
Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau

Abstract
1. Introduction
2. APP Metabolic Products Including Aβ Facilitated the Influx of Ca2+ into the Neurons of AD Animals and Patients
3. Ca2+ Transporters on the Surface of the Nerve Cell Membrane Are Responsible for Promoting the Influx of Ca2+ during the Course of AD Development and Progression
4. ER Is an Important Reservoir to Elevate the Levels of Ca2+ in the Neurons of AD
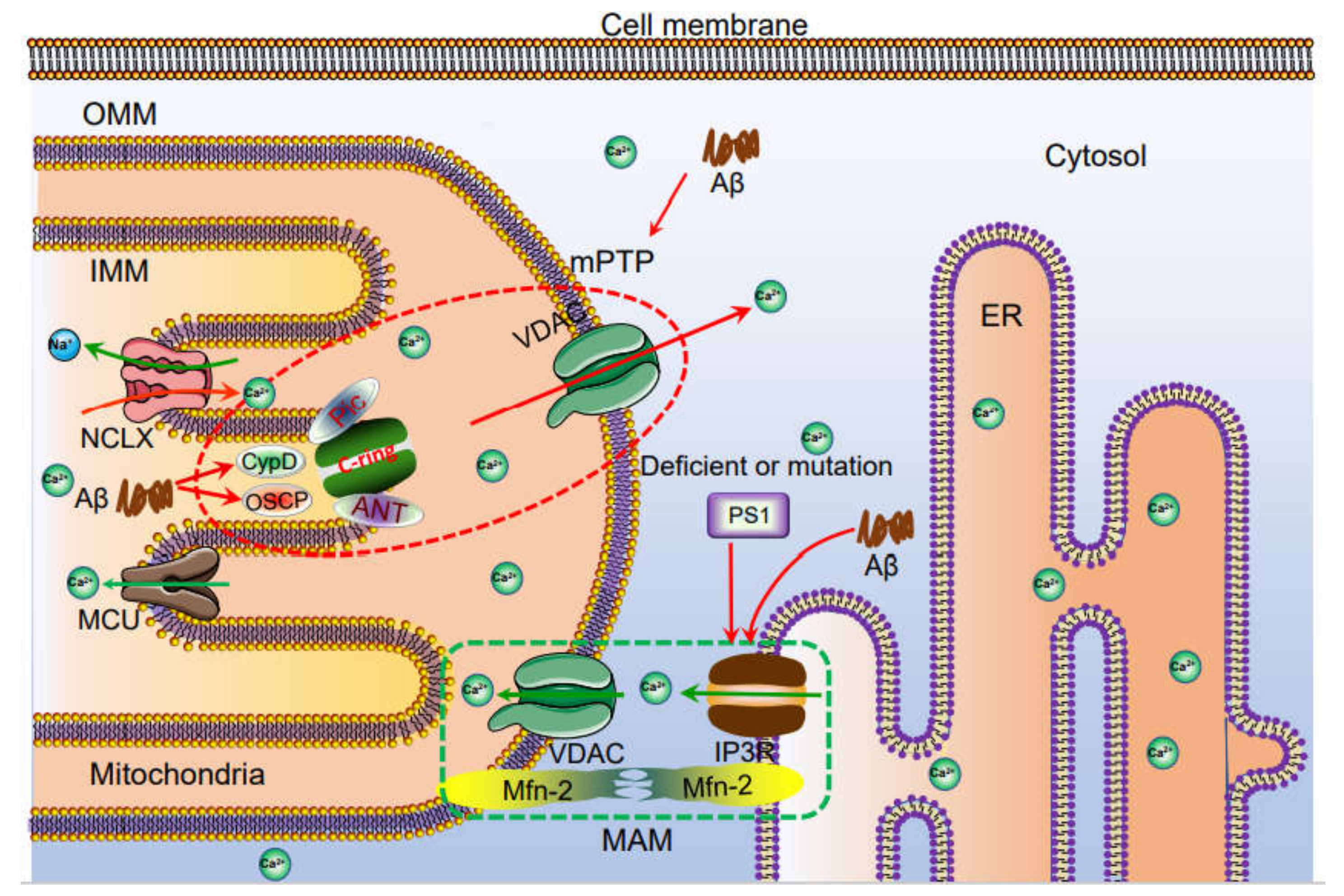
5. Mitochondria and Lysosomes Also Act as Important Organelles for Regulating the Dyshomeostasis of Ca2+ during the Development and Progression of AD
6. The Roles of Ca2+ in the Production and Deposition of Aβ during the Course of AD Development and Progression
7. Ca2+ Transporters on the Cell Membrane Are Potentially Contributed to the Role of Aβ in the Pathogenesis of AD
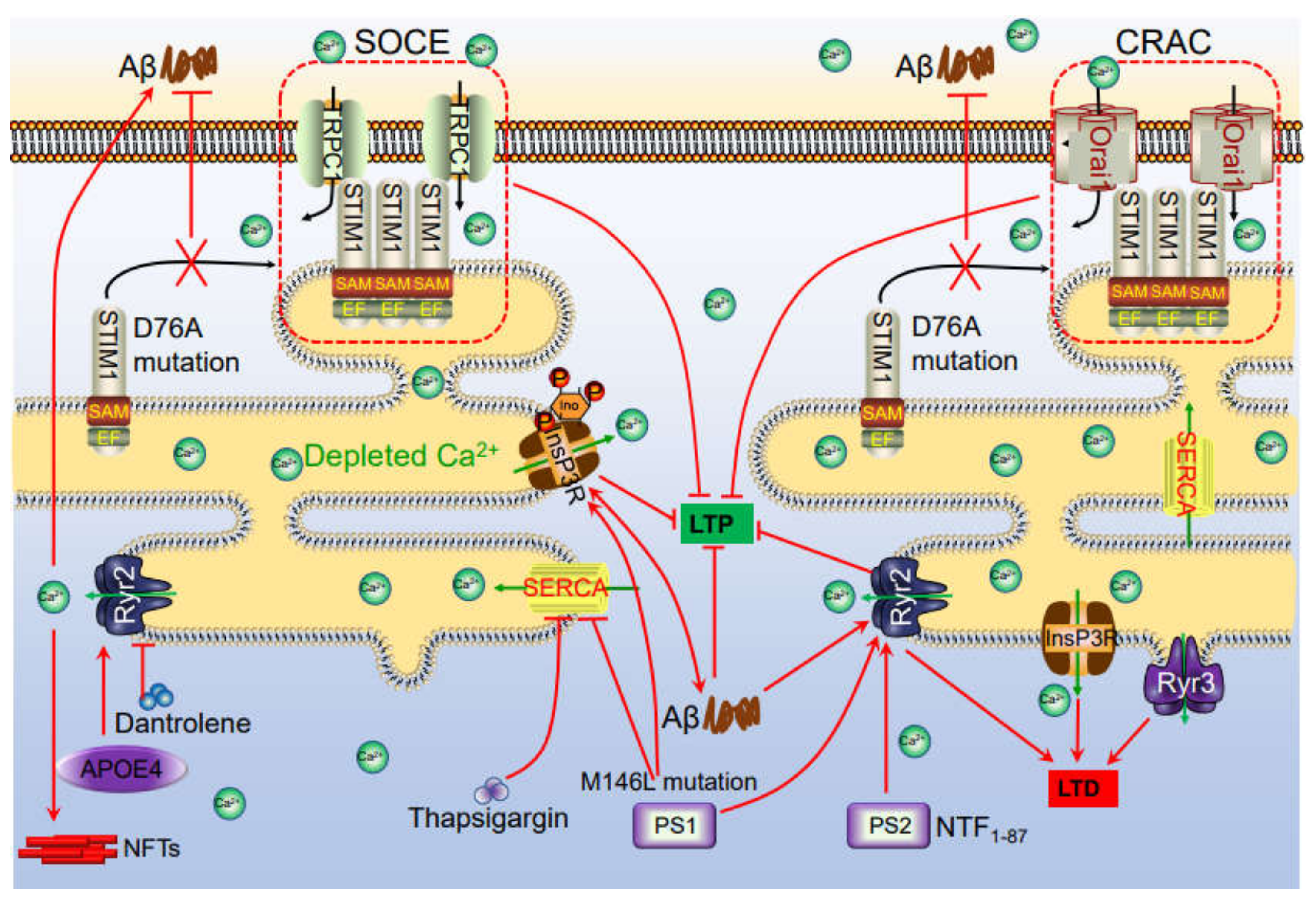
8. Ca2+ Leakage from ER Modulates the Production and Deposition of Aβ via Activating Ca2+ Transporters on ER
9. Ca2+ Transporters on the Membranes of Mitochondria Are Also Involved in Regulating the Production and Deposition of Aβ during the Course of AD Development and Progression
10. The Roles of Ca2+ in Regulating the Phosphorylation of Tau
11. Ca2+ Accelerates the Cognitive Decline Associated with AD
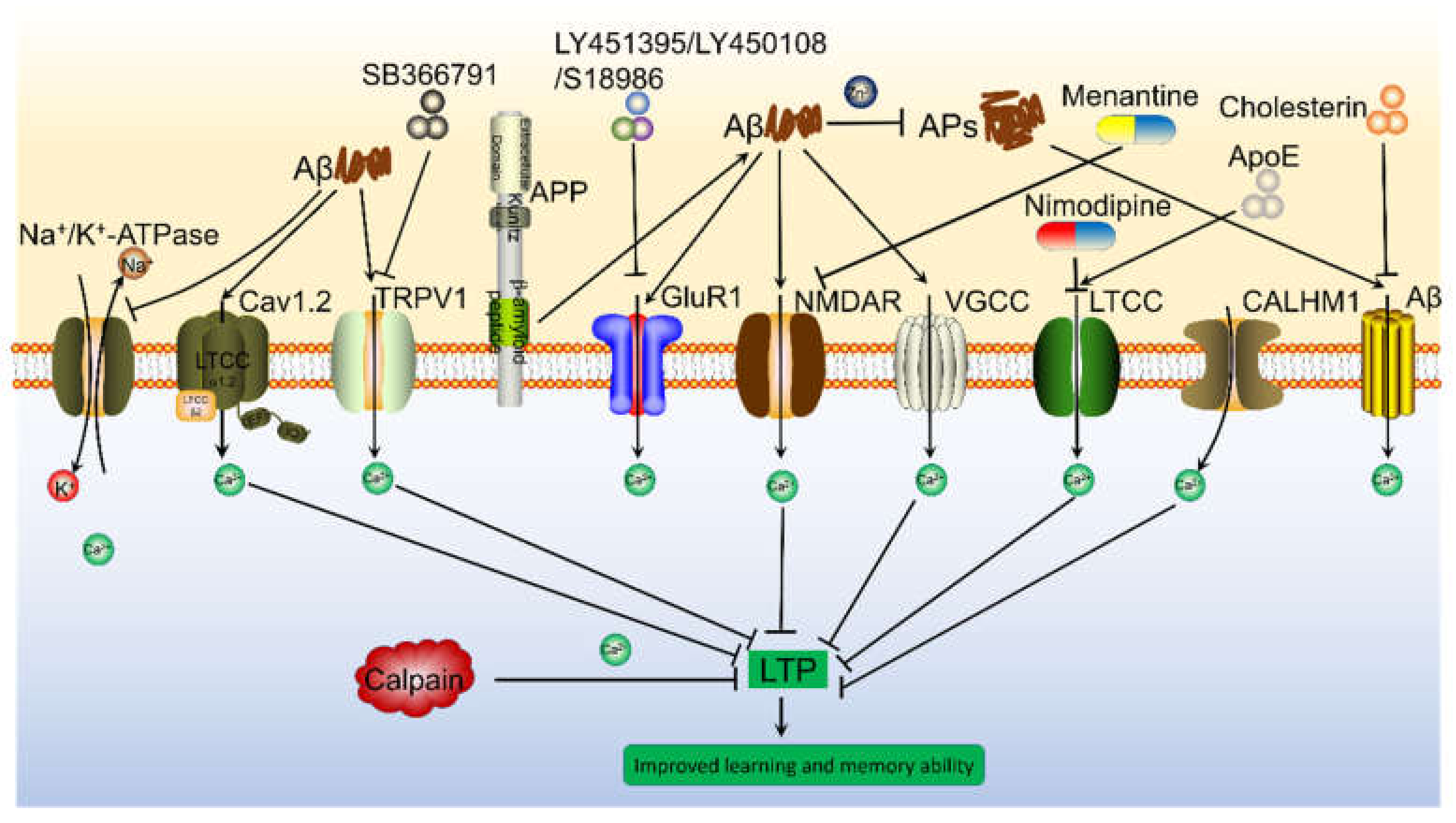
12. Transporters on the Cell Membrane Mediated the Effects of Ca2+ on Inducing the Cognitive Decline of AD
13. Ca2+ Transporters on ER Are also Involved in Impairing the Memory of AD
14. Ca2+ Transporters on Mitochondria and Lysosomes Potentially Contribute to the Memory Loss of AD
15. The Roles of Ca2+ in Synaptic Plasticity
16. Ca2+ Transporters on the Cell Membrane Are Involved in Regulating the Synaptic Plasticity
17. ER Transporters Are Responsible for Releasing Ca2+ from Internal Stores, Leading to Regulate the Synaptic Plasticity
18. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Elgh, E.; Åstot, A.L.; Fagerlund, M.; Eriksson, S.; Olsson, T.; Näsman, B. Cognitive Dysfunction, Hippocampal Atrophy and Glucocorticoid Feedback in Alzheimer’s Disease. Biol. Psychiatry 2006, 59, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Almenar-Queralt, A.; Leblanc, J.F.; Roberts, E.A.; Goldstein, L.S.B. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nat. Cell Biol. 2001, 414, 643–648. [Google Scholar] [CrossRef]
- Mazanetz, M.P.; Fischer, P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A Mouse Model of Amyloid Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss In Vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef]
- Heyman, A.; Wilkinson, W.E.; Stafford, J.A.; Helms, M.J.; Sigmon, A.H.; Weinberg, T. Alzheimer’s disease: A study of epidemiological aspects. Ann. Neurol. 1984, 15, 335–341. [Google Scholar] [CrossRef]
- Patterson, C.; Feightner, J.W.; Garcia, A.; Hsiung, G.-Y.R.; Macknight, C.; Sadovnick, A.D. Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. Can. Med. Assoc. J. 2008, 178, 548–556. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- O’Donoghue, M.C.; Murphy, S.E.; Zamboni, G.; Nobre, A.C.; Mackay, C.E. APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: A review. Cortex 2018, 104, 103–123. [Google Scholar] [CrossRef]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- Sinha, S.; Lieberburg, I. Cellular mechanisms of beta -amyloid production and secretion. Proc. Natl. Acad. Sci. USA 1999, 96, 11049–11053. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Z.-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Neec, L.; Prince, J.; Govonid, S.; Racchie, M.; Tanzi, R.E.; Alkon, D.L. Calcium Responses in Fibroblasts from Asymptomatic Members of Alzheimer’s Disease Families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef]
- Yu, J.-T.; Chang, R.C.-C.; Tan, L. Calcium dysregulation in Alzheimer’s disease: From mechanisms to therapeutic opportunities. Prog. Neurobiol. 2009, 89, 240–255. [Google Scholar] [CrossRef]
- Cao, L.-L.; Guan, P.-P.; Liang, Y.-Y.; Huang, X.-S.; Wang, P. Calcium Ions Stimulate the Hyperphosphorylation of Tau by Activating Microsomal Prostaglandin E Synthase 1. Front. Aging Neurosci. 2019, 11, 108. [Google Scholar] [CrossRef]
