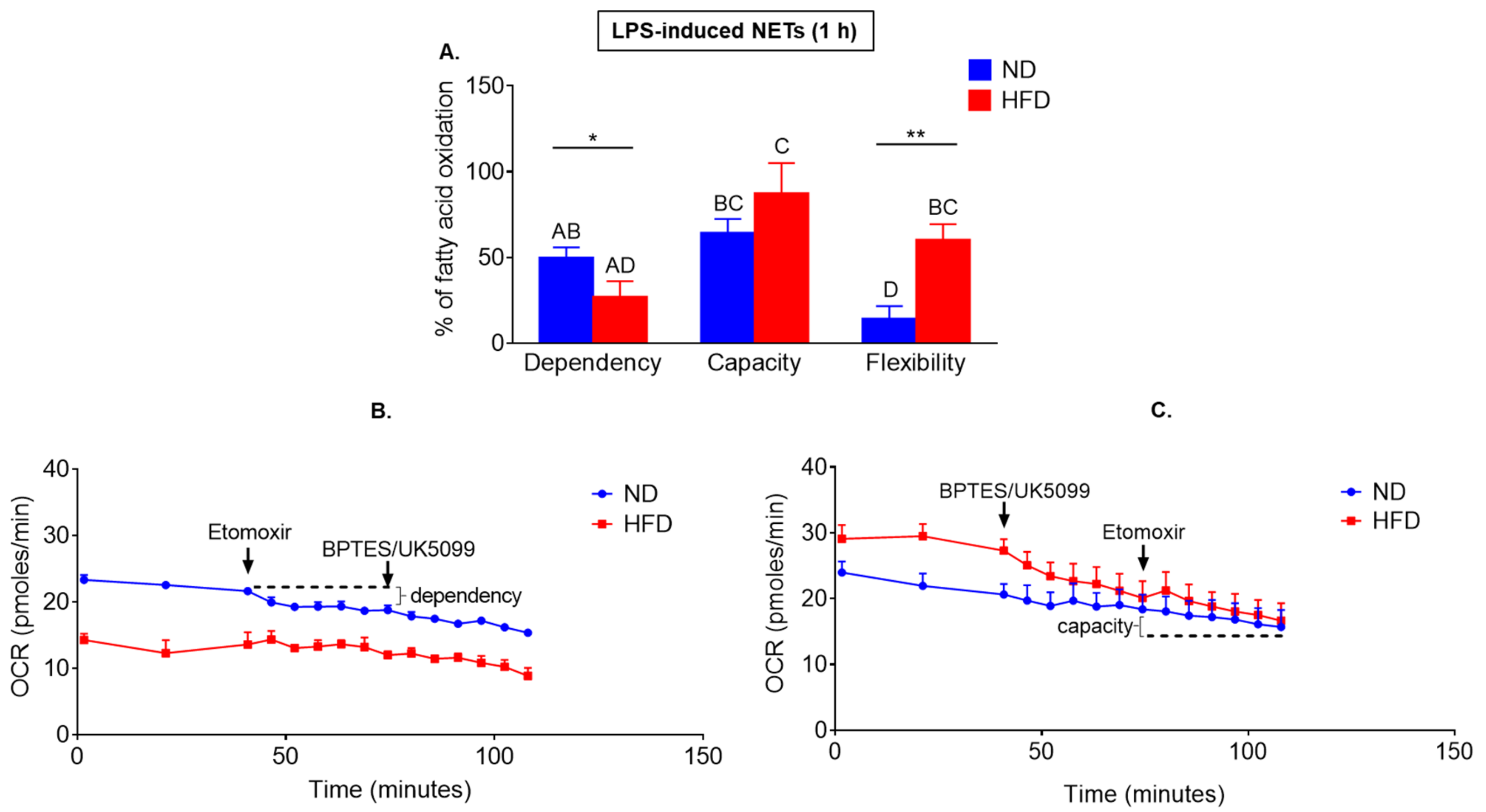
2.4. Neutrophils of Obese Mice Are More Prone to Switch Their Metabolism between Glucose/Glutamine and Fatty Acid Oxidation Pathways
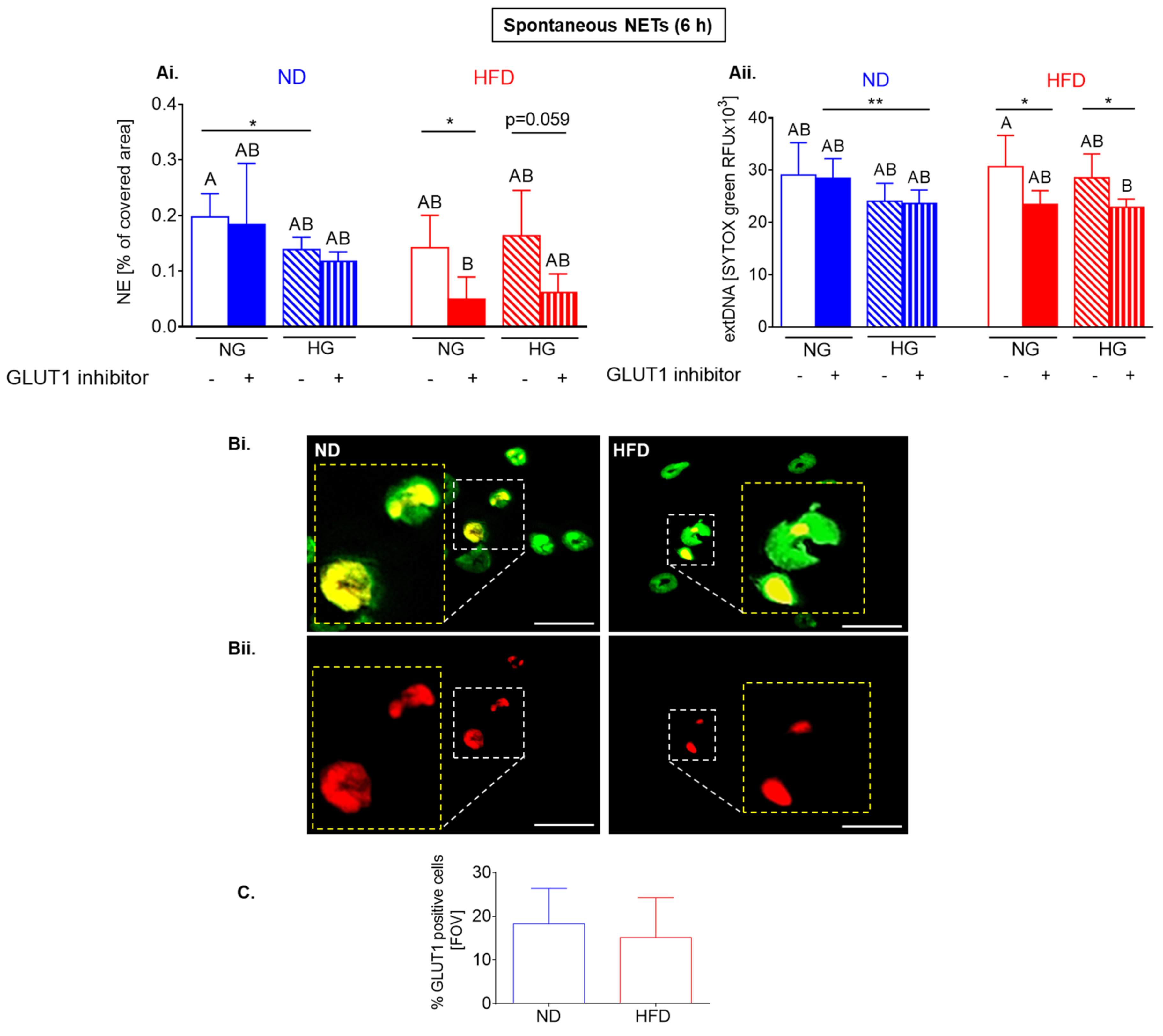
2.5. Upon Longer Incubation, Spontaneously Released NETs Differentially Depend on Glycolysis in HFD and ND Mice
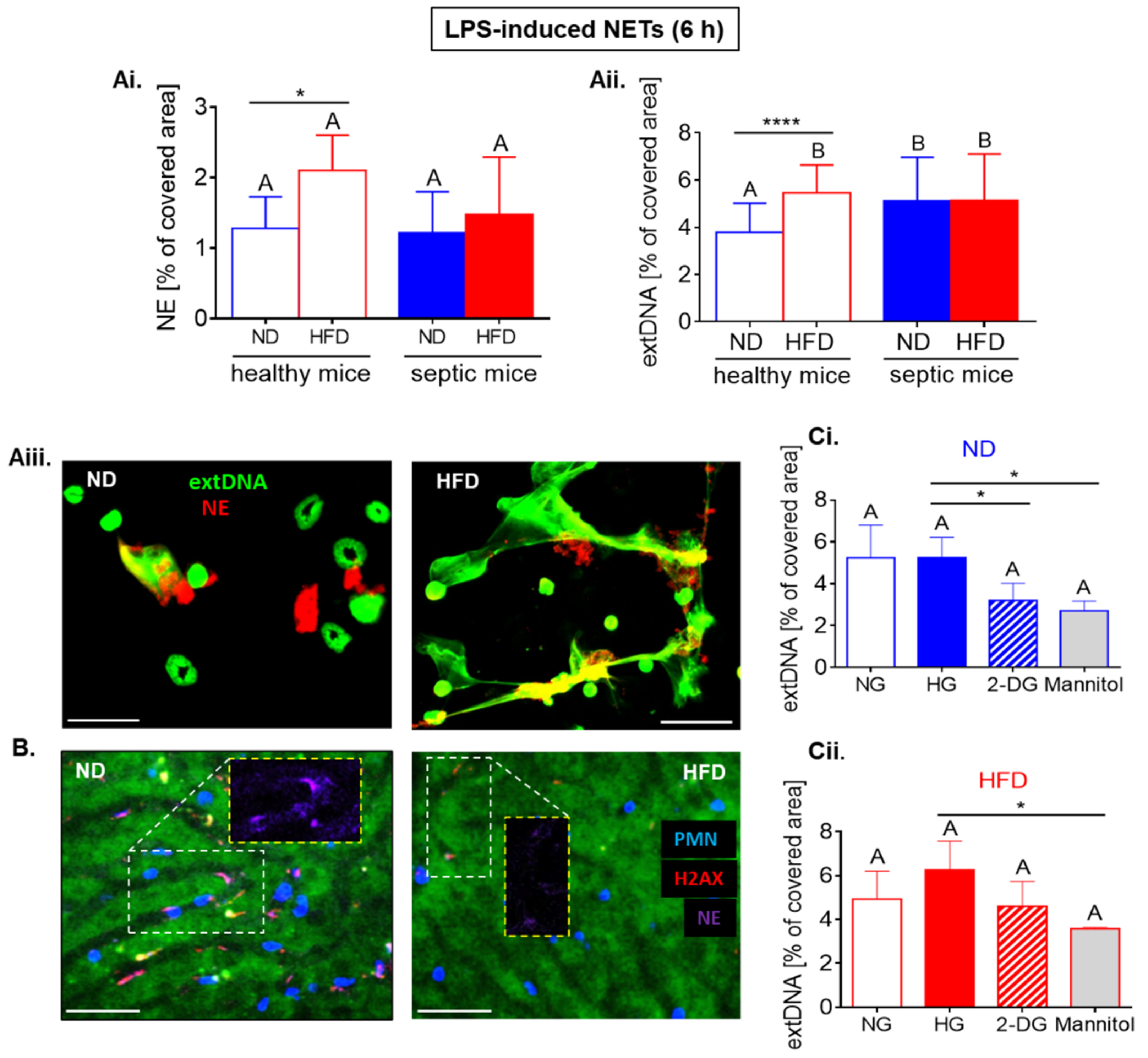
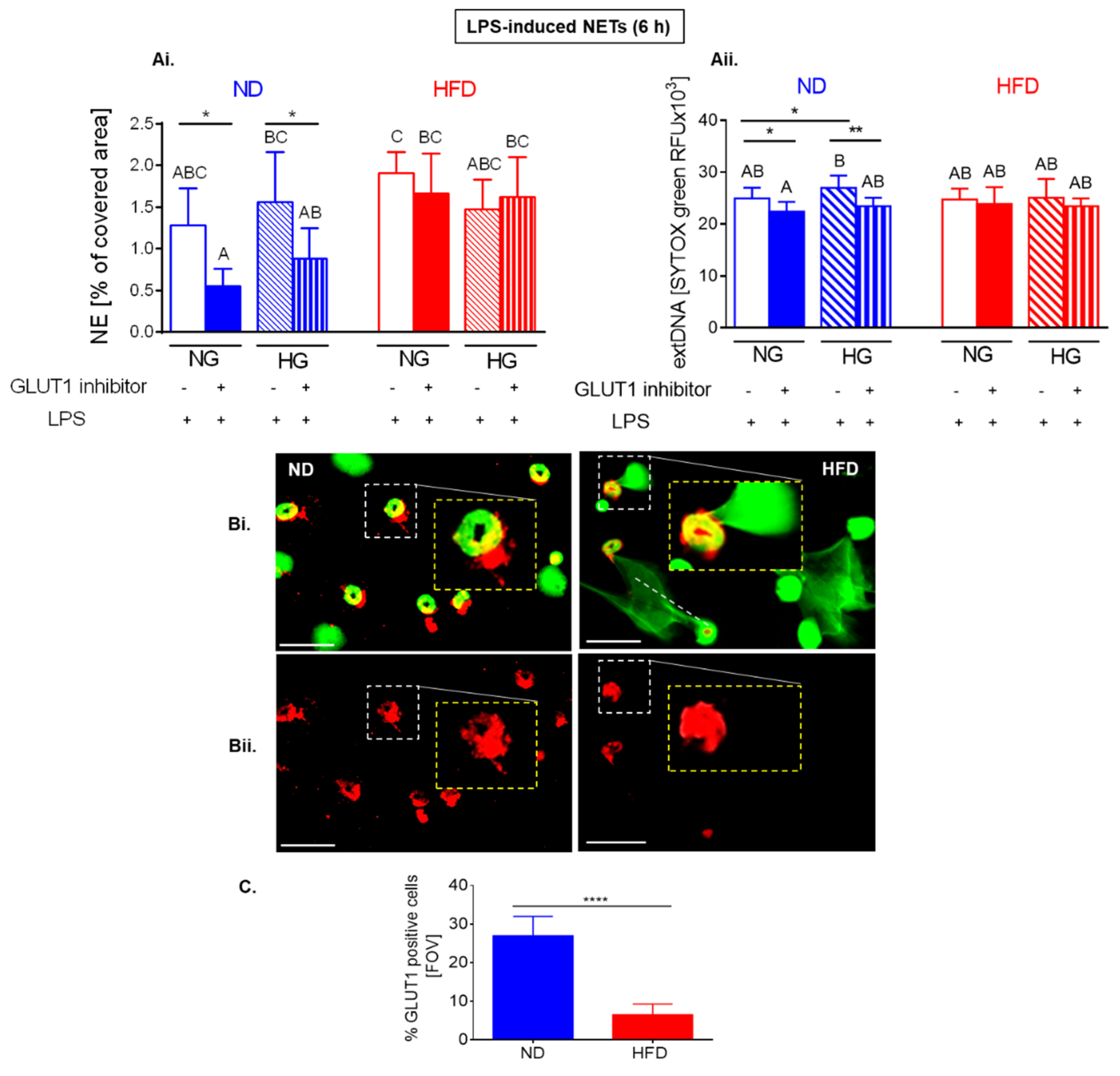
2.6. Upon Longer LPS Stimulation Neutrophils of HFD Mice Release NETs Independently of Glycolysis
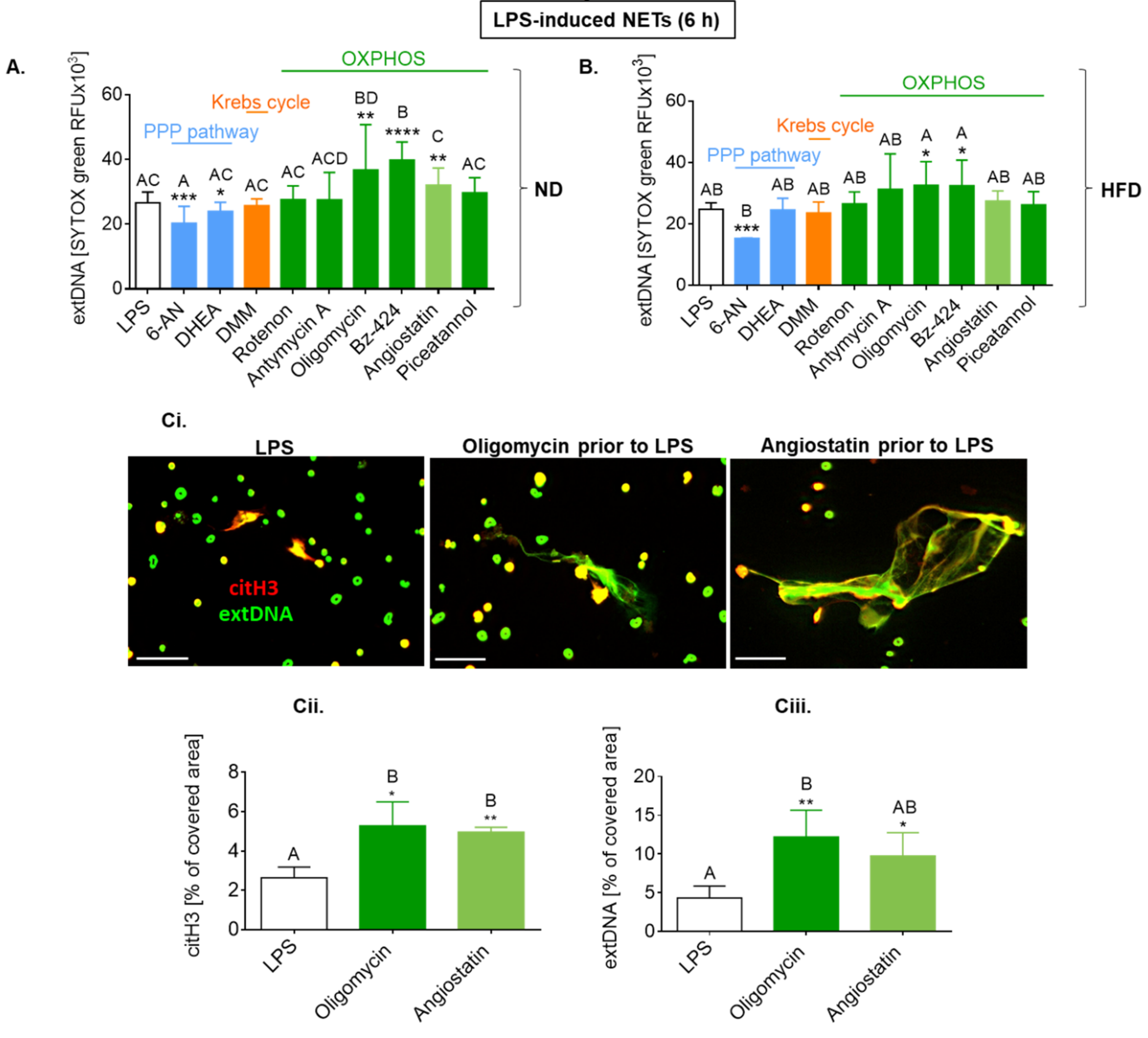
2.7. Upon Longer LPS Stimulation, NET Release Also Depends on PPP but Not Citrate Cycle and Inversely on Oxidative Phosphorylation
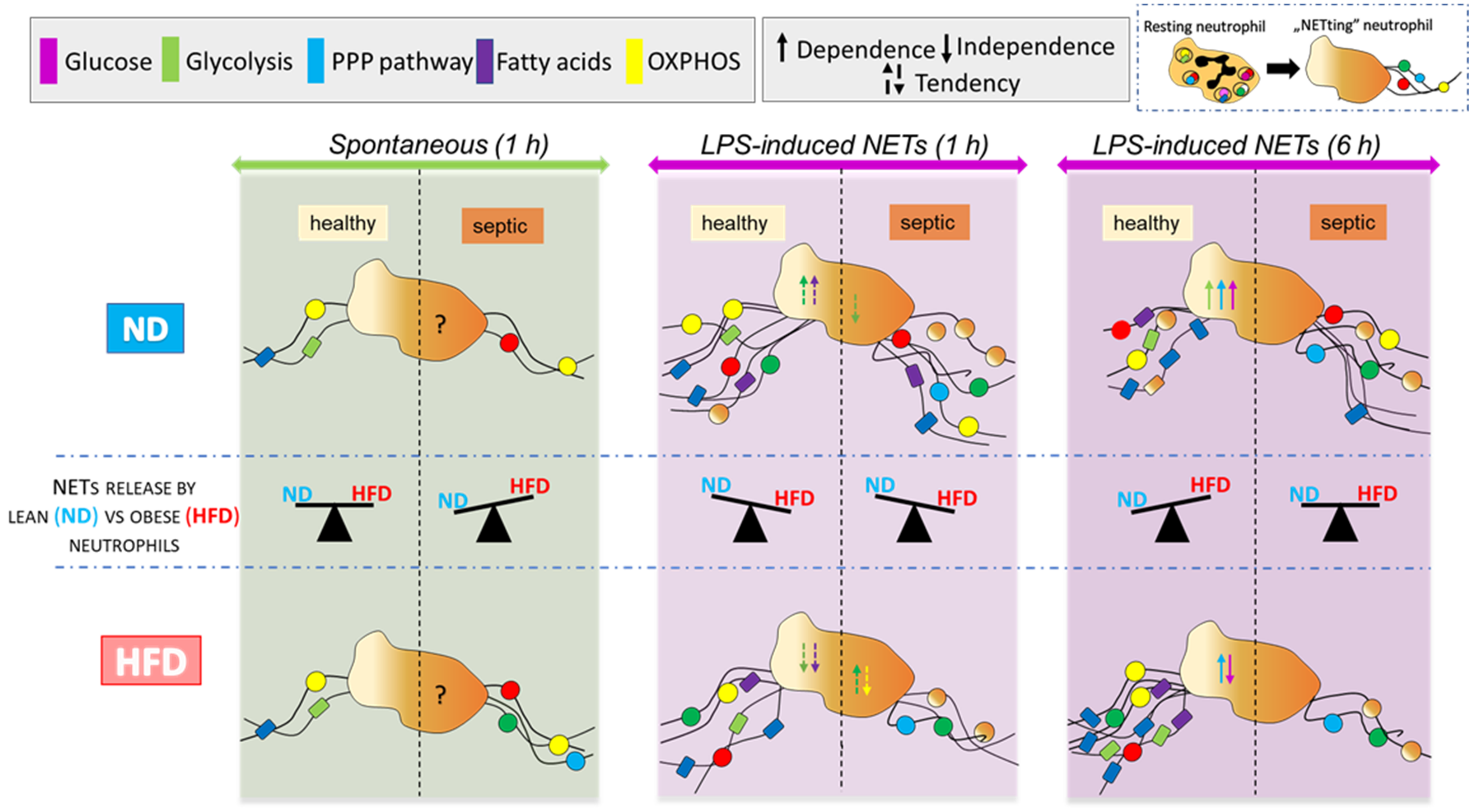
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Induction of Systemic Inflammation/Endotoxemia
4.3. Neutrophil Isolation
4.4. Metabolic Pathway Inhibitors
4.5. Quantification of NETs by Fluorescence Spectrophotometry
4.6. Quantification and Visualization of NETs by Fluorescence/Confocal Microscopy
4.7. Glucose Transporter (GLUT1) Immunostaining
4.8. Mitochondrial Tracker Green and Janus Green B Staining
4.9. Seahorse Analysis
4.9.1. Seahorse XF Measurement of ECAR and OCR
4.9.2. Seahorse XF Glycolysis Stress Test Assay
4.9.3. Seahorse XF Mito Fuel Flex Test
4.10. Staining of NETs In Vivo with Intravital Microscopy
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pillon, N.J.; Loos, R.J.F.; Marshall, S.M.; Zierath, J.R. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef] [PubMed]
- Controlling the Global Obesity Epidemic. Available online: https://www.who.int/activities/controlling-the-global-obesity-epidemic (accessed on 4 May 2021).
- Prescott, H.C.; Chang, V.W.; O’Brien, J.M., Jr.; Langa, K.M.; Iwashyna, T.J. Obesity and One-Year Outcomes in Older Americans with Severe Sepsis. Crit. Care Med. 2014, 23, 1–7. [Google Scholar] [CrossRef]
- Khan, M.; Patrick, A.L.; Fox-Robichaud, A.E. Development of a murine model of early sepsis in diet-induced obesity. Biomed. Res. Int. 2014, 2014, 719853. [Google Scholar] [CrossRef]
- Siegl, D.; Annecke, T.; Johnson, B.L.; Schlag, C.; Martignoni, A.; Huber, N.; Conzen, P.; Caldwell, C.C.; Tschöp, J. Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 2014, 121, 98–114. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Nowell, M.; Lahni, P.; O’Connor, M.; Hake, P.W.; Zingarelli, B. Short-Term High Fat Feeding Increases Organ Injury and Mortality After Polymicrobial Sepsis. Obesity 2012, 176, 100–106. [Google Scholar] [CrossRef]
