
Whole Blood Transcriptome Characterization of 3xTg-AD Mouse and Its Modulation by Transcranial Direct Current Stimulation (tDCS)

 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
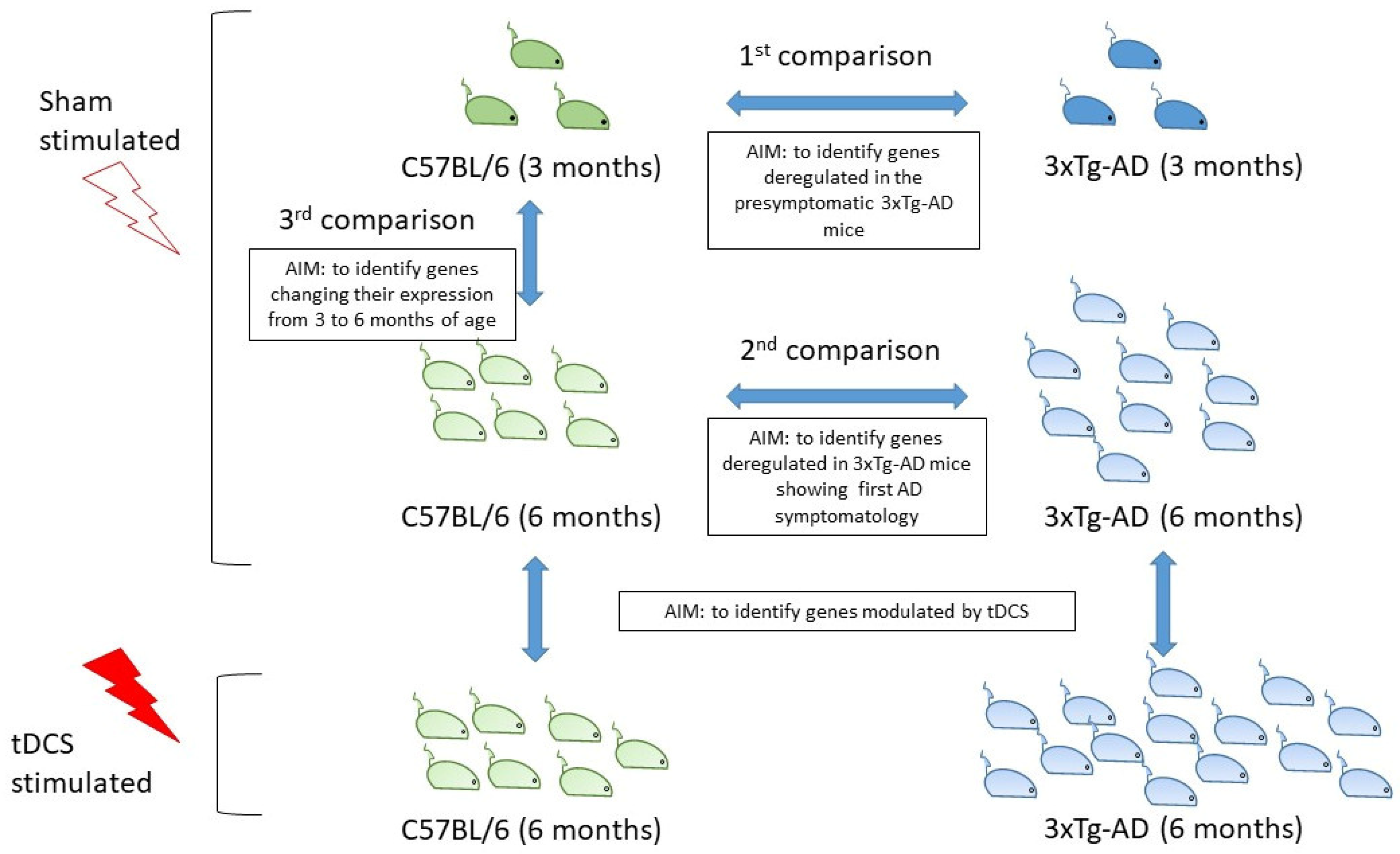
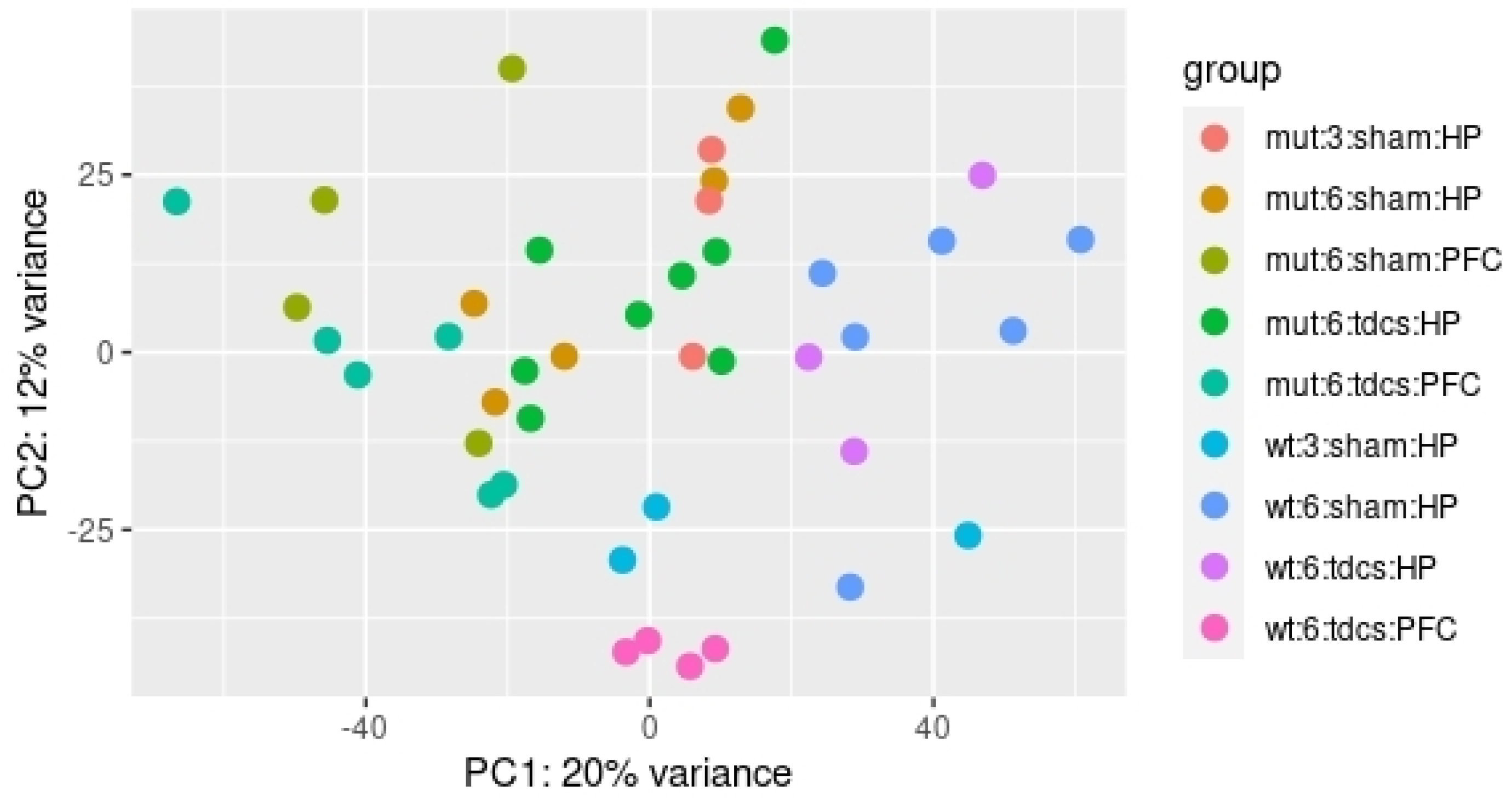
2.1. Differential Expression Analyses
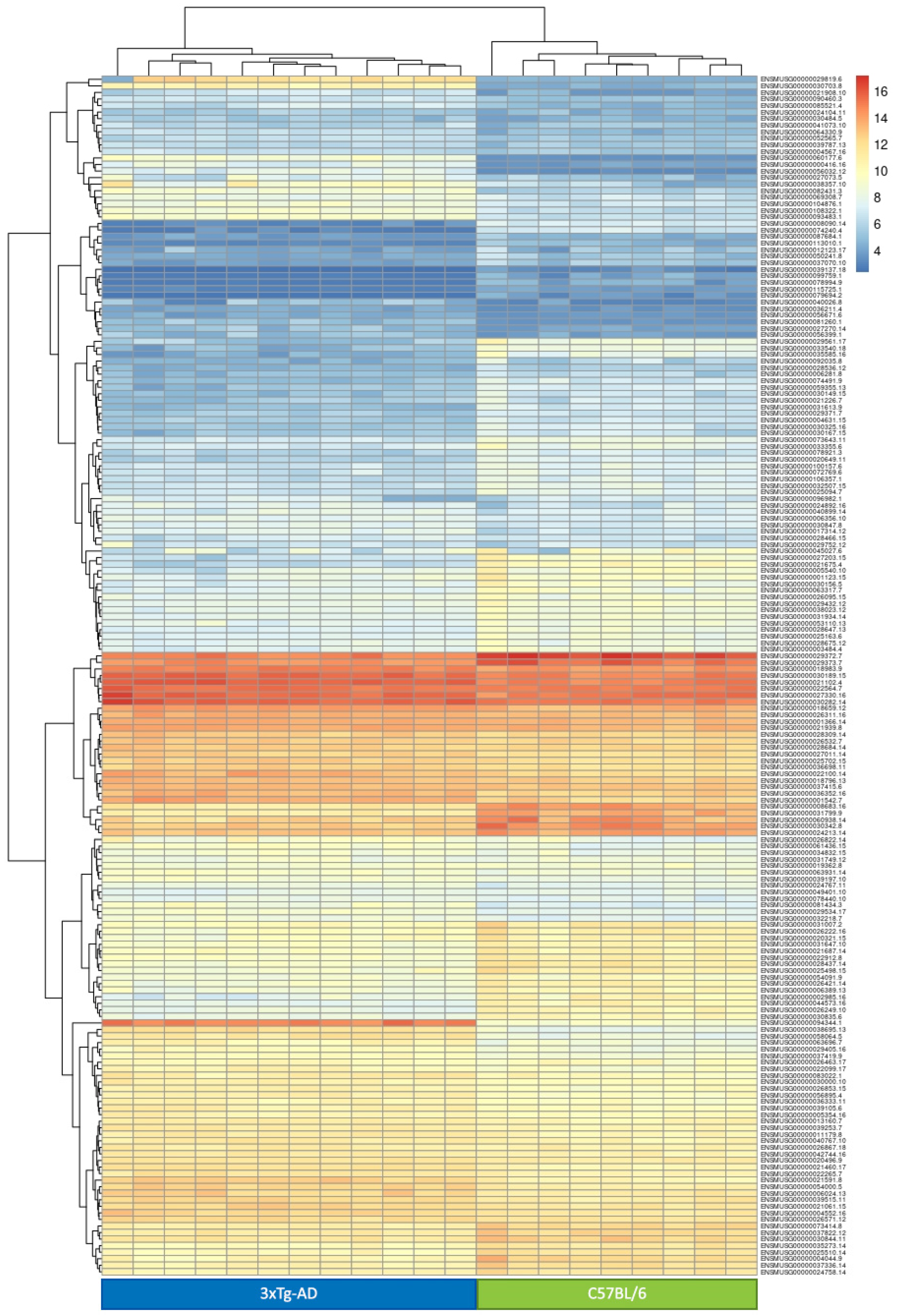
- 3-month-old (3 mo.) 3xTg-AD vs. 3 mo. C57BL/6 mice. Of the 15,613 genes studied, 69 were not analyzed in this comparison because of a total read count equal to zero. This analysis identified 470 genes (193 up- and 277 down-regulated) deregulated in 3xTg-AD mice at the pre-symptomatic stage, mostly representing the effect of the specific genetic background of this strain. The DEG with the highest log2 fold change was the neuropeptide Y gene (Npy). This gene, which is usually poorly expressed in whole blood [23,24], was not detected in the 3 mo. C57BL/6 (WT mice), while it was expressed at very high levels in 3 mo. 3xTg-AD mice (normalized mean number of reads: 6295 ± 2565). Among the top ten DEGs, Npy, Serpine2 and Acp1 genes were previously found associated at different levels with AD [25,26,27,28,29,30]. DEGs in 3 mo. 3xTg-AD mice were significantly enriched (FDR < 0.05) for genes belonging to the biological processes of leukocyte chemotaxis, blood coagulation, response to lipopolysaccharide, cellular metal ion homeostasis, positive regulation of hemopoiesis, regulation of cell-cell adhesion, negative regulation of secretion by cell, and regulation of blood coagulation (Supplementary Table S2).
