1. Introduction
Breast cancer (BC) is the most common malignant tumor in women representing 25% of the total number of cancer cases in women with an annual increase of 793,700 new cases and 197,600 deaths only in developed countries [
1,
2].
The presence of germline mutations in the
BRCA1 and
BRCA2 genes significantly increases the risk of developing breast and ovarian cancer [
3,
4,
5], however only 25% of the risk of familial cancer can be attributed to pathogenetic mutations localized in the coding regions of these two genes. Most of the risk is probably due to a combination of high, moderate, and low risk variants which may also include non-coding variants located in regulatory regions [
4].
In clinical practice, during the past years, the mutational analysis in patients with Hereditary Breast and Ovarian Cancer (HBOC) has been mostly limited to the coding regions and to the intron-exon junctions of
BRCA1 and
BRCA2, precluding the identification of mutations in the non-coding and/or regulatory regions and in other genes that can confer a high or moderate risk to the disease. Under these conditions, mutation screening is negative in 80% of the cases analyzed [
6].
Segregation analysis generated no evidence for further dominant genes with a risk penetrance profile comparable to
BRCA1 or
BRCA2 and this has been corroborated by linkage studies [
7,
8]. Over the years, efforts have been made to identify new intermediate-penetrance predisposition genes, although it is difficult to predict how many they might be and what proportion of the total familial risk is attributable to them. The known intermediate-penetrance genes are all involved in DNA repair or have a role in BRCA1 or BRCA2 pathway, so these genes represent plausible candidates and are the most intensively investigated [
9]. In addition, in recent years, the interest in investigating the effect that regulatory variants could have on cancer risk has increased [
10].
Generally, gene expression is controlled at various levels by key regulatory elements located in promoters, introns, in “long-range enhancers” and in Untranslated Regions (5′and 3′ UTR). This is also true for
BRCA1 and
BRCA2; in fact, it has been shown that the activity of the 5′UTR and of the
BRCA1/2 exon-flanking regions is controlled by a self-regulation mechanism and by an increasing number of transcription factors that act as activators or repressors. Interestingly, germline variants of 5′UTR causing a significant change in promoter activity have been identified in such regions [
11,
12,
13].
Similarly, germline variants localized in consensus regions for “RNA binding protein” and miRNA located in 3′UTR of
BRCA1 [
14,
15] and
PTEN [
16,
17] have been associated with breast cancer risk.
Therefore, germline regulatory variants can be associated with a deregulation of the transcriptional activity and modify the physiological expression both of BRCA genes and of other genes that can affect the risk of breast cancer [
13].
With the progression of DNA sequencing technologies, it is possible to analyze in each patient multiple genomic regions by simultaneously screening multiple target genes.
An ever-increasing number of variants of unknown functional and clinical significance (VUS) have been identified in both coding and non-coding regions. Therefore, it is absolutely necessary to establish their risk level in carrier patients, for more effective genetic counselling.
In 2008, the International Agency for Research on Cancer (IARC) proposed to classify the missense variants on the basis of their probable pathogenicity, calculated by multifactorial models, in five risk classes: Class 1 (non-pathogenic or of no clinical significance), Class 2 (likely non-pathogenic or of little clinical significance), Class 3 (uncertain), Class 4 (likely pathogenic), and Class 5 (definitely pathogenic) [
18].
Recently, the indications of the American College of Medical Genetics (ACMG) have been increasingly followed by several clinicians and research groups. They have recommended a five-tier classification system where variants can be classified as: ‘Pathogenic’, ‘Likely pathogenic’, ‘Uncertain significance’, ‘Likely benign’, and ‘Benign’ [
19].
In this work we used this classification that, among those available, is the most complete and is based on an algorithm that takes into account data on penetrance, clinical information of carriers, and scientific evidence.
To classify VUSs, alternative methods have also been developed. They do not directly evaluate the role of the variant in the development of cancer, but the structural and functional modifications of the proteins of interest by providing indirect evidence of the pathogenicity of the variant [
20,
21]. Both in vitro functional assays and in silico predictors can be used [
20].
Recently the international consortium ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) has become in charge of reviewing and comparing all the available and useful functional tests in order to define the functional meaning of a variant and to contribute to the interpretation of clinical data [
22,
23,
24,
25].
In this study involving five Italian hospital centers (Pisa, Cagliari, Varese, Modena, and Bari), a genomic screening was conducted on 450 Breast and/or Ovarian Cancer patients with cancer family history in at least two generations and wild type for germinal mutations in the coding regions and/or for large rearrangements in BRCA1/2 genes.
The analysis was carried out by Next Generation Sequencing (NGS) using a panel that covers the coding regions and the intron-exon junctions of 10 genes mainly involved in DNA repair pathways: ATM, BRIP1, CDH1, CHECK2, PALB2, PTEN, RAD51C, RAD51D, STK11, TP53. For 120 of these patients, the screening was extended to RAD50 and NBN genes and to 5′UTR and 3′UTR of all the genes listed above including the known regulatory regions of BRCA1 and BRCA2. We hypothesized that variants located in the regulatory regions of the main BC predisposition genes can contribute to tumor development in a manner comparable to the variants located in coding regions. To support this hypothesis, we compared the average age of disease onset between the coding and the non-coding variant carriers with the same cancer type. Moreover, we analyzed the proportion of cases with a tumor onset under the age of 40 between the two groups and we verified any variant association in more aggressive tumours.
2. Results
2.1. Summary Overview of Germline Variants Found
Mutational screening was performed for 450 patients in the coding regions and 50 bp flanking intronic sequences of 10 genes: ATM, BRIP1, CDH1, CHECK2, PALB2, PTEN, RAD51C, RAD51D, STK11, TP53. For 120 of these patients, the analysis was extended to two additional genes, RAD50 and NBN, and to the regulatory regions of BRCA1 and BRCA2, 5′UTR, and 3′UTR for all 12 genes listed above.
We identified 88 coding variants in 91 patients distributed as follows:
- -
16 class 4 (Likely-pathogenic) and 19 class 5 (Pathogenic) variants in 39 patients (8.7%)
- -
37 class 3 (VUS) in 41 patients (9.1%)
- -
16 Likely-benign variants in 19 patients (4.2%), but all of them potentially classifiable as VUS, according to ACMG classification, because of interpretation conflicts.