- Stranberg, L.; Verdrengh, M.; Enge, M.; Andersson, N.; Amu, S.; Önnheim, K.; Benrick, A.; Brisslert, M.; Bylund, J.; Bokarewa, M.; et al. Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS ONE 2009, 4, e7605. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Nowell, M.; Lahni, P.; Shen, H.; Shanmukhappa, S.K.; Zingarelli, B. Obesity enhances sepsis induced liver inflammation and injury in mice. Obesity 2016, 176, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–20. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Santocki, M.; Kolaczkowska, E. On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little. Cells 2020, 9, 2079. [Google Scholar] [CrossRef]
- Cichon, I.; Ortmann, W.; Santocki, M.; Opydo-Chanek, M.; Kolaczkowska, E. Scrutinizing Mechanisms of the ‘Obesity Paradox in Sepsis’: Obesity Is Accompanied by Diminished Formation of Neutrophil Extracellular Traps (NETs) Due to Restricted Neutrophil–Platelet Interactions. Cells 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Maratou, E.; Dimitriadis, G.; Kollias, A.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Raptis, S.A. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur. J. Clin. Investig. 2007, 37, 282–290. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.P.; Brody, S.L.; Zhou, Z.; Bernstein, M.; Arch, R.; Link, D.; Mueckler, M. Regulation of lipopolysaccharide-induced increases in neutrophil glucose uptake. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, 845–851. [Google Scholar] [CrossRef]
- Sadiku, P.; Willson, J.A.; Ryan, E.M.; Sammut, D.; Coelho, P.; Watts, E.R.; Grecian, R.; Young, J.M.; Bewley, M.; Arienti, S.; et al. Neutrophils Fuel Effective Immune Responses through Gluconeogenesis and Glycogenesis. Cell Metab. 2021, 33, 411–423.e4. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.; Toepfner, N.; Bashant, K.R.; Guck, J.; Ashcroft, M.; Farahi, N.; Chilvers, E.R. Metabolic profiling of human eosinophils. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Bao, Y.; Ledderose, C.; Seier, T.; Graf, A.F.; Brix, B.; Chong, E.; Junger, W.G. Mitochondria regulate Neutrophil activation by generating ATP for Autocrine Purinergic signaling. J. Biol. Chem. 2014, 289, 26794–26803. [Google Scholar] [CrossRef]
- Zanoni, I.; Gioia, M.D.; Broggi, A.; Spreafico, R.; Kumar, S.; Dikshit, M. Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e5. [Google Scholar] [CrossRef] [PubMed]