- Cao, L.-L.; Guan, P.-P.; Liang, Y.-Y.; Huang, X.-S.; Wang, P. Cyclooxygenase-2 is Essential for Mediating the Effects of Calcium Ions on Stimulating Phosphorylation of Tau at the Sites of Ser 396 and Ser 404. J. Alzheimer’s Dis. 2019, 68, 1095–1111. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mandelkow, E.-M. A Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta BBA Bioenerg. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, D.; Farrelly, O.; Miles, L.; Li, F.; Kim, S.E.; Lo, T.Y.; Wang, F.; Li, T.; Thompson-Peer, K.L.; et al. The Mechanosensitive Ion Channel Piezo Inhibits Axon Regeneration. Neuron 2019, 102, 373–389.e6. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C.; Golanov, E.; Yamamoto, S.; Yee, F.; Ericson, H.; Yoo, H.; Inturrisi, C.E.; Reis, D.J. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nat. Cell Biol. 1993, 363, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.-P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis In Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef]
- MacManus, A.; Ramsden, M.; Murray, M.; Henderson, Z.; Pearson, H.A.; Campbell, V.A. Enhancement of 45Ca2+ Influx and Voltage-dependent Ca2+ Channel Activity by β-Amyloid-(1–40) in Rat Cortical Synaptosomes and Cultured Cortical Neurons. J. Biol. Chem. 2000, 275, 4713–4718. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Li, L.; Tsai, H.-J.; Li, L.; Wang, X.-M. Icariin Inhibits the Increased Inward Calcium Currents Induced by Amyloid-β25-35 Peptide in CA1 Pyramidal Neurons of Neonatal Rat Hippocampal Slice. Am. J. Chin. Med. 2010, 38, 113–125. [Google Scholar] [CrossRef]
- Yallampalli, S.; Micci, M.-A.; Taglialatela, G. Ascorbic acid prevents β-amyloid-induced intracellular calcium increase and cell death in PC12 cells. Neurosci. Lett. 1998, 251, 105–108. [Google Scholar] [CrossRef]
- Ekinci, F.J.; Linsley, M.-D.; Shea, T.B. β-Amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Mol. Brain Res. 2000, 76, 389–395. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 1584–1590. [Google Scholar] [CrossRef]