In the intron-exon junctions, 13 variants in 23 patients were identified (5.1%).
The extension of the screening in 120 patients to the known regulatory regions of BRCA1/2 and to 5′UTR, 3′UTR for all the genes analyzed, allowed us to identify another 51 variants in 39 patients (32.5%).
2.2. Coding Variants
A total of 88 coding variants were identified: 35 Pathogenic or Likely-pathogenic variants, 37 VUS, and 16 Likely-benign variants.
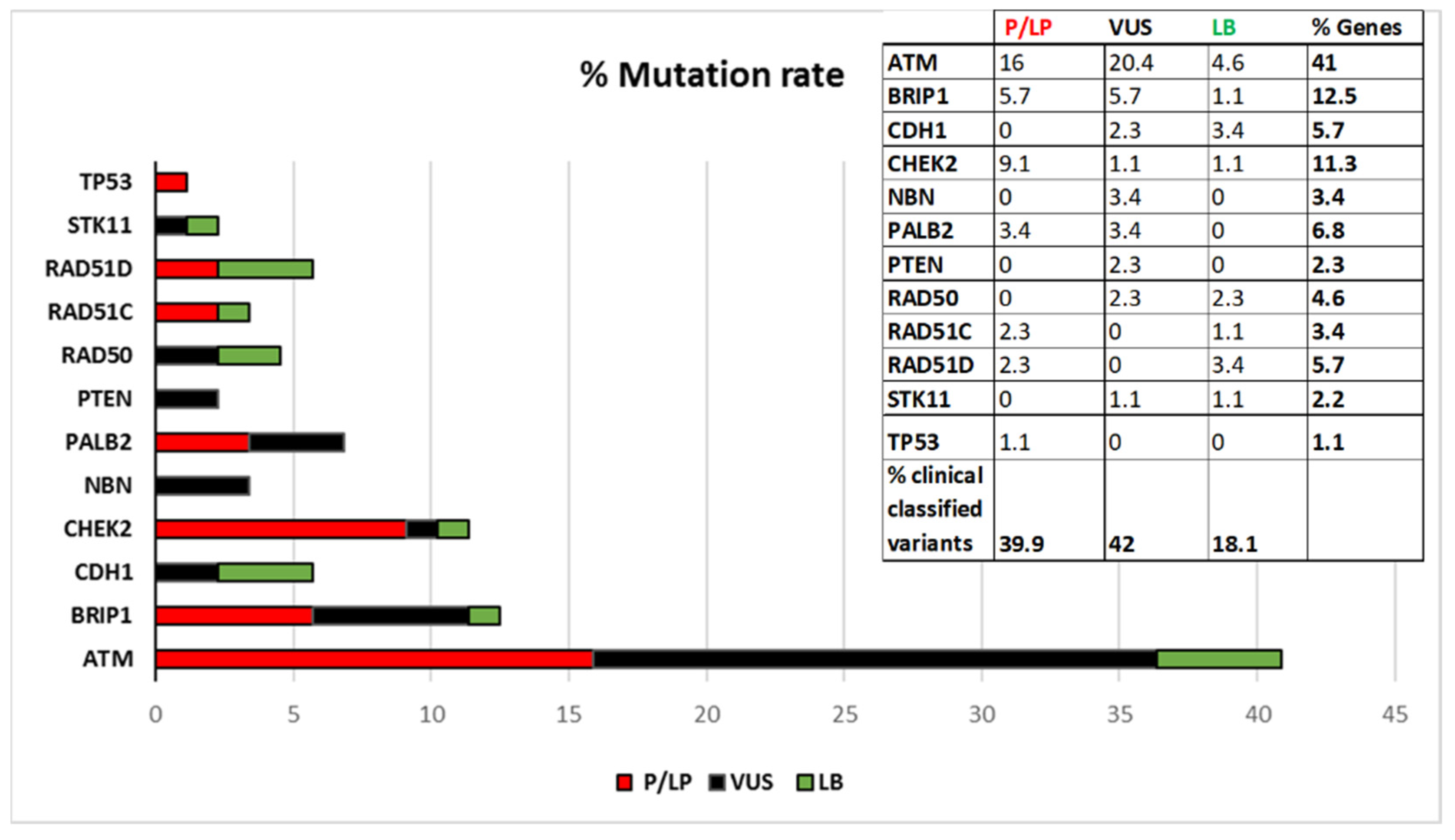
Thirty out of 35 Pathogenic or Likely-pathogenic identified coding variants were encoded in dbSNP and 27 were already known in literature or reported in databases such as IARC, LOVD, MGeND, ClinVar Miner with more clinical information. Five variants have never been registered: c.8368delA in ATM, c.507delT and c.1430delC in CHEK2, c.1565dupC in PALB2, c.397_398delCA in RAD51C. The 35 class 4 and 5 variants are distributed in the following genes: 14 in ATM, 5 in BRIP1, 8 in CHEK2, 3 in PALB2, 2 in RAD51C, 2 in RAD51D, and 1 in TP53.
Thirty-four out of 37 VUS identified were encoded in dbSNP and 25 of these were already known and reported in databases previously cited. The following 3 VUSs were completely new: c.2317A > G in ATM, c.3029A > G in BRIP1 and c.17_18insGCG in PTEN. Eighteen of the 37 VUSs identified are in ATM, 5 in BRIP1, 2 in CDH1, 1 in CHEK2, 3 in NBN, 3 in PALB2, 2 in PTEN, 2 in RAD50, and 1 in STK11.
Fifteen out of 16 Likely-benign variants found had a dbSNP entrie and 12 of these were encoded in databases. One variant had never been registered (c.252T > C in RAD51D). The 16 Likely-benign variants are distributed as follows: 4 in ATM, 1 in BRIP1, 3 in CDH1, 1 in CHEK2, 2 in RAD50, 1 in RAD51C, 3 in RAD51D, and 1 in STK11.
Figure 1 shows the percentage of distributions of the variants stratified by gene, according to the current ACMG classification. As shown in
Figure 1 ATM is the gene with the highest mutation rate.