- Six, E.; Lagresle-Peyrou, C.; Susini, S.; De Chappedelaine, C.; Sigrist, N.; Sadek, H.; Chouteau, M.; Cagnard, N.; Fontenay, M.; Hermine, O.; et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. 2015, 6, e1856. [Google Scholar] [CrossRef]
- Jones, R.; McDonald, K.E.; Willson, J.A.; Ghesquière, B.; Sammut, D.; Daniel, E.; Harris, A.J.; Lewis, A.; Thompson, A.A.R.; Dickinson, R.S.; et al. Mutations in succinate dehydrogenase B (SDHB) enhance neutrophil survival independent of HIF-1α expression. Blood 2016, 127, 2641–2644. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Yu, X.; Tan, J.; Diamond, S.L. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J. Thromb. Haemost. 2018, 16, 316–329. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.B.; López-Villegas, E.O.; Sánchez-García, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef]
- Awasthi, D.; Nagarkoti, S.; Sadaf, S.; Chandra, T.; Kumar, S.; Dikshit, M. Glycolysis dependent lactate formation in neutrophils: A metabolic link between NOX-dependent and independent NETosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1–9. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; De Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-Acetate-induced neutrophil extracellular trap (NET) formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef]
- Keshari, R.S.; Verma, A.; Barthwal, M.K.; Dikshit, M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J. Cell. Biochem. 2013, 114, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Black, A.R.; Black, J.D. Protein kinase C signaling and cell cycle regulation. Front. Immunol. 2012, 3, 1–17. [Google Scholar] [CrossRef]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzym. Res. 2011, 2011, 1–26. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front. Immunol. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D. Dynamic NETosis is carried by live Neutrophils in Human and Mouse Bacterial Abscesses and during Severe Gram-Positive Infection. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Neubert, E.; Senger-Sander, S.N.; Manzke, V.S.; Busse, J.; Polo, E.; Scheidmann, S.E.F.; Schön, M.P.; Kruss, S.; Erpenbeck, L. Serum and serum albumin inhibit in vitro formation of Neutrophil Extracellular Traps (NETs). Front. Immunol. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef]
- Kamoshida, G.; Kikuchi-Ueda, T.; Nishida, S.; Tansho-Nagakawa, S.; Kikuchi, H.; Ubagai, T.; Ono, Y. Spontaneous formation of neutrophil extracellular traps in serum-free culture conditions. FEBS Open Bio 2017, 7, 877–886. [Google Scholar] [CrossRef]