- 6-month-old (6 mo.) 3xTg-AD vs. 6 mo. C57BL/6 mice. Of the 15,613 genes selected, only one was not analyzed in this comparison because of a total read count equal to zero. This comparison identified 1551 DEGs (912 up- and 639 down-regulated) deregulated in 3xTg-AD mice at a stage where they show the first symptoms of AD pathology. Thus, observed differences in gene expression can be either a direct consequence of the genetic background of this strain, or a secondary effect of the appearance of first AD symptoms. Among the top ten DEGs, those with the highest log2 fold change is the kallikrein 1-related peptidase b22 (Klk1b22). The identified DEGs were significantly enriched for genes belonging to the REACTOME pathway of immunoregulatory interactions between a lymphoid and a non-lymphoid cell (Supplementary Table S2).
- 6 mo. C57BL/6 vs. 3 mo. C57BL/6 mice. Of the 15,613 genes selected, 18 were not analyzed in this comparison because of a total read count equal to zero. This comparison identified 271 DEGs (214 up- and 57 down-regulated) changing their expression from 3 to 6 months of age. Among DEGs, the expression of Ddit4 and Folr1 has been previously shown to be age-related and associated with AD [31,32,33,34]. Pathway enrichment analysis revealed a significant enrichment for genes belonging to the keratinocyte differentiation pathway (Supplementary Table S2).