- Hermes, M.; Eichhoff, G.; Garaschuk, O. Intracellular calcium signalling in Alzheimer’s disease. J. Cell. Mol. Med. 2009, 14, 30–41. [Google Scholar] [CrossRef]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef]
- Mäkinen, S.; Van Groen, T.; Clarke, J.; Thornell, A.; Corbett, D.; Hiltunen, M.; Soininen, H.; Jolkkonen, J. Coaccumulation of Calcium and β-Amyloid in the Thalamus after Transient Middle Cerebral Artery Occlusion in Rats. Br. J. Pharmacol. 2007, 28, 263–268. [Google Scholar] [CrossRef]
- Arispe, N.; Diaz, J.; Durell, S.R.; Shafrir, Y.; Guy, H.R. Polyhistidine Peptide Inhibitor of the Aβ Calcium Channel Potently Blocks the Aβ-Induced Calcium Response in Cells. Theoretical Modeling Suggests a Cooperative Binding Process. Biochemistry 2010, 49, 7847–7853. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid β protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J. 2001, 15, 2433–2444. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Amyloid pores from pathogenic mutations. Nat. Cell Biol. 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.M.; Petre, B.M.; Wall, J.S.; Simon, M.N.; Walz, T.; Lansbury, P.T. Mixtures of Wild-type and a Pathogenic (E22G) Form of Aβ40 in Vitro Accumulate Protofibrils, Including Amyloid Pores. J. Mol. Biol. 2003, 332, 795–808. [Google Scholar] [CrossRef]
- Durell, S.; Guy, H.; Arispe, N.; Rojas, E.; Pollard, H. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys. J. 1994, 67, 2137–2145. [Google Scholar] [CrossRef]
- Jang, H.; Ma, B.; Lal, R.; Nussinov, R. Models of Toxic β-Sheet Channels of Protegrin-1 Suggest a Common Subunit Organization Motif Shared with Toxic Alzheimer β-Amyloid Ion Channels. Biophys. J. 2008, 95, 4631–4642. [Google Scholar] [CrossRef]
- Pollard, H.B.; Rojas, E.; Arispe, N. A New Hypothesis for the Mechanism of Amyloid Toxicity, Based on the Calcium Channel Activity of Amyloid β Protein (AβP) in Phospholipid Bilayer Membranes. Ann. N. Y. Acad. Sci. 1993, 695, 165–168. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different Conformations of Amyloid beta Induce Neurotoxicity by Distinct Mechanisms in Human Cortical Neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef]