- Palvinskaya, T.; Antkowiak, M.; Burg, E.; Lenox, C.C.; Ubags, N.; Cramer, A.; Rincón, M.; Dixon, A.E.; Fessler, M.B.; Poynter, M.E.; et al. Effects of acute and chronic low density lipoprotein exposure on neutrophil function. Pulm. Pharmacol. Ther. 2013, 26, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Wierusz-Wysocka, B.; Wysocki, H.; Wykrętowicz, A.; Klimas, R. The influence of increasing glucose concentrations on selected functions of polymorphonuclear neutrophils. Acta Diabetol. Lat. 1988, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Alba-Loureiro, T.C.; Munhoz, C.D.; Martins, J.O.; Cerchiaro, G.A.; Scavone, C.; Curi, R.; Sannomiya, P. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz. J. Med. Biol. Res. 2007, 40, 1037–1044. [Google Scholar] [CrossRef]
- Perner, A.; Nielsen, S.E.; Rask-Madsen, J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. 2003, 29, 642–645. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Kummer, U.; Zobeley, J.; Brasen, J.C.; Fahmy, R.; Kindzelskii, A.L.; Petty, A.R.; Clark, A.J.; Petty, H.R. Elevated glucose concentrations promote receptor-independent activation of adherent human neutrophils: An experimental and computational approach. Biophys. J. 2007, 92, 2597–2607. [Google Scholar] [CrossRef]
- Bartels, M.; Murphy, K.; Rieter, E.; Bruin, M. Understanding chronic neutropenia: Life is short. Br. J. Haematol. 2016, 172, 157–169. [Google Scholar] [CrossRef]
- Kordonowy, L.L.; Burg, E.; Lenox, C.C.; Gauthier, L.M.; Petty, J.M.; Antkowiak, M.; Palvinskaya, T.; Ubags, N.; Rincón, M.; Dixon, A.E.; et al. Obesity is associated with neutrophil dysfunction and attenuation of murine acute lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 120–127. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M.; Biophys, B.; Author, A. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 726–734. [Google Scholar] [CrossRef]
- Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil extracellular trap formation: Physiology, pathology, and pharmacology. Biomolecules 2019, 9, 365. [Google Scholar] [CrossRef]
- Li, R.H.L.; Ng, G.; Tablin, F. Lipopolysaccharide-induced neutrophil extracellular trap formation in canine neutrophils is dependent on histone H3 citrullination by peptidylarginine deiminase. Vet. Immunol. Immunopathol. 2017, 193–194, 29–37. [Google Scholar] [CrossRef]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef]