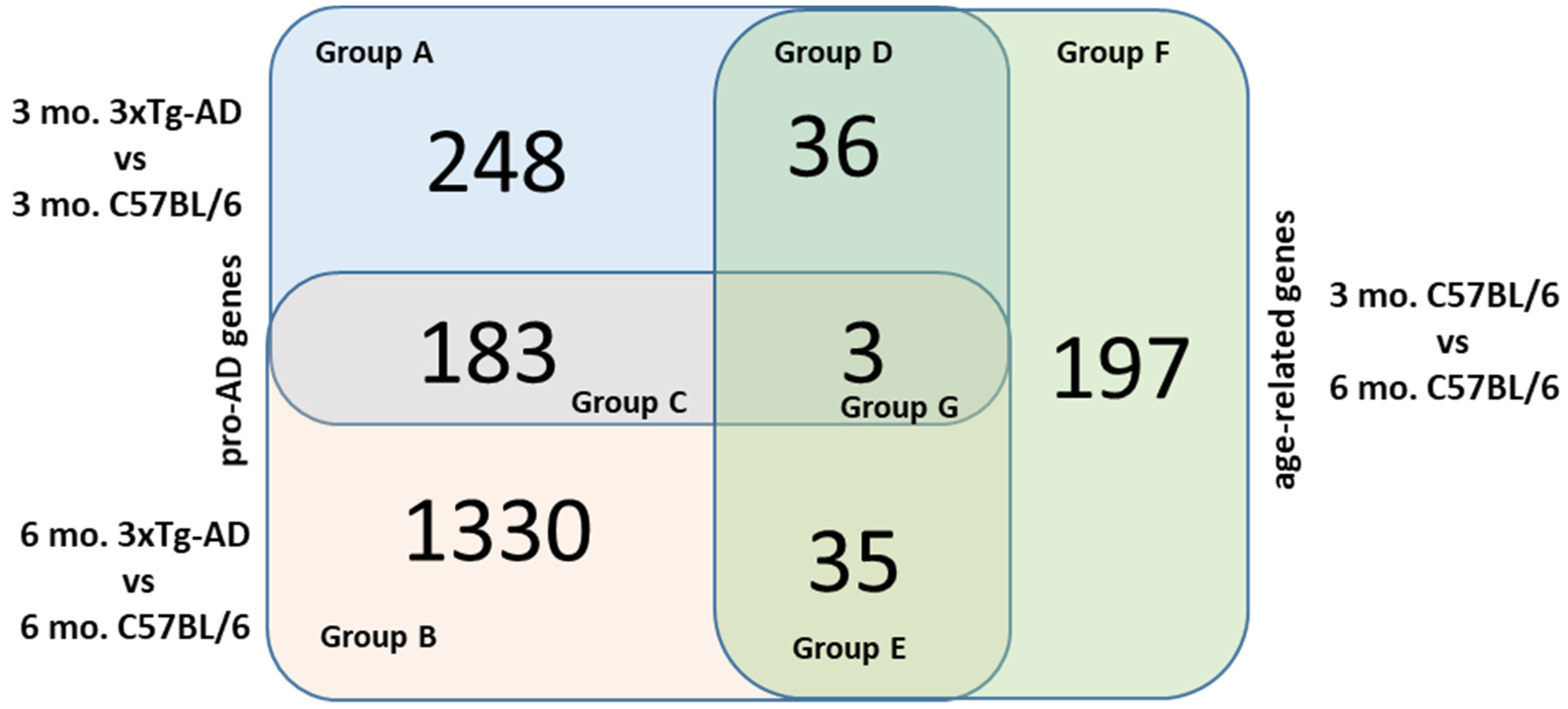
2.2. Venn Diagram Analysis
2.2.1. Group C
2.2.2. Group D
2.2.3. Group E
2.2.4. Groups A, B and F
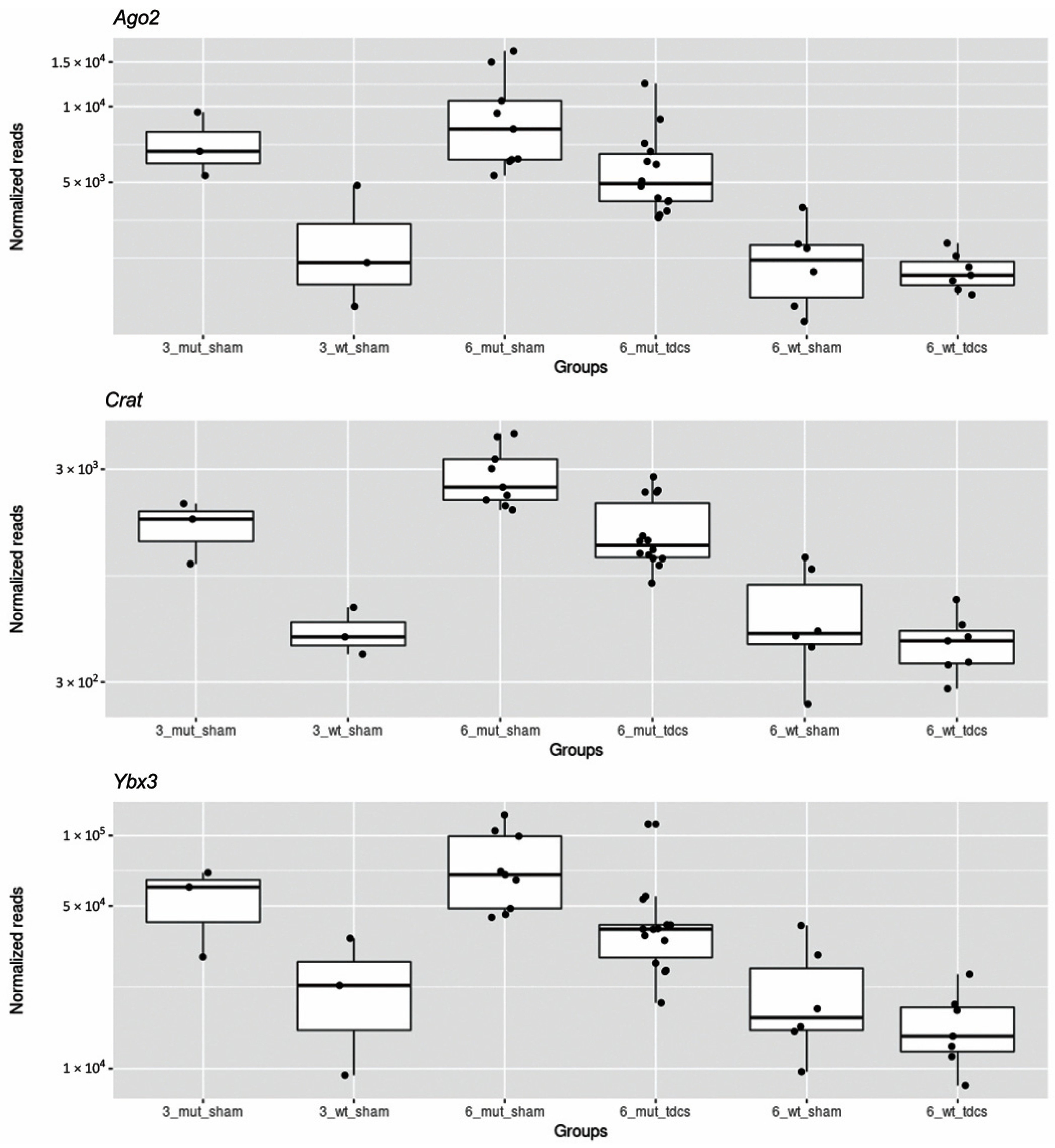
2.3. tDCS Effects on Whole Blood Transcriptome
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. tDCS Protocol
4.3. RNA-Seq on Blood Samples
4.4. Whole Transcriptome Analysis
4.4.1. Identification of Differentially Expressed Genes
4.4.2. Random Test Statistics on Venn Diagram
4.4.3. Over-Representation Analysis (ORA)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organisation Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 1 June 2021).