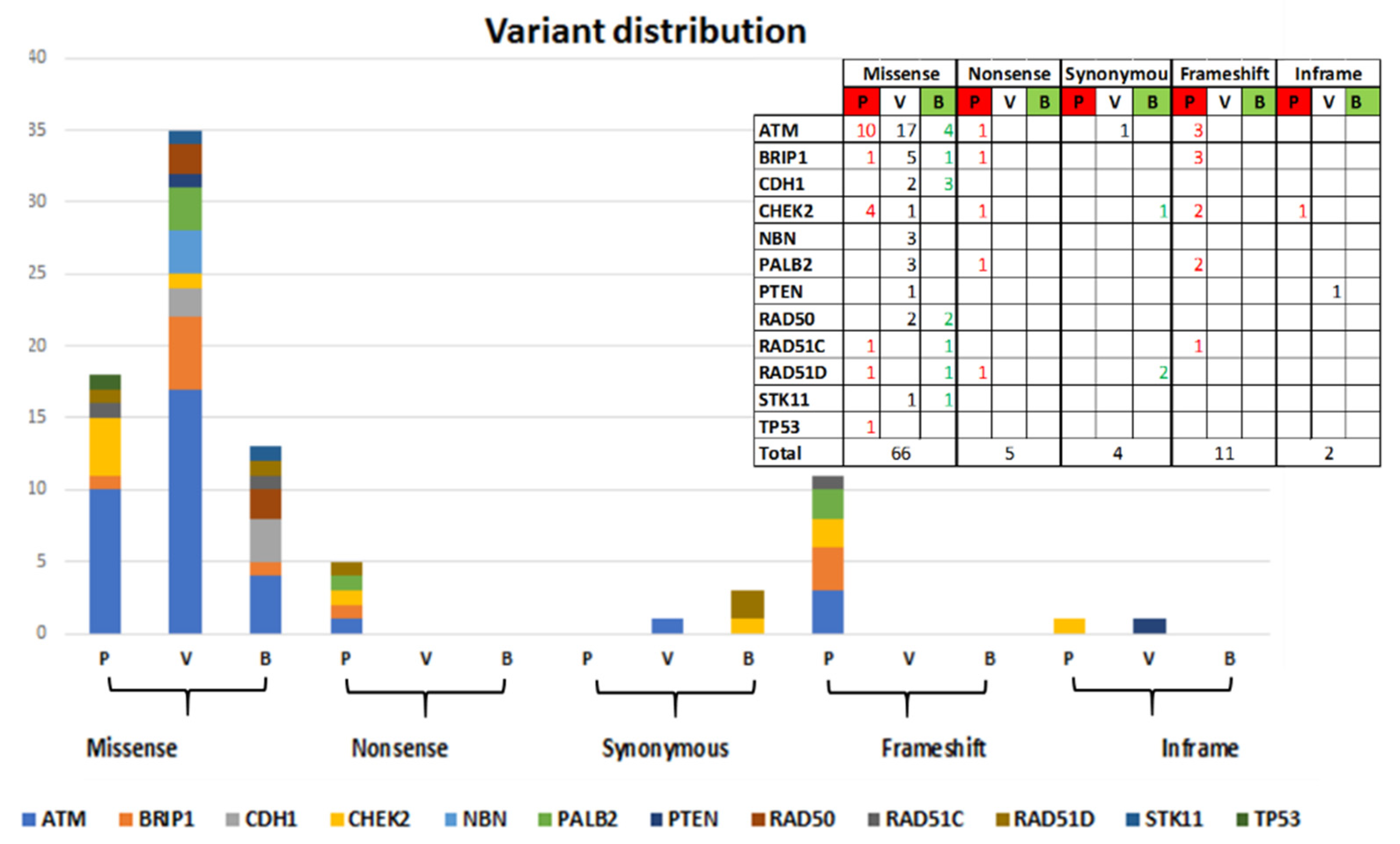
Sixty-six out of 88 identified variants are missense variants (of which 18 Pathogenic and Likely-pathogenic, 35 VUS and 13 Likely-benign), 5 Nonsense variants (all Pathogenic), 4 Synonymous variants (1 VUS and 3 Likely-benign), 11 frameshift variants (all Pathogenic), and 2 in-frame variants (1VUS and 1 Pathogenic). The distribution by gene is shown in
Figure 2.
The predictive analysis on the effect of amino acid change on the function of the protein was conducted with PROVEAN and Polyphen2 software, as described in Materials and Methods. Specifically, the information for each identified variant is shown in
Table S1 in Supplementary Materials, where it is also reported whether the variant has already been identified and registered in database by other groups.
2.3. Demographics and Clinical Features of Patiens with Coding Variants
The average age of tumor diagnosis for 91 patients with coding variants is 44. 8.8% (8) of the patients have a bilateral BC, 2.2% (2) have both breast and ovarian cancer, 4.4% (4) have ovarian cancer, 2.2% (2) have multiple tumors (in addition to breast cancers) and the remaining percentage, 82.4% (75) of total cases, has a BC. Among the latter patients, 16% (12) have Triple-Negative Breast Cancer (TNBC).
Their family histories are distributed as follows: 48.3% (44) have Hereditary Breast Cancer (HBC), 12.1% (11) have Hereditary Breast and Ovarian Cancer (HBOC), 18.7% (17) have Early Onset Breast Cancer (EOBC), and 20.9% (19) have other Hereditary Breast Cancer Syndromes (HBCS) (
Table 1).