- Neubert, E.; Meyer, D.; Kruss, S.; Erpenbeck, L. The power from within-Understanding the driving forces of neutrophil extracellular trap formation. J. Cell Sci. 2020, 133, jcs241075. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Morotomi-Yano, K.; Sakugawa, T.; Saitoh, H.; Yano, K. ichi Nanosecond pulsed electric fields induce extracellular release of chromosomal DNA and histone citrullination in neutrophil-differentiated HL-60 cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef]
- Maueröder, C.; Mahajan, A.; Paulus, S.; Gößwein, S.; Hahn, J.; Kienhöfer, D.; Biermann, M.H.; Tripal, P.; Friedrich, R.P.; Munoz, L.E.; et al. Ménage-à-trois: The ratio of bicarbonate to CO2 and the pH regulate the capacity of neutrophils to form NETs. Front. Immunol. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Lin, R.C.; Zhang, Y.; Pradhan, K.; Li, L. TICAM2-related pathway mediates neutrophil exhaustion. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Hong, C.W. Current understanding in neutrophil differentiation and heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Dalby, M.J.; Aviello, G.; Ross, A.W.; Walker, A.W.; Barrett, P.; Morgan, P.J. Diet induced obesity is independent of metabolic endotoxemia and TLR4 signalling, but markedly increases hypothalamic expression of the acute phase protein, SerpinA3N. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Geng, S.; Yuan, R.; Diao, N.; Upchurch, Z.; Li, L. Super-low Dose Endotoxin Pre-conditioning Exacerbates Sepsis Mortality. EBioMedicine 2015, 2, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Liao, J.K. A Mouse Model of Diet-Induced Obesity and Insulin Resistance. Methods Mol. Biol. 2012, 821, 421–433. [Google Scholar] [CrossRef]
- Mashimo, H.; Ohguro, N.; Nomura, S.; Hashida, N.; Nakai, K.; Tano, Y. Neutrophil chemotaxis and local expression of interleukin-10 in the tolerance of endotoxin-induced uveitis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5450–5457. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, B.A. Tolerance and exhaustion: Defining mechanisms of T cell disfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: Implications for their use as bioenergetic biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef]