- Pini, L.; Pievani, M.; Bocchetta, M.; Altomare, D.; Bosco, P.; Cavedo, E.; Galluzzi, S.; Marizzoni, M.; Frisoni, G.B. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res. Rev. 2016, 30, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Mufson, E.J.; Mahady, L.; Waters, D.; Counts, S.E.; Perez, S.E.; DeKosky, S.T.; Ginsberg, S.D.; Ikonomovic, M.D.; Scheff, S.W.; Binder, L.I. Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience 2015, 309, 51–67. [Google Scholar] [CrossRef]
- Dennis, E.L.; Thompson, P.M. Functional brain connectivity using fMRI in aging and Alzheimer’s disease. Neuropsychol. Rev. 2014, 24, 49–62. [Google Scholar] [CrossRef]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef]
- Chiquita, S.; Ribeiro, M.; Castelhano, J.; Oliveira, F.; Sereno, J.; Batista, M.; Abrunhosa, A.; Rodrigues-Neves, A.C.; Carecho, R.; Baptista, F.; et al. A longitudinal multimodal in vivo molecular imaging study of the 3xTg-AD mouse model shows progressive early hippocampal and taurine loss. Hum. Mol. Genet. 2019, 28, 2174–2188. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, Z.; Liang, Z.; Sun, S.; Dai, C.-L.; Lee, M.H.; LaFerla, F.M.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; et al. Brain gene expression of a sporadic (icv-STZ Mouse) and a familial mouse model (3xTg-AD mouse) of Alzheimer’s disease. PLoS ONE 2012, 7, e51432. [Google Scholar] [CrossRef]
- Gatta, V.; D’Aurora, M.; Granzotto, A.; Stuppia, L.; Sensi, S.L. Early and sustained altered expression of aging-related genes in young 3xTg-AD mice. Cell Death Dis. 2014, 5, e1054. [Google Scholar] [CrossRef]
- Ochi, S.; Iga, J.-I.; Funahashi, Y.; Yoshino, Y.; Yamazaki, K.; Kumon, H.; Mori, H.; Ozaki, Y.; Mori, T.; Ueno, S.-I. Identifying Blood Transcriptome Biomarkers of Alzheimer’s Disease Using Transgenic Mice. Mol. Neurobiol. 2020, 57, 4941–4951. [Google Scholar] [CrossRef] [PubMed]
- Cavaleiro, C.; Martins, J.; Gonçalves, J.; Castelo-Branco, M. Memory and Cognition-Related Neuroplasticity Enhancement by Transcranial Direct Current Stimulation in Rodents: A Systematic Review. Neural Plast. 2020, 2020, 4795267. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Holland, P.; Frens, M.A.; Donchin, O. Impact of Transcranial Direct Current Stimulation (tDCS) on Neuronal Functions. Front. Neurosci. 2016, 10, 550. [Google Scholar] [CrossRef]
- Thair, H.; Holloway, A.L.; Newport, R.; Smith, A.D. Transcranial Direct Current Stimulation (tDCS): A Beginner’s Guide for Design and Implementation. Front. Neurosci. 2017, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Podda, M.V.; Cocco, S.; Mastrodonato, A.; Fusco, S.; Leone, L.; Barbati, S.A.; Colussi, C.; Ripoli, C.; Grassi, C. Anodal transcranial direct current stimulation boosts synaptic plasticity and memory in mice via epigenetic regulation of Bdnf expression. Sci. Rep. 2016, 6, 22180. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, M.; Unverricht-Yeboah, M.; Keuters, M.H.; Pikhovych, A.; Hucklenbroich, J.; Vay, S.U.; Blaschke, S.; Ladwig, A.; Walter, H.L.; Beiderbeck, M.; et al. Transcranial Current Stimulation Alters the Expression of Immune-Mediating Genes. Front. Cell. Neurosci. 2019, 13, 461. [Google Scholar] [CrossRef]
- Holmes, B.; Jung, S.H.; Lu, J.; Wagner, J.A.; Rubbi, L.; Pellegrini, M.; Jankord, R. Transcriptomic Modification in the Cerebral Cortex following Noninvasive Brain Stimulation: RNA-Sequencing Approach. Neural Plast. 2016, 2016, 5942980. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, C.A.; Choi, J.; Baldwin, T.M.; Bolden, J.E.; Fairfax, K.A.; Robinson, A.J.; Biben, C.; Morgan, C.; Ramsay, K.; Ng, A.P.; et al. Haemopedia: An Expression Atlas of Murine Hematopoietic Cells. Stem Cell Rep. 2016, 7, 571–582. [Google Scholar] [CrossRef]
- Haemosphere. Available online: https://www.haemosphere.org/expression/show?geneId=ENSMUSG00000029819 (accessed on 1 June 2021).