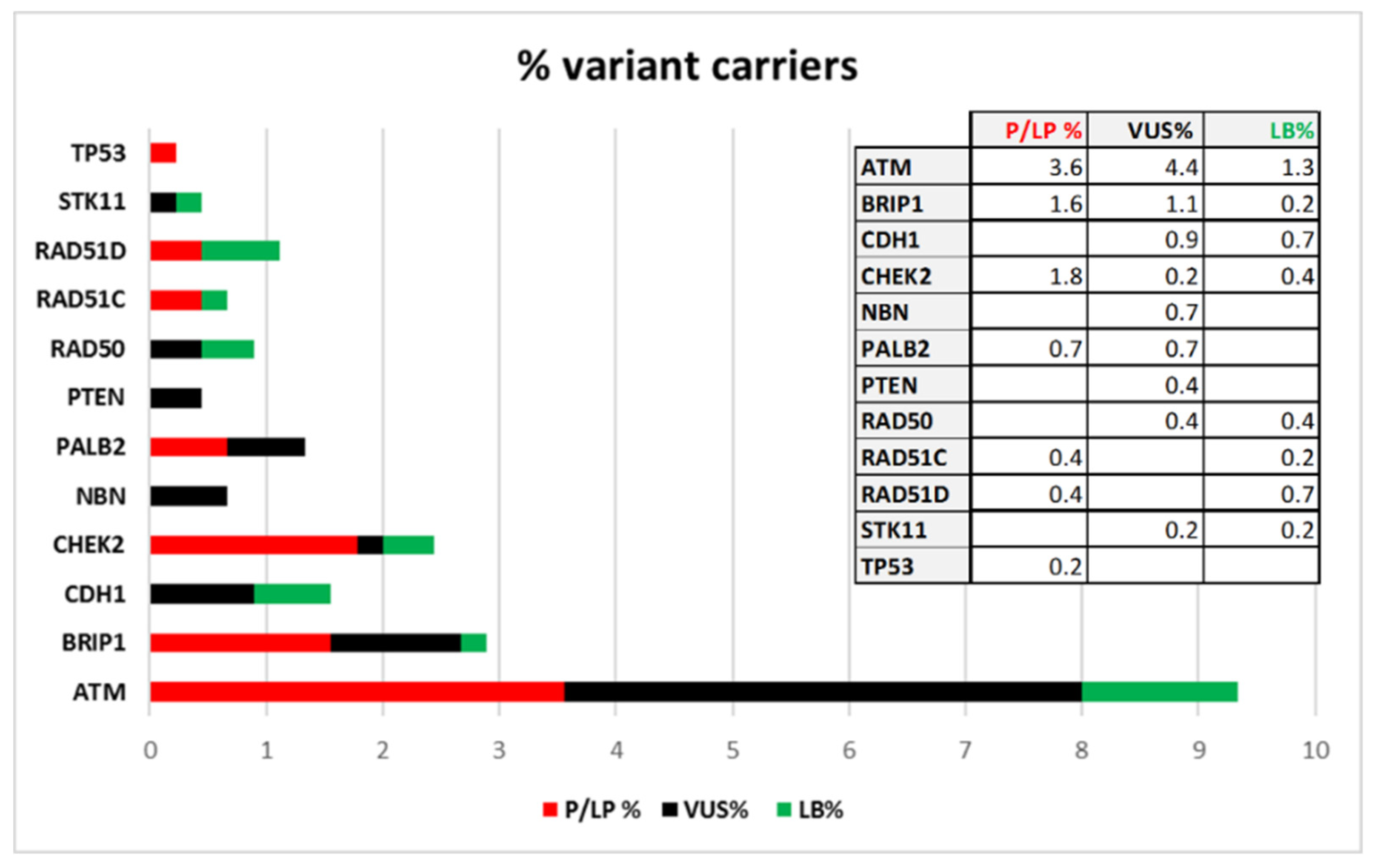
Thirty-nine patients are carriers of variants classified as “Pathogenic” or “Likely-pathogenic”, 41 patients are carriers of VUS, while “Likely-benign” variants have been identified in 19 patients. If we consider the total number of patients screened in the coding regions (450) the percentage of class 4 and 5 variant carriers is 8.7% and it is distributed in this way: 3.6% have variants in
ATM, 1.6% in
BRIP1, 1.8% in
CHEK2, 0.7% in
PALB2, 0.4% in
RAD51C, 0.4% in
RAD51D, and 0.2% in
TP53. In
Figure 3 the frequencies of patients with Pathogenic and Likely-pathogenic, VUS and Likely-benign variants are reported.
Among the 75 patients with BC, we identified 82 variants of which 30 Pathogenic and Likely-pathogenic, broken down as follows: 13 in ATM gene, 5 in BRIP1, 2 in PALB2, 6 in CHEK2, 2 in RAD51C, 1 in RAD51D and 1 in TP53. In these same patients we identified 38 VUS: 15 in ATM, 6 in BRIP1, 4 in CDH1, 3 in NBN, 3 in PALB2, 2 in CHEK2, 2 in PTEN, 1 in RAD50, 1 in RAD51D and 1 in STK11. The 14 remaining variants found are classified as Likely-benign and distributed as follows: 6 in ATM, 3 in RAD51D, 2 in CDH1 and 3 respectively in BRIP1, RAD50, and RAD51C genes.
Among the four cases of patients with ovarian cancer, we found a Pathogenic variant in RAD51C, 2 VUSs, respectively in ATM and RAD50 genes, and a Likely-benign variant in STK11.
Among the 12 cases of multiple tumors in coding variant carriers (9 cases of bilateral breast cancer, 2 of breast and ovarian cancer, and 1 case of breast tumor associated with pancreatic cancer), 9 have Pathogenic or Likely-pathogenic variants, 1 VUS and 2 Likely-benign variants. The statistical analysis has found a significant association of these cases with Pathogenic variant carriers (χ2-test, p < 0.01) in comparison with the cases of single tumors.
Three out of nine patients with Pathogenic variants have variants in
ATM, 4 in
CHEK2, 1 in
BRIP1 and 1 in
PALB2 (
Table 2). Three of these nine variants are new variants never reported in dbSNP. One case has a VUS in
ATM gene and two patients with multiple tumors have Likely-benign variants in CDH1 and
CHEK2 genes. Frequency of coding variants in
CHEK2 gene is significant in these patients (Fisher test,
p < 0.05).
By analyzing cases of more aggressive tumors such as the 12 Triple-negative breast cancer patients (TNBC) (
Table 3), all with G2 or G3 infiltrating ductal carcinoma, we identified 6 Pathogenic and Likely-pathogenic variants: 1 in
ATM, 1 in
BRIP1, 2 in
CHEK2, 1 in
RAD51D and 1 in
TP53. In 6 patients, 4 VUSs (3 in ATM and 1 in
PTEN never recorded before) and 2 Likely-benign variants were identified. The latter all have a conserved evolutionary profile (GERP > 3.8) and a conflict of interpretation according to Varsome and ClinVar.