- Tackenberg, H.; Möller, S.; Filippi, M.-D.; Laskay, T. The Small GTPase Cdc42 Negatively Regulates the Formation of Neutrophil Extracellular Traps by Engaging Mitochondria. Front. Immunol. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Hear. Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M. Neutrophil extracelullar traps (NETs): Double-edged swords of innate immunity. J. Immunol. 2013, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Bowers, N.L.; Helton, E.S.; Huijbregts, R.P.H.; Goepfert, P.A.; Heath, S.L.; Hel, Z. Immune Suppression by Neutrophils in HIV-1 Infection: Role of PD-L1/PD-1 Pathway. PLoS Pathog. 2014, 10, e1003993. [Google Scholar] [CrossRef]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer Hassane. Clin. Cancer Res. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Osuch, S.; Laskus, T.; Berak, H.; Perlejewski, K.; Metzner, K.J.; Paciorek, M.; Radkowski, M.; Caraballo Cortés, K. Decrease of T-cells exhaustion markers programmed cell death-1 and T-cell immunoglobulin and mucin domain-containing protein 3 and plasma IL-10 levels after successful treatment of chronic hepatitis C. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, S.; Hu, Y.; Yang, Z.; Li, J.; Liu, X.; Deng, L.; Wang, Y.; Zhang, X.; Jiang, T.; et al. Targeting PD-1 and Tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous dendritic/tumor vaccines. J. Immunother. 2016, 39, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Mcnab, F.W.; Berry, M.P.R.; Graham, C.M.; Bloch, S.A.A.; Oni, T.; Wilkinson, K.A.; Wilkinson, R.J.; Kon, O.M.; Banchereau, J.; Chaussabel, D.; et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur. J. Immunol. 2011, 41, 1941–1947. [Google Scholar] [CrossRef]
- Pedersen, O.; Kahn, C.R.; Kahn, B.B. Divergent regulation of the Glut 1 and Glut 4 glucose transporters in isolated adipocytes from Zucker rats. J. Clin. Investig. 1992, 89, 1964–1973. [Google Scholar] [CrossRef]
- Moriguchi, S.; Kalo, M.; Sakai, K.; Yamamoto, S.; Shimizu, E. Decreased mitogen response of splenic lymphocytes in obese Zucker rats is associated with the decreased expression of glucose transporter 1 (GLUT-1). Am. J. Clin. Nutr. 1998, 67, 1124–1129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Mudaliar, S.; Barzin, A.; Macievic, J.A.; Edelman, S.V.; Park, K.S.; Henry, R.R. Skeletal Muscle GLUT1 Transporter Protein Expression and Basal Leg Glucose Uptake Are Reduced in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Köhler, E.; Barrach, H.J.; Neubert, D. Inhibition of NADP dependent oxidoreductases by the 6-aminonicotinamide analogue of NADP. FEBS Lett. 1970, 6, 225–228. [Google Scholar] [CrossRef]