- Zou, D.; Li, R.; Huang, X.; Chen, G.; Liu, Y.; Meng, Y.; Wang, Y.; Wu, Y.; Mao, Y. Identification of molecular correlations of RBM8A with autophagy in Alzheimer’s disease. Aging (Albany. N. Y.) 2019, 11, 11673–11685. [Google Scholar] [CrossRef]
- Bi Oh, S.; Suh, N.; Kim, I.; Lee, J.-Y. Impacts of aging and amyloid-β deposition on plasminogen activators and plasminogen activator inhibitor-1 in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 2015, 1597, 159–167. [Google Scholar] [CrossRef]
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 991–1001. [Google Scholar] [CrossRef]
- Spencer, B.; Potkar, R.; Metcalf, J.; Thrin, I.; Adame, A.; Rockenstein, E.; Masliah, E. Systemic Central Nervous System (CNS)-targeted Delivery of Neuropeptide Y (NPY) Reduces Neurodegeneration and Increases Neural Precursor Cell Proliferation in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2016, 291, 1905–1920. [Google Scholar] [CrossRef]
- Duarte-Neves, J.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol. Dis. 2016, 95, 210–224. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Lustig, A.; Carter, A.; Sankar, M.; Daimon, C.M.; Premont, R.T.; Etienne, H.; van Gastel, J.; Azmi, A.; Janssens, J.; et al. Genomic deletion of GIT2 induces a premature age-related thymic dysfunction and systemic immune system disruption. Aging (Albany. N. Y.) 2017, 9, 706–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ju, X.; Cheng, Y.; Guo, X.; Wen, T. Identifying Tmem59 related gene regulatory network of mouse neural stem cell from a compendium of expression profiles. BMC Syst. Biol. 2011, 5, 152. [Google Scholar] [CrossRef]
- Woo, J.-M.; Park, S.J.; Kang, H.I.; Kim, B.G.; Shim, S.B.; Jee, S.W.; Lee, S.H.; Sin, J.S.; Bae, C.J.; Jang, M.K.; et al. Characterization of changes in global gene expression in the brain of neuron-specific enolase/human Tau23 transgenic mice in response to overexpression of Tau protein. Int. J. Mol. Med. 2010, 25, 667–675. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, H.-S.; Ahn, J.H.; Park, J.H.; Won, M.-H.; Lee, C.-H. Age-dependent changes in the protein expression levels of Redd1 and mTOR in the gerbil hippocampus during normal aging. Mol. Med. Rep. 2016, 13, 2409–2414. [Google Scholar] [CrossRef]
- Xapelli, S.; Agasse, F.; Ferreira, R.; Silva, A.P.; Malva, J.O. Neuropeptide Y as an endogenous antiepileptic, neuroprotective and pro-neurogenic peptide. Recent Pat. CNS Drug Discov. 2006, 1, 315–324. [Google Scholar] [CrossRef]
- Croce, N.; Gelfo, F.; Ciotti, M.T.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. NPY modulates miR-30a-5p and BDNF in opposite direction in an in vitro model of Alzheimer disease: A possible role in neuroprotection? Mol. Cell. Biochem. 2013, 376, 189–195. [Google Scholar] [CrossRef]
- Pain, S.; Vergote, J.; Gulhan, Z.; Bodard, S.; Chalon, S.; Gaillard, A. Inflammatory process in Parkinson disease: Neuroprotection by neuropeptide Y. Fundam. Clin. Pharmacol. 2019, 33, 544–548. [Google Scholar] [CrossRef]
- Gøtzsche, C.R.; Woldbye, D.P.D. The role of NPY in learning and memory. Neuropeptides 2016, 55, 79–89. [Google Scholar] [CrossRef]
- Do, K.; Laing, B.T.; Landry, T.; Bunner, W.; Mersaud, N.; Matsubara, T.; Li, P.; Yuan, Y.; Lu, Q.; Huang, H. The effects of exercise on hypothalamic neurodegeneration of Alzheimer’s disease mouse model. PLoS ONE 2018, 13, e0190205. [Google Scholar] [CrossRef]
- Mahar, I.; Albuquerque, M.S.; Mondragon-Rodriguez, S.; Cavanagh, C.; Davoli, M.A.; Chabot, J.-G.; Williams, S.; Mechawar, N.; Quirion, R.; Krantic, S. Phenotypic Alterations in Hippocampal NPY- and PV-Expressing Interneurons in a Presymptomatic Transgenic Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 327. [Google Scholar] [CrossRef]
- Ramos, B.; Baglietto-Vargas, D.; del Rio, J.C.; Moreno-Gonzalez, I.; Santa-Maria, C.; Jimenez, S.; Caballero, C.; Lopez-Tellez, J.F.; Khan, Z.U.; Ruano, D.; et al. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Diez, M.; Danner, S.; Frey, P.; Sommer, B.; Staufenbiel, M.; Wiederhold, K.-H.; Hökfelt, T. Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing beta-amyloid precursor protein (APP) with the Swedish double mutation (APP23). Neurobiol. Dis. 2003, 14, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Diez, M.; Koistinaho, J.; Kahn, K.; Games, D.; Hökfelt, T. Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F beta-amyloid precursor protein--initial observations. Neuroscience 2000, 100, 259–286. [Google Scholar] [CrossRef]
- Beal, M.F.; Mazurek, M.F.; Chattha, G.K.; Svendsen, C.N.; Bird, E.D.; Martin, J.B. Neuropeptide Y immunoreactivity is reduced in cerebral cortex in Alzheimer’s disease. Ann. Neurol. 1986, 20, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Proto, C.; Romualdi, D.; Cento, R.M.; Spada, R.S.; Di Mento, G.; Ferri, R.; Lanzone, A. Plasma levels of neuropeptides in Alzheimer’s disease. Gynecol. Endocrinol. 2006, 22, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.; Onishi, H.; Hashimoto, H.; Kai, T.; Yamagami, S. Plasma neuropeptide Y is reduced in patients with Alzheimer’s disease. Neurosci. Lett. 1995, 198, 149–151. [Google Scholar] [CrossRef]
- Zheng, X.; Demirci, F.Y.; Barmada, M.M.; Richardson, G.A.; Lopez, O.L.; Sweet, R.A.; Kamboh, M.I.; Feingold, E. A rare duplication on chromosome 16p11.2 is identified in patients with psychosis in Alzheimer’s disease. PLoS ONE 2014, 9, e111462. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Mann, D.; Richard, F.; Tian, J.; Shi, J.; Thaker, U.; Merrot, S.; Harris, J.; Frigard, B.; Iwatsubo, T.; et al. Is there a relation between APOE expression and brain amyloid load in Alzheimer’s disease? J. Neurol. Neurosurg. Psychiatry 2005, 76, 928–933. [Google Scholar] [CrossRef][Green Version]