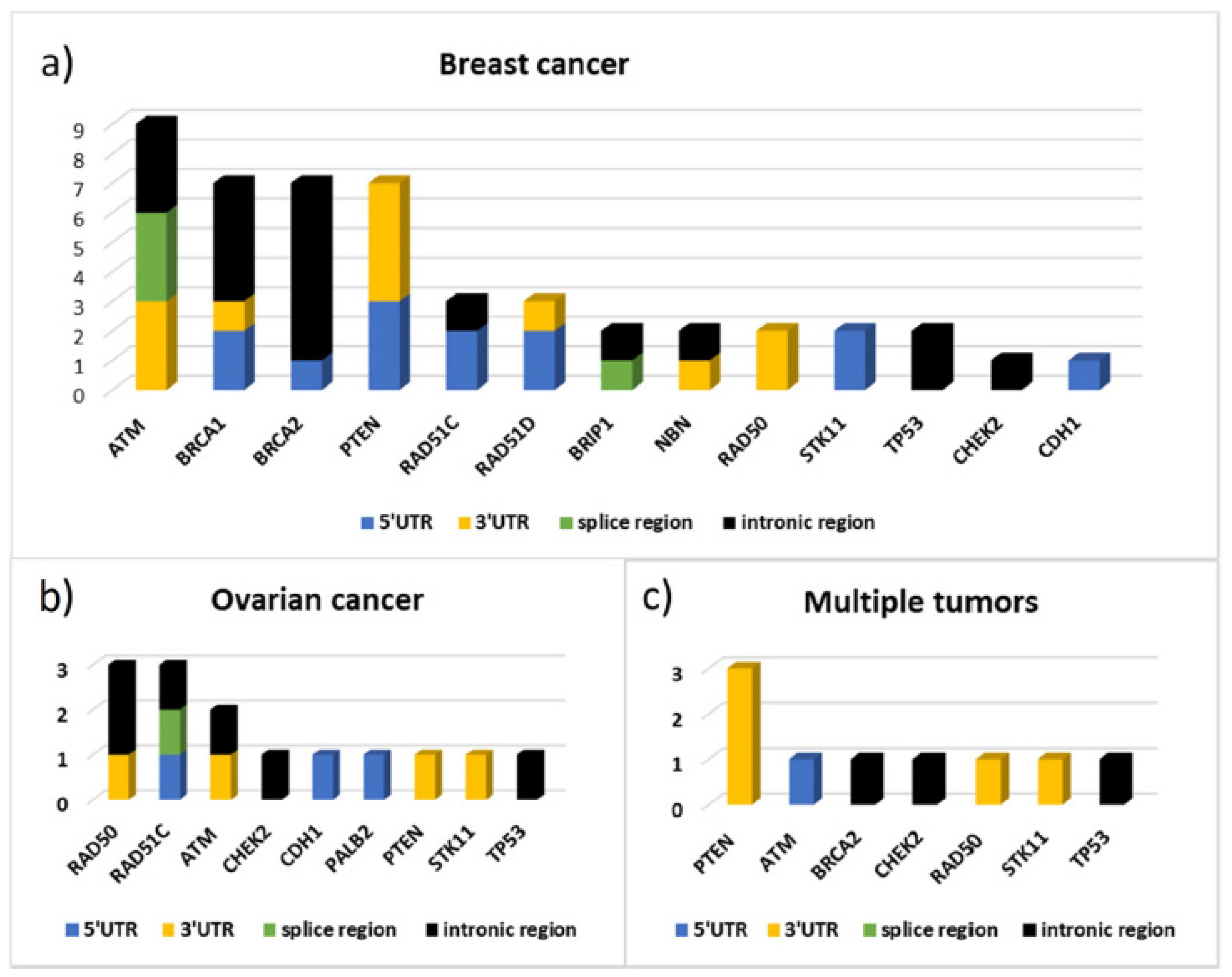
2.4. Non-Coding and/or Regulatory Variants
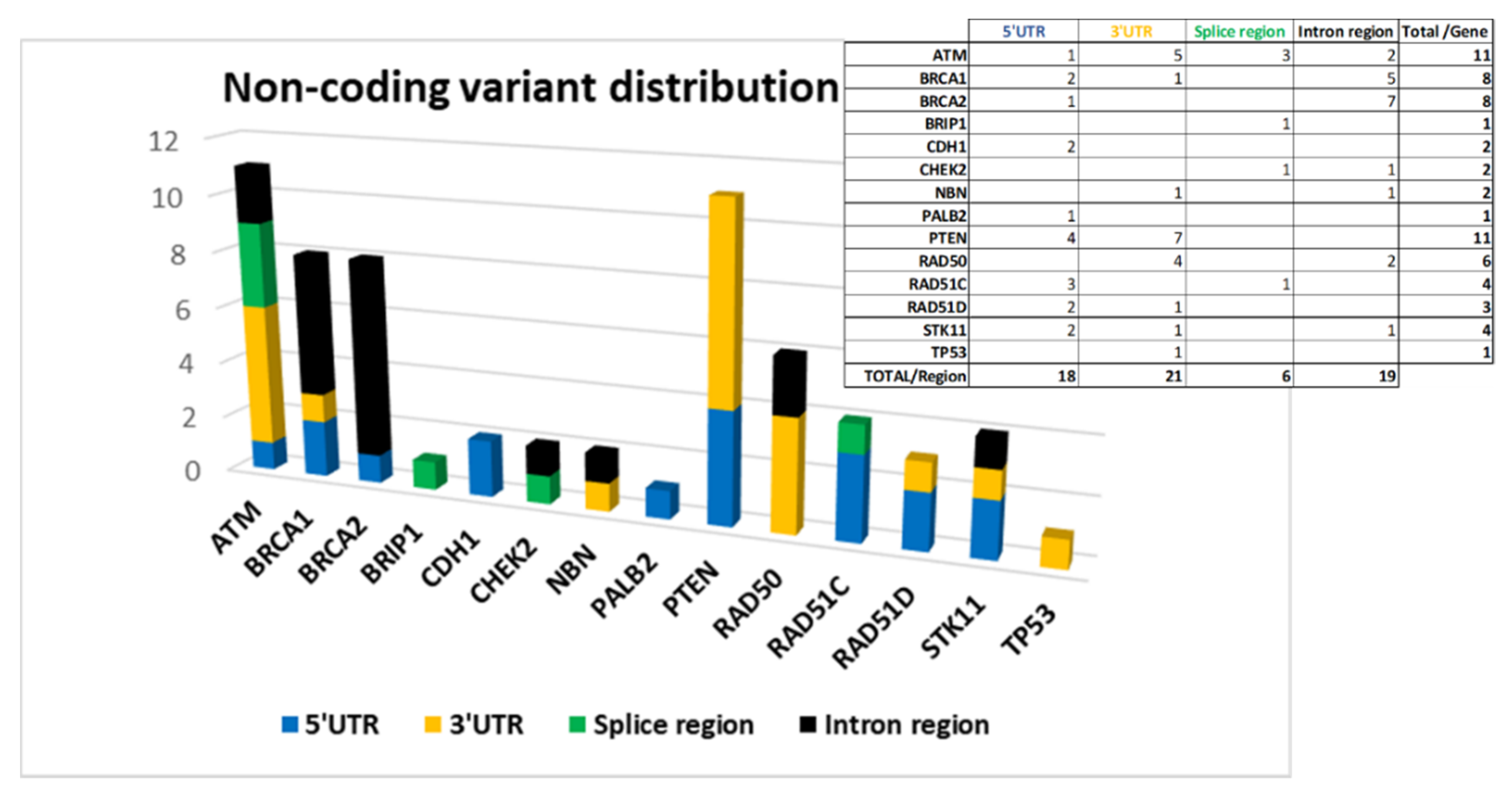
The screening of non-coding regions has allowed us to identify 64 variants. Forty-six of these have a dbSNP code and only 24 are known variants already registered in databases by other groups.
The 64 variants are distributed as follows: 18 are located at 5′UTR, 21 at 3′UTR, 6 in splice site regions, and 19 in intron regions.
Figure 4 shows the distribution of the variants by gene divided according to their location.
ATM,
BRCA1,
BRCA2, and
PTEN are among the genes in which we have found more non-coding variants (
Figure 4).
The variant putative effect on gene expression, was carried out by using different software (described in Materials and Methods) depending on variant position. The results are reported in
Table S2a–d in Supplementary Materials.
An unexpectedly high number of variants in 5′ UTR of
PTEN (4) and
RAD51C (3) was found and, according to our analysis, all of them are localized in TF binding regions (
Table S2a/Supplementary Materials).
The in-silico analysis performed by ‘TFbind’ and ‘Promo’ software, has identified TF consensus sequences in the regions where the 5′UTR of
BRCA1 (c.-676T > A) and
BRCA2 (c.-1193C > T) variants are localized (
Table S2a/Supplementary Materials).
In 3′UTR, the greatest contribution is due to variants located in
ATM,
PTEN, and
RAD50 genes (
Figure 4) on which putative miRNA-binding regions have been identified by ‘TargetScanHuman’ and ‘miRDB’ software (
Table S2b/Supplementary Materials).
In splicing regions, we identified six variants and only one, c.904+5G > T, was predicted to inactivate the wild-type donor site in the intron 6 of the RAD51C gene, and therefore is classified by the ACMG criteria as ‘Pathogenic’.
Three variants were in
ATM but only the c.7788+ 8G > T, localized in intron 52, was predicted by ‘Human Splicing Finder’ as a possible modifier of an exonic ESE site with a potential alteration of splicing. The remaining 2 in
CHEK2 and
BRIP1 genes were predicted neutral on splicing (
Table S2c/Supplementary materials).
In the remaining intronic regions, variants in
ATM,
BRCA1,
BRCA2,