- Jimenez, P.T.; Frolova, A.I.; Chi, M.M.; Grindler, N.M.; Willcockson, A.R.; Reynolds, K.A.; Zhao, Q.; Moley, K.H. DHEA-mediated inhibition of the pentose phosphate pathway alters oocyte lipid metabolism in mice. Endocrinology 2013, 154, 4835–4844. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M.; Karnovsky, M.L.; Karnovsky, M.I. Glycogen Accumulation in Polymorphonuclear Leukocytes, and Other Intracellular Alternations that Occur DuringInflammation. J. Cell Biol. 1982, 95, 933–942. [Google Scholar] [CrossRef]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W.; Glick, G.D. Identification and validation of the mitochondrial F1F 0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem. Biol. 2005, 12, 485–496. [Google Scholar] [CrossRef]
- Aulakh, G.K.; Balachandran, Y.; Liu, L.; Singh, B. Angiostatin inhibits activation and migration of neutrophils. Cell Tissue Res. 2014, 355, 375–396. [Google Scholar] [CrossRef]
- Benelli, R.; Morini, M.; Carrozzino, F.; Ferrari, N.; Minghelli, S.; Santi, L.; Cassatella, M.; Noonan, D.M.; Albini, A. Neutrophils as a key cellular target for angiostatin: Implications for regulation of angiogenesis and inflammation. FASEB J. 2002, 16, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Cao, M.; Liu, Y.; He, Y.; Wang, Y.; Yang, C.; Wang, W.; Du, Y.; Zhou, M.; Gao, F. Mitochondrial F1Fo-ATP synthase translocates to cell surface in hepatocytes and has high activity in tumor-like acidic and hypoxic environment. Acta Biochim. Biophys. Sin. 2010, 42, 530–537. [Google Scholar] [CrossRef]
- Sadiku, P.; Willson, J.A.; Dickinson, R.S.; Murphy, F.; Harris, A.J.; Lewis, A.; Sammut, D.; Mirchandani, A.S.; Ryan, E.; Watts, E.R.; et al. Prolyl hydroxylase 2 inactivation enhances glycogen storage and promotes excessive neutrophilic responses. J. Clin. Investig. 2017, 127, 3407–3420. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.; Pearce, E. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Lakschevitz, F.S.; Visser, M.B.; Sun, C.; Glogauer, M. Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell. Mol. Immunol. 2015, 12, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Injarabian, L.; Devin, A.; Ransac, S.; Marteyn, B.S. Neutrophil metabolic shift during their lifecycle: Impact on their survival and activation. Int. J. Mol. Sci. 2020, 21, 287. [Google Scholar] [CrossRef]
- Reithofer, M.; Karacs, J.; Strobl, J.; Kitzmüller, C.; Polak, D.; Seif, K.; Kamalov, M.; Becker, C.F.W.; Greiner, G.; Schmetterer, K.; et al. Alum triggers infiltration of human neutrophils ex vivo and causes lysosomal destabilization and mitochondrial membrane potential-dependent NET-formation. FASEB J. 2020, 34, 14024–14041. [Google Scholar] [CrossRef]