- Wang, C.; Yu, J.-T.; Wang, H.-F.; Jiang, T.; Tan, C.-C.; Meng, X.-F.; Soares, H.D.; Tan, L. Meta-analysis of peripheral blood apolipoprotein E levels in Alzheimer’s disease. PLoS ONE 2014, 9, e89041. [Google Scholar] [CrossRef]
- Gupta, V.B.; Wilson, A.C.; Burnham, S.; Hone, E.; Pedrini, S.; Laws, S.M.; Lim, W.L.F.; Rembach, A.; Rainey-Smith, S.; Ames, D.; et al. Follow-up plasma apolipoprotein E levels in the Australian Imaging, Biomarkers and Lifestyle Flagship Study of Ageing (AIBL) cohort. Alzheimer’s Res. Ther. 2015, 7, 16. [Google Scholar] [CrossRef]
- Teng, E.; Chow, N.; Hwang, K.S.; Thompson, P.M.; Gylys, K.H.; Cole, G.M.; Jack, C.R.; Shaw, L.M.; Trojanowski, J.Q.; Soares, H.D.; et al. Low plasma ApoE levels are associated with smaller hippocampal size in the Alzheimer’s disease neuroimaging initiative cohort. Dement. Geriatr. Cogn. Disord. 2015, 39, 154–166. [Google Scholar] [CrossRef]
- Wolters, F.J.; Koudstaal, P.J.; Hofman, A.; van Duijn, C.M.; Ikram, M.A. Serum apolipoprotein E is associated with long-term risk of Alzheimer’s disease: The Rotterdam Study. Neurosci. Lett. 2016, 617, 139–142. [Google Scholar] [CrossRef]
- Akiyama, H.; Ikeda, K.; Kondo, H.; McGeer, P.L. Thrombin accumulation in brains of patients with Alzheimer’s disease. Neurosci. Lett. 1992, 146, 152–154. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol. Aging 2015, 36, 608–617. [Google Scholar] [CrossRef]
- Gupta, A.; Watkins, A.; Thomas, P.; Majer, R.; Habubi, N.; Morris, G.; Pansari, K. Coagulation and inflammatory markers in Alzheimer’s and vascular dementia. Int. J. Clin. Pract. 2005, 59, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Canobbio, I.; Visconte, C.; Oliviero, B.; Guidetti, G.; Zarà, M.; Pula, G.; Torti, M. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer’s disease. Cell. Signal. 2016, 28, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Jarre, A.; Gowert, N.S.; Donner, L.; Münzer, P.; Klier, M.; Borst, O.; Schaller, M.; Lang, F.; Korth, C.; Elvers, M. Pre-activated blood platelets and a pro-thrombotic phenotype in APP23 mice modeling Alzheimer’s disease. Cell. Signal. 2014, 26, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Suidan, G.L.; Singh, P.K.; Patel-Hett, S.; Chen, Z.-L.; Volfson, D.; Yamamoto-Imoto, H.; Norris, E.H.; Bell, R.D.; Strickland, S. Abnormal clotting of the intrinsic/contact pathway in Alzheimer disease patients is related to cognitive ability. Blood Adv. 2018, 2, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Song, J.-X.; Wang, S.-F.; Cai, C.-Z.; Li, M.; Lu, J.-H. Selective autophagy: The new player in the fight against neurodegenerative diseases? Brain Res. Bull. 2018, 137, 79–90. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef]
- Kritsilis, M.; V Rizou, S.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.-G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar] [CrossRef]
- Wang, W.; Bu, B.; Xie, M.; Zhang, M.; Yu, Z.; Tao, D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009, 89, 1–17. [Google Scholar] [CrossRef]
- Seward, M.E.; Swanson, E.; Norambuena, A.; Reimann, A.; Cochran, J.N.; Li, R.; Roberson, E.D.; Bloom, G.S. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 2013, 126, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Casadesus, G.; Nunomura, A.; Zhu, X.; Castellani, R.J.; Richardson, S.L.; Perry, G.; Felsher, D.W.; Petersen, R.B.; Smith, M.A. The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am. J. Pathol. 2009, 174, 891–897. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Smith, M.A.; Perry, G.; Whitehouse, P.J.; Taniguchi, T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Res. 1998, 780, 260–269. [Google Scholar] [CrossRef]
- Su, J.H.; Deng, G.; Cotman, C.W. Bax protein expression is increased in Alzheimer’s brain: Correlations with DNA damage, Bcl-2 expression, and brain pathology. J. Neuropathol. Exp. Neurol. 1997, 56, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kwon, Y.-A.; Ahn, I.S.; Kim, S.; Kim, S.; Jo, S.A.; Kim, D.K. Overexpression of Cell Cycle Proteins of Peripheral Lymphocytes in Patients with Alzheimer’s Disease. Psychiatry Investig. 2016, 13, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Koo, H.; Han, S.W.; Paulus, W.; Nitsche, M.A.; Kim, Y.-H.; Yoon, J.A.; Shin, Y.-I. Repeated anodal transcranial direct current stimulation induces neural plasticity-associated gene expression in the rat cortex and hippocampus. Restor. Neurol. Neurosci. 2017, 35, 137–146. [Google Scholar] [CrossRef]
- Smith, C.M.; Hayamizu, T.F.; Finger, J.H.; Bello, S.M.; McCright, I.J.; Xu, J.; Baldarelli, R.M.; Beal, J.S.; Campbell, J.; Corbani, L.E.; et al. The mouse Gene Expression Database (GXD): 2019 update. Nucleic Acids Res. 2019, 47, D774–D779. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Lane, H.-Y.; Lin, C.-H. Brain Stimulation in Alzheimer’s Disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhou, F.; Cao, X.; Yang, J.; Bai, Y.; Yan, X.; Cao, J.; Qi, J. Abnormal circadian locomotor rhythms and Per gene expression in six-month-old triple transgenic mice model of Alzheimer’s disease. Neurosci. Lett. 2018, 676, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Piedrahita, D.; Hernández, I.; López-Tobón, A.; Fedorov, D.; Obara, B.; Manjunath, B.S.; Boudreau, R.L.; Davidson, B.; Laferla, F.; Gallego-Gómez, J.C.; et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J. Neurosci. 2010, 30, 13966–13976. [Google Scholar] [CrossRef]