CHEK2,
NBN,
RAD50,
STK11, and
TP53 genes were identified. The possible presence of enhancer regions, compatible with bonds to transcription factors, was analyzed by the software mentioned above. The four variants identified in intron 2 of
BRCA1 (c.81-3510_81-3505delCTTTTT; c.81-3790G > C; c.81-4100A > C; c.80 + 247C > T) deserve attention. In fact, all four variants identified in this study map in two conserved enhancer regions, that have already been demonstrated to be involved in gene expression regulation (
Table S2d/Supplementary Materials) [
26].
In conclusion, we found only one Pathogenic variant in the RAD51C gene, whereas the large majority were classified as VUSs (50), 2 as Likely-benign and 11 as Benign variants.
The high number of VUSs is due to the fact that very few studies have analyzed these non-coding regions, and for this reason the classification is based only on in silico prediction.
2.5. Deep Intronic Regions
The panel of 9071 probes used was designed to optimize coverage of exonic regions, 50 bp flanking each intron-exon junction, 5′UTR, 3′UTR of all the genes of interest and the known regulatory regions of
BRCA1 and
BRCA2. However, the position of the probes managed to cover additional regions which led us to identify another nine variants in intronic regions (all with a coverage ≥ 50X) that proved to be interesting for the predictions obtained by ‘TFbind’ and ‘Promo’ software and for the information obtained from rVarBase and ENCODE (
Table S2e/Supplementary Materials). Two variants are in the intron 61 of
ATM, one in the intron 16 of
BRIP1, two in intron 2 respectively of
RAD51C and
RAD51D genes and four variants are in the intron 1 of
TP53. Eight out of nine variants are classified as VUS and four are not registered in dbSNP.
2.6. Demographics and Clinical Features of Patiens with Non-Coding Variants
Non-coding variants were found in 64 patients (excluding two cases of co-occurrence of Pathogenic coding variants). According to this data, in these patients there is a significant increase in both ovarian cancer (χ2-test,
p < 0.05) and HBOC families (χ2-test,
p < 0.01), compared to patients with variants in coding regions (
Table 1/
Table 4).