- Miller, E.S.; Koebel, D.A.; Sonnenfeld, G. The Metabolic Stresser 2-Deoxy-D-Glucose (2-DG) Enhances LPS-Stimulated Cytokine Production in Mice. Brain Behav. Immun. 1993, 7, 317–325. [Google Scholar] [CrossRef]
- Huang, H.; Liu, T.; Rose, J.L.; Stevens, R.L.; Hoyt, D.G. Sensitivity of mice to lipopolysaccharide is increased by a high saturated fat and cholesterol diet. J. Inflamm. 2007, 4, 1–11. [Google Scholar] [CrossRef]
- Delano, M.J.; Kelly-Scumpia, K.M.; Thayer, T.C.; Winfield, R.D.; Scumpia, P.O.; Cuenca, A.G.; Harrington, P.B.; O’Malley, K.A.; Warner, E.; Gabrilovich, S.; et al. Neutrophil Mobilization from the Bone Marrow during Polymicrobial Sepsis Is Dependent on CXCL12 Signaling. J. Immunol. 2011, 187, 911–918. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Gruhn, A.L.; Kudryasheva, G.; Günay, G.; Meyer, D.; Busse, J.; Neubert, E.; Schön, M.P.; Rehfeldt, F.; Kruss, S. Effect of Adhesion and Substrate Elasticity on Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Moura, M.B.; Van Houten, B. Bioenergetic analysis of intact mammalian cells using the seahorse XF24 Extracellular flux analyzer and a luciferase ATP assay. Methods Mol. Biol. 2014, 1105, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Repp, B.; Biagosch, C.; Terrile, C.; Prokisch, H. Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using Seahorse XF96 analyzer. Methods Mol. Biol. 2017, 1567, 217–230. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cichon, I.; Ortmann, W.; Kolaczkowska, E. Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (NETs) Differ in Obesity and Systemic Inflammation. Int. J. Mol. Sci. 2021, 22, 7718. https://doi.org/10.3390/ijms22147718
Cichon I, Ortmann W, Kolaczkowska E. Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (NETs) Differ in Obesity and Systemic Inflammation. International Journal of Molecular Sciences. 2021; 22(14):7718. https://doi.org/10.3390/ijms22147718
Chicago/Turabian StyleCichon, Iwona, Weronika Ortmann, and Elzbieta Kolaczkowska. 2021. "Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (NETs) Differ in Obesity and Systemic Inflammation" International Journal of Molecular Sciences 22, no. 14: 7718. https://doi.org/10.3390/ijms22147718
APA StyleCichon, I., Ortmann, W., & Kolaczkowska, E. (2021). Metabolic Pathways Involved in Formation of Spontaneous and Lipopolysaccharide-Induced Neutrophil Extracellular Traps (NETs) Differ in Obesity and Systemic Inflammation. International Journal of Molecular Sciences, 22(14), 7718. https://doi.org/10.3390/ijms22147718