- Melkonyan, M.M.; Hunanyan, L.; Lourhmati, A.; Layer, N.; Beer-Hammer, S.; Yenkoyan, K.; Schwab, M.; Danielyan, L. Neuroprotective, Neurogenic, and Amyloid Beta Reducing Effect of a Novel Alpha 2-Adrenoblocker, Mesedin, on Astroglia and Neuronal Progenitors upon Hypoxia and Glutamate Exposure. Int. J. Mol. Sci. 2017, 19, 9. [Google Scholar] [CrossRef]
- Dissanayaka, T.D.; Zoghi, M.; Farrell, M.; Egan, G.F.; Jaberzadeh, S. Sham transcranial electrical stimulation and its effects on corticospinal excitability: A systematic review and meta-analysis. Rev. Neurosci. 2018, 29, 223–232. [Google Scholar] [CrossRef]
- Barbati, S.A.; Cocco, S.; Longo, V.; Spinelli, M.; Gironi, K.; Mattera, A.; Paciello, F.; Colussi, C.; Podda, M.V.; Grassi, C. Enhancing Plasticity Mechanisms in the Mouse Motor Cortex by Anodal Transcranial Direct-Current Stimulation: The Contribution of Nitric Oxide Signaling. Cereb. Cortex 2020, 30, 2972–2985. [Google Scholar] [CrossRef]
- Cocco, S.; Rinaudo, M.; Fusco, S.; Longo, V.; Gironi, K.; Renna, P.; Aceto, G.; Mastrodonato, A.; Li Puma, D.D.; Podda, M.V.; et al. Plasma BDNF Levels Following Transcranial Direct Current Stimulation Allow Prediction of Synaptic Plasticity and Memory Deficits in 3xTg-AD Mice. Front. cell Dev. Biol. 2020, 8, 541. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 1997. [Google Scholar]
- Hardwick, S.A.; Chen, W.Y.; Wong, T.; Deveson, I.W.; Blackburn, J.; Andersen, S.B.; Nielsen, L.K.; Mattick, J.S.; Mercer, T.R. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods 2016, 13, 792–798. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Age | Stimulation | Number of Mice 1 |
|---|---|---|---|
| 3xTg-AD | 3 months old | sham | 3 (HP) |
| 6 months old | sham | 9 (5 HP, 4 PFC) | |
| tDCS | 14 (8 HP, 6 PFC) | ||
| C57BL/6 | 3 months old | sham | 3 (HP) |
| 6 months old | sham | 6 (HP) | |
| tDCS | 7 (3 HP, 4 PFC) |
| Gene Name | Base Mean | log2 Fold Change | log2 Fold SE | p Value | FDR | Official Full Name |
|---|---|---|---|---|---|---|
| 1st Comparison: 3 mo. 3xTg-AD vs. 3 mo. C57BL/6 mice | ||||||
| Npy | 3148 | 16.02 | 1.95 | 1.36 × 10−33 | 1.66 × 10−29 | neuropeptide Y |
| Gdpd3 | 1025 | 13.24 | 1.87 | 2.48 × 10−30 | 1.52 × 10−26 | glycerophosphodiester phosphodiesterase domain containing 3 |
| Dut | 552 | −4.55 | 0.49 | 1.66 × 10−23 | 6.76 × 10−20 | deoxyuridine triphosphatase |
| Gm11942 | 29,125 | 9.49 | 0.63 | 4.35 × 10−23 | 1.33 × 10−19 | predicted gene 11,942 b22 |
| F2rl2 | 711 | −4.88 | 0.53 | 7.00 × 10−23 | 1.71 × 10−19 | coagulation factor II (thrombin) receptor-like 2 |
| Klk1b22 | 272 | 11.94 | 1.87 | 9.40 × 10−23 | 1.92 × 10−19 | kallikrein 1-related peptidase |
| Tpm4 | 6746 | −3.75 | 0.40 | 2.79 × 10−22 | 4.87 × 10−19 | tropomyosin 4 |
| Serpine2 | 798 | −3.12 | 0.37 | 4.13 × 10−19 | 6.31 × 10−16 | serine (or cysteine) peptidase inhibitor, clade E, member 2 |
| Acp1 | 597 | −3.65 | 0.44 | 3.29 × 10−18 | 4.47 × 10−15 | acid phosphatase 1, soluble |
| Josd2 | 2113 | 4.65 | 0.49 | 3.91 × 10−15 | 4.78 × 10−12 | Josephin domain containing 2 |
| 2nd Comparison: 6 mo. 3xTg-AD vs. 6 mo. C57BL/6 mice | ||||||
| Klk1b22 | 770 | 12.65 | 0.90 | 9.89 × 10−87 | 1.42 × 10−82 | kallikrein 1-related peptidase b22 |
| Gm11942 | 35,739 | 8.60 | 0.35 | 3.51 × 10−78 | 2.53 × 10−74 | predicted gene 11,942 |
| Aa465934 | 665 | 4.77 | 0.25 | 1.05 × 10−70 | 5.03 × 10−67 | lncRNA gene |
| Bc018473 | 297 | 11.30 | 0.91 | 8.18 × 10−66 | 2.94 × 10−62 | cDNA sequence BC018473 |
| Gdpd3 | 2066 | 9.60 | 0.48 | 3.58 × 10−65 | 1.03 × 10−61 | glycerophosphodiester phosphodiesterase domain containing |
| Cttnbp2 | 326 | 5.89 | 0.39 | 1.91 × 10−58 | 4.57 × 10−55 | cortactin Binding Protein 2 |
| Josd2 | 5004 | 5.96 | 0.32 | 9.44 × 10−57 | 1.94 × 10−53 | Josephin domain containing 2 |
| Fgfrl1 | 85 | −7.98 | 0.68 | 7.04 × 10−44 | 1.27 × 10−40 | 3fibroblast growth factor receptor-like 1 |
| Gm48905 | 151 | −7.56 | 0.57 | 1.60 × 10−38 | 2.55 × 10−35 | predicted gene 48,905 |
| 4930526A20rik | 129 | 5.30 | 0.39 | 1.56 × 10−36 | 2.24 × 10−33 | pseudogene |
| 3rd Comparison: 6 mo. C57BL/6 vs. 3 mo. C57BL/6 mice | ||||||
| Evpl | 47 | 9.50 | 1.32 | 1.03 × 10−16 | 1.45 × 10−12 | envoplakin |
| Slc13a2 | 73 | 4.33 | 0.57 | 4.59 × 10−14 | 3.23 × 10−10 | solute carrier family 13 (sodium-dependent dicarboxylate transporter), member 2 |
| Ddit4 | 157 | −2.96 | 0.44 | 4.77 × 10−13 | 2.24 × 10−9 | DNA-damage-inducible transcript 4 |
| Folr1 | 23 | 7.27 | 1.32 | 6.63 × 10−11 | 2.25 × 10−7 | folate receptor 1 (adult) |
| Acsm1 | 32 | 8.71 | 1.45 | 7.98 × 10−11 | 2.25 × 10−7 | acyl-CoA synthetase medium-chain family member 1 |
| Cyp4a12B | 109 | 6.82 | 1.08 | 5.43 × 10−10 | 1.28 × 10−6 | cytochrome P450, family 4, subfamily a, polypeptide 12B |
| Gm16136 | 39 | 9.07 | 1.53 | 3.55 × 10−9 | 7.14 × 10−6 | predicted gene 16,136 |
| B3gnt8 | 936 | 2.19 | 0.34 | 4.18 × 10−9 | 7.36 × 10−6 | UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 8 |
| Ces2f | 16 | 7.40 | 1.42 | 9.79 × 10−9 | 1.53 × 10−5 | carboxylesterase 2F |
| Ddr1 | 23 | 7.28 | 1.41 | 2.00 × 10−8 | 2.56 × 10−5 | discoidin domain receptor family, member 1 |
| Pathways Enriched among Down-Regulated Genes of Group C | |||||||
| Gene Set | Description | Size | Expect | Ratio | pValue | FDR | Gene List |
| GO:0007596 | blood coagulation | 117 | 13.99 | 64.33 | 1.1 × 10−5 | 0.034 | Apoe, F2rl2, Dmtn, Pros1, Serpine2, Pf4, Cd9, Hpse, Mpig6b |
| GO:0030193 | regulation of blood coagulation | 50 | 0.60 | 10.04 | 2.7 × 10−5 | 0.045 | Apoe, Dmtn, Pros1, Serpine2, Cd9, Hpse |
| Pathways Enriched among Down-Regulated Genes of Group D | |||||||
| Gene Set | Description | Size | Expect | Ratio | pValue | FDR | Gene List |