The mean age of tumor onset for these patients is 44.4. If we compare the coding and non-coding variant carriers in each proband group with the same kind of cancer, the average age of disease onset is completely comparable (
Table 1/
Table 4). In particular, if we compare the 64 non-coding variant carriers to the 39 Pathogenic coding variant carriers, there is not a statistically significant difference in the proportion of cases with a tumor onset under age 40 (
Table 5, χ2-test,
p > 0.05).
Figure 5 shows the distribution by gene and by gene location of the variants identified respectively in 43 patients with BC, in 12 patients with ovarian cancer, and in 9 cases of patients with multiple tumors (seven with bilateral cancer, one with breast and ovarian cancer, and one with breast and pancreatic cancer). From this data emerges the high frequency of variants identified in the
ATM gene, as well as in
BRCA1 and
BRCA2, in patients with BC and the high contribution of variants localized in the 3′UTR of
PTEN in patients with multiple tumors. In particular, three out of nine variants found in these patients are localized in
PTEN 3′UTR and the bioinformatics predictions found, for all three, consensus sequences compatible with miRNA binding sites (
Figure 5,
Table S2b/Supplementary Materials). In patients with ovarian cancer, instead, variants were identified mainly in the
RAD50 and
RAD51C genes.
Among the 12 cases of TNBC, all with G2 or G3 infiltrating ductal carcinoma, 4 variants out of 19 identified are on the
PTEN gene: one located at 5′UTR and the others at 3′UTR. Other interesting variants, for the type of feedback we obtained through prediction analysis, are the variants in the 5′UTR of
RAD51D and
STK11 genes. The complete list of variants found in these patients is shown in
Table 6. Four variants are in intronic regions of
BRCA2. Furthermore, among all the non-coding variants filtered in this work, the only variant in the
RAD51C gene classified as “Pathogenic” by ACMG criteria was found in one of these patients.
Twelve patients with multiple variants in non-coding regions were filtered to assess whether the presence of multiple mutations could affect tumor pathogenicity. Ten out of 12 filtered patients have BC and 6 of these have TNBC. According to our data, there is a statistically significant association between TNBC and multiple variants in non-coding regions (χ2-test,
p < 0.05). In fact, 50% of BC patients with multiple variants in non-coding regions have TNBC, while, among the 52 BC non-coding variants carriers, only 12 (23.1%) have TNBC (χ2-test,
p < 0.05;
Table 7).
3. Discussion
This study regards the analysis of coding regions and 50 bp flanking intron–exon junction of 10 BC predisposition genes in a cohort of 450 HBOC patients. For a portion of these patients (120) the screening was extended to additional two genes (NBN and RAD50), to the 5′UTR and 3′UTR of each gene analyzed and to the known regulatory regions of BRCA1 and BRCA2.Eighty eight coding variants were identified in 91/450 patients (20.2%): 35 of these were classified as pathogenic (class 5) or likely-pathogenic (class 4) and found in 39 patients (8.7%), 37 were classified as VUSs in 41 patients (9.1%) and 16 as “Likely-benign” in 19 patients (4.2%).
ATM was the more mutated gene (16 carriers), followed by CHEK2 (8 carriers), BRIP1 (7 carriers), PALB2 (3 carriers). RAD51C and RAD51D were found mutated in 2 patients/each and TP53 in one patient only.
Thirteen variants in the intron-exon junctions were identified in 23/450 patients (5.1%) and 51 non-coding variants in 39 out of 120 patients analyzed for this purpose (32.5%).
Our results support what was recently published by the Breast Cancer Association Consortium in a larger and more geographically heterogeneous population consisting of 60,466 women. With a panel of 34 putative susceptibility genes, a significant association between the increased risk of breast cancer and truncating mutations in
ATM,
CHEK2,
PALB2,
BARD1,
RAD51C,
RAD51D, and
TP53 was identified [
27]. Similar results were found through a screening of 65,057 white woman with breast cancer where a high or moderately increased risk of disease was found in relation with pathogenic mutations in
ATM,
CHEK2,
PALB2, and
RAD51D [
28].
ATM,
CHEK2,
PALB2, and
TP53 were confirmed as breast cancer predisposition genes also by the German Consortium for Hereditary Breast and Ovarian Cancer, through a screening of eight cancer predisposition genes conducted on 5589 breast cancer patients negative for pathogenic
BRCA1/2 mutations [
29]. An American retrospective study performed on 35,000 women who underwent genetic testing through a 25-gene hereditary cancer panel found the same correlation [
30].
The analysis of non-coding regions reports a consistent number of variants never registered previously (18/64 variants).
In details, in the 5′UTR 2 variants were found, one in BRCA1 (c.-676t > a) and one in BRCA2 (c.-1193c > t). Both were registered in BRCA-exchange and LOVD database as VUSs. The in-silico analysis, conducted by ‘TFbind’ and ‘Promo Prediction’, has identified putative consensus sequences for Elk-1, NF-AT1, STAT4 transcription factors, for the first variant, and NRF2, GATA1/2, USF for the second.
Whereas the 5′UTR and 3′UTR of the
BRCA1/2 genes have been two of the most intensely investigated non–coding regions over the last few years [
12,
13,
31,
32,
33,
34], this is the first fully comprehensive study of 5′and 3′UTRs of so called “minor genes for breast/ovarian cancer susceptibility” and a number of variants were also identified in the 5′UTRs: 4 in the
PTEN gene and 3 in the
RAD51C gene and the in-silico predictions obtained regarding their involvement in TF binding are interesting (
Table S2a in Supplementary Materials).
In the 3′UTRs of genes other than BRCA1/2, the greatest contribution was due to variants located in the
ATM (5),
PTEN (7) and
RAD50 (4) genes. The use of software such as ‘TargetScanHuman’ and ‘miRDB’ has made it possible to identify in those regions binding sites for miRNAs (
Table S2b in Supplementary Materials).
The variants localized in splice regions mainly affect the
ATM (3) gene and, in particular the c.7788+8G >T variant, localized in intron 52 of
ATM, could modify an exonic ESE site (
Table S2c in Supplementary Materials). Another variant of interest is the c.904+5G >T of
RAD51C, classified by ACMG criteria as ‘Pathogenic’.