| GO:0051591 | response to cAMP | 66 | 0.11 | 35.96 | 4.0 × 10−6 | 0.0330 | Dusp1, Fos, Fosb, Junb |
| Pathways Enriched among Down-Regulated Genes of Group A | |||||||
| Gene Set | Description | Size | Expect | Ratio | pValue | FDR | Gene List |
| R-MMU-449147 | Signaling by Interleukins | 247 | 3.01 | 4.65 | 01.9 × 10−6 | 0.008 | Lck, Il17ra, Psmb1, Peli1, Vamp2, Stat5b, Psmb8, Il1r2, Il1b, Nfkbib, Il15, Pik3cb, Osm, Mapk1 |
| Pathways Enriched Among Up-Regulated Genes of Group B | |||||||
| Gene Set | Description | Size | Expect | Ratio | pValue | FDR | Gene List |
| GO:0061912 | selective autophagy | 42 | 2.03 | 4.93 | 2.3 × 10−5 | 0.042 | Sqstm1, Cdc37, Sptlc1, Prkn, Rb1cc1, Optn, Becn1, Wdr81, Mapk3, Clec16a |
| GO:0010466 | negative regulation of peptidase activity | 161 | 7.78 | 2.70 | 3.1 × 10−5 | 0.043 | Bcl2l1, Birc5, Arrb1, Rffl, Cast, Pak2, Birc6, Crim1, C3, Dnajb6, Ints1, Rps6ka3, Ngp, Ltf, Dhcr24, Gpi1, Fcmr, Usp14, Wfdc21, Gpx1, Wfdc3 |
| Pathways Enriched among DEGs of Group F | |||||||
| Gene Set | Description | Size | Expect | Ratio | pValue | FDR | Gene List |
| GO:0030216 | keratinocyte differentiation | 102 | 1.32 | 8.3 | 7.7 × 10−8 | 0.0006 | Grhl1, Cers3, Ptgs2, Evpl, Kdf1, Lats1, Lce1l, Dsp, Lce1a1, Cyp26b1, Lce1a2 |
| GO:0030036 | actin cytoskeleton organization | 485 | 6.27 | 3.0 | 1.6 × 10−5 | 0.033 | Scin, Tnfaip1, Myo1b, Cobl, Dpysl3, Epb41l5, Epb41l4b, Fgr, Dlc1, Sorbs2, Cgnl1, Kif23, Kank1, Arhgef5, Rhov, Prr5, Lats1, Tacstd2, Gm14137 |
| R-MMU-211945 | Phase I—Functionalization of compounds | 73 | 0.94 | 7.4 | 4.2 × 10−5 | 0.048 | Fdxr, Aldh3a1, Aldh1a7, Adh7, Ces2f, Cyp26b1, Cyp4a12b |
| GO:0050891 | multicellular organismal water homeostasis | 32 | 0.41 | 12.1 | 5.1 × 10−5 | 0.048 | Grhl1, Tfap2b, Aqp3, Kdf1, Cyp26b1 |
| GO:0045104 | intermediate filament cytoskeleton organization | 34 | 0.44 | 11.4 | 6.9 × 10−5 | 0.048 | Krt18, Dst, Pkp1, Evpl, Dsp |
| Genes | Base Mean | log2 Fold Change | log2 Fold SE | p Value | FDR | Official Full Name |
|---|---|---|---|---|---|---|
| Pabpc1 | 363,466 | −1.17 | 0.25 | 2.0 × 10−6 | 0.010 | poly(A) binding protein, cytoplasmic 1 |
| Crat | 1846 | −0.86 | 0.20 | 1.4 × 10−5 | 0.022 | carnitine acetyltransferase |
| Casp4 | 113 | 1.33 | 0.28 | 1.1 × 10−5 | 0.022 | caspase 4, apoptosis-related cysteine peptidase |
| Il13ra1 | 181 | 1.15 | 0.27 | 3.6 × 10−5 | 0.042 | interleukin 13 receptor, alpha 1 |
| Septin1 | 928 | −0.81 | 0.21 | 8.3 × 10−5 | 0.049 | septin 1 |
| Ccr1 | 262 | 1.33 | 0.31 | 5.9 × 10−5 | 0.049 | chemokine (C-C motif) receptor 1 |
| Ybx3 | 48,855 | −0.79 | 0.21 | 7.7 × 10−5 | 0.049 | Y box protein 3 |
| Ptp4a3 | 35,887 | −0.75 | 0.19 | 8.1 × 10−5 | 0.049 | protein tyrosine phosphatase 4a3 |
| Ap1b1 | 458 | −0.65 | 0.17 | 1.1 × 10−4 | 0.049 | adaptor protein complex AP-1, beta 1 subunit |
| Tmpo | 362 | 0.57 | 0.14 | 1.2 × 10−4 | 0.049 | thymopoietin |
| Fcgr3 | 583 | 0.96 | 0.24 | 1.1 × 10−4 | 0.049 | Fc receptor, IgG, low affinity III |
| Stfa2l1 | 521 | 1.35 | 0.33 | 1.3 × 10−4 | 0.049 | stefin A2 |
| Cd52 | 6685 | 0.53 | 0.14 | 2.6 × 10−4 | 0.049 | CD52 antigen |
| Cbfa2t3 | 366 | −0.72 | 0.20 | 2.6 × 10−4 | 0.049 | CBFA2/RUNX1 translocation partner 3 |
| Ogdh | 1902 | −0.70 | 0.19 | 2.3 × 10−4 | 0.049 | oxoglutarate (alpha-ketoglutarate) dehydrogenase (lipoamide) |
| Retnlg | 9116 | 1.36 | 0.34 | 1.6 × 10−4 | 0.049 | resistin-like gamma |
| Il1r2 | 262 | 2.04 | 0.52 | 2.6 × 10−4 | 0.049 | interleukin 1 receptor, type II |
| Vim | 948 | 0.81 | 0.21 | 2.2 × 10−4 | 0.049 | vimentin |
| Tlr13 | 263 | 0.93 | 0.24 | 2.4 × 10−4 | 0.049 | toll-like receptor 13 |
| Ago2 | 6349 | −0.67 | 0.18 | 1.9× 10−4 | 0.049 | argonaute RISC catalytic subunit 2 |
| Rnf123 | 285 | −0.94 | 0.26 | 1.9 × 10−4 | 0.049 | ring finger protein 123 |
| Mical3 | 269 | −0.85 | 0.23 | 2.0 × 10−4 | 0.049 | microtubule associated monooxygenase, calponin and LIM domain containing 3 |
| Cstdc4 | 472 | 1.40 | 0.35 | 1.7 × 10−4 | 0.049 | predicted gene 5483 |
| Sirpb1b | 133 | 1.19 | 0.31 | 1.8 × 10−4 | 0.049 | signal-regulatory protein beta 1B |
| Mrpl33 | 1057 | 0.85 | 0.22 | 2.0 × 10−4 | 0.049 | mitochondrial ribosomal protein L33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magri, C.; Vitali, E.; Cocco, S.; Giacopuzzi, E.; Rinaudo, M.; Martini, P.; Barbon, A.; Grassi, C.; Gennarelli, M. Whole Blood Transcriptome Characterization of 3xTg-AD Mouse and Its Modulation by Transcranial Direct Current Stimulation (tDCS). Int. J. Mol. Sci. 2021, 22, 7629. https://doi.org/10.3390/ijms22147629
Magri C, Vitali E, Cocco S, Giacopuzzi E, Rinaudo M, Martini P, Barbon A, Grassi C, Gennarelli M. Whole Blood Transcriptome Characterization of 3xTg-AD Mouse and Its Modulation by Transcranial Direct Current Stimulation (tDCS). International Journal of Molecular Sciences. 2021; 22(14):7629. https://doi.org/10.3390/ijms22147629
Chicago/Turabian StyleMagri, Chiara, Erika Vitali, Sara Cocco, Edoardo Giacopuzzi, Marco Rinaudo, Paolo Martini, Alessandro Barbon, Claudio Grassi, and Massimo Gennarelli. 2021. "Whole Blood Transcriptome Characterization of 3xTg-AD Mouse and Its Modulation by Transcranial Direct Current Stimulation (tDCS)" International Journal of Molecular Sciences 22, no. 14: 7629. https://doi.org/10.3390/ijms22147629
APA StyleMagri, C., Vitali, E., Cocco, S., Giacopuzzi, E., Rinaudo, M., Martini, P., Barbon, A., Grassi, C., & Gennarelli, M. (2021). Whole Blood Transcriptome Characterization of 3xTg-AD Mouse and Its Modulation by Transcranial Direct Current Stimulation (tDCS). International Journal of Molecular Sciences, 22(14), 7629. https://doi.org/10.3390/ijms22147629