According to our data, variants in the intronic regions have been identified mainly in the BRCA1 (5), BRCA2 (7), ATM (2), and RAD50 (2) genes.
Four out of 5
BRCA1 variants are localized in intron 2 (c.81-3510_81-3505delCTTTTT; c.81-3790G > C; c.81-4100A > C; c.80+247C > T) and the in-silico predictive analysis led us to identify TF binding sites in those regions (
Table S2d/Supplementary Materials). Those variants deserve to be studied in depth through a functional assay because in
BRCA1 intron 2, two conserved enhancer regions (CNS1 and CNS2) are localized and involved in the regulation of gene expression [
26].
Eleven out of 12 intronic variants found in
BRCA1/2 genes could be classified as ‘deep intronic’. The deep intronic regions of the BRCA genes are intensely been studied in the recent years. These investigations have led to the identification of variants capable to modify the expression level or the functionality of the BRCA1 protein [
35].
Finally, we incidentally found two new variants in deep intronic regions of the ATM and TP53 genes in two patients with TNBC (due to the fact that additional regions that were not of our interest were included in the panel design). The variants are respectively in intron 61 of ATM (c.8850+389G > A) and intron 1 of TP53(c.-29+475C > A) and are both predicted as TF binding regions. This result underlines how important is to broaden screening to deeper intronic regions (not only in BRCA genes).
By analyzing the characteristics of patients for the type of variants identified, distinguishing between coding variant carriers versus non-coding variant carriers, we can make some interesting considerations.
The average age of breast cancer onset in coding variants carriers is 44 and most of them have breast cancer only family history as shown in
Table 1.
A possible association of
ATM-BRIP1-PALB2 and a significant association of
CHEK2 pathogenic/likely-pathogenic coding variants with multiple tumors, including bilateral breast cancer, was observed. These data are in agreement with the findings of German Consortium for Hereditary Breast and Ovarian Cancer, that demonstrated a significant association of
CHEK2 deleterious variants with bilateral breast cancer [
29].
Among the 12 cases of TNBC patients, 6 were carriers of pathogenic coding variants: 2 in
CHEK2 and the remaining 4 in the
ATM,
BRIP1,
RAD51D and
TP53 genes respectively (
Table 3).
Studies conducted on a larger number of TNBC patients have come to concordant results regarding the association of these cases with pathogenic variants in the
PALB2,
BRIP1,
RAD51C, and
RAD51D genes [
30,
36,
37,
38]. Our results partially agree with these data, having identified pathogenic variants in TNBC patients with
ATM,
RAD51D,
BRIP1, and
TP53 variants. It is likely that the analysis of a higher number of TNBC cases could have led us to a greater degree of concordance.
However, it is certainly very interesting that 50% of the TNBC cases analyzed had pathogenic coding variants: it is a higher proportion compared to that obtained in other cohorts of TNBC patients filtered in the population solely on the basis of their tumor phenotype [
36,
37]. It is probable that the higher percentage registered in our cases depends on the double filtering, based not only on patient tumor phenotype but also on the patient breast cancer family history.
It is known that some TNBCs BRCAWT show the so-called ‘BRCAness’ phenotype, exhibiting clinical and pathological properties similar to BRCA-mutated tumors such as the homologous recombination deficiency and the sensitivity to DNA damaging agents [
39,
40]. Therefore, our data could be of interest at a therapeutic level. In fact, the data supports what we have already reported in Spugnesi et al. [
38] where germline mutations in DNA repair genes, in BRCA1/2 WT tumors, were associated with a group of TNBC patients who responded to anthracyclines/taxanes neoadjuvant therapy. It is becoming increasingly evident that genes involved in DNA repair could become response biomarkers for DNA-damaging therapy in these patients [
41].
Although we cannot make any consideration about the variants identified in ovarian cancer, having only analyzed four patients with ovarian cancer and two with breast and ovarian cancer, the detection of pathogenic variants in
RAD51C and
ATM is in agreement with what is reported in a case-control study performed on 2051 women with ovarian cancer. The authors of this paper identified an increased risk for ovarian cancer associated with
MSH6,
RAD51C,
TP53, and
ATM [
42].
Analyzing the characteristics of non-coding variants carriers, we recorded an average age of disease onset of 44.4 absolutely similar to the average age of disease onset in coding variant carriers for each proband’s group with the same cancer type. Furthermore, there is not a statistically significant difference in the proportion of cases with a tumor onset under the age of 40 between the two groups (χ2-test,
p > 0.05;
Table 5).
A statistically significant increase in cases of ovarian cancer and of HBOC families in non-coding variant carriers compared to coding variant carriers was found (
Table 1/
Table 4). This is an interesting observation but very preliminary considering the limited number of samples analyzed, and it needs to be further investigated.
One of the results that emerges clearly when analyzing the characteristics of non-coding variant carriers, is the relationship between variants in 3′UTR of
PTEN and multiple tumors or TNBC tumor phenotype. This result is in agreement with what has already been described in previous studies in which the deregulation of
PTEN expression, via binding sites for RNA-binding-protein and miRNA located in 3′UTR, was associated with aggressive phenotype and poor outcome for BC patients [
16,
17]. Moreover, four out of seven
BRCA2 deep intronic variants identified (c.9257-3610G > A, c.681+697C > T, c.317-1021A > G, c.317-512A > T) have been found in TNBC patients.
Finally, by analyzing the 12 patients carrying multiple non-coding variants we observed a statistically significant association with a TNBC phenotype (χ2-test, p < 0.05).
This work has the merit of having highlighted certain variants never recorded in the coding and regulatory regions which are worthy of further study. It also underlines the importance, increasingly evident in recent years, of also screening the regulatory regions in patients with clearly familial tumors with no relevant variation in the coding sequences of the main breast/ovarian cancer predisposition genes. Certainly, in the future it will be necessary to perform a segregation analysis in order to understand the real involvement of certain genomic regions in breast and ovarian cancer predisposition. This work surely could help to pave the way for other in-depth projects on the functional meaning of the variants identified, by providing, thanks to the many notes obtained, a lot of useful information for a better understanding of their clinical significance.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}