- Lee, G.; Pollard, H.B.; Arispe, N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-β-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides 2002, 23, 1249–1263. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proc. Natl. Acad. Sci. USA 1996, 93, 1710–1715. [Google Scholar] [CrossRef]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Rhee, S.K.; Quist, A.P.; Lal, R. Amyloid β Protein-(1–42) Forms Calcium-permeable, Zn2+-sensitive Channel. J. Biol. Chem. 1998, 273, 13379–13382. [Google Scholar] [CrossRef]
- Arispe, N.; Doh, M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AβP (1–40) and (1–42) peptides. FASEB J. 2002, 16, 1526–1536. [Google Scholar] [CrossRef]
- Kawahara, M.; Kuroda, Y. Intracellular Calcium Changes in Neuronal Cells Induced by Alzheimer’s β-Amyloid Protein Are Blocked by Estradiol and Cholesterol. Cell. Mol. Neurobiol. 2001, 21, 1–13. [Google Scholar] [CrossRef]
- Barger, S.W.; Fiscus, R.R.; Ruth, P.; Hofmann, F.; Mattson, M.P. Role of Cyclic GMP in the Regulation of Neuronal Calcium and Survival by Secreted Forms of β-Amyloid Precursor. J. Neurochem. 2002, 64, 2087–2096. [Google Scholar] [CrossRef]
- Guo, Q.; Robinson, N.; Mattson, M.P. Secreted β-Amyloid Precursor Protein Counteracts the Proapoptotic Action of Mutant Presenilin-1 by Activation of NF-κB and Stabilization of Calcium Homeostasis. J. Biol. Chem. 1998, 273, 12341–12351. [Google Scholar] [CrossRef]
- Murray, J.N.; Igwe, O.J. Regulation of ?-amyloid precursor protein and inositol 1,4,5-trisphosphate receptor gene expression during differentiation of a human neuronal cell line. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 351–363. [Google Scholar] [CrossRef]
- Cao, X.; Südhof, T.C. A Transcriptively Active Complex of APP with Fe65 and Histone Acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef]
- Leissring, M.A.; Murphy, M.P.; Mead, T.R.; Akbari, Y.; Sugarman, M.C.; Jannatipour, M.; Anliker, B.; Müller, U.; Saftig, P.; De Strooper, B.; et al. A physiologic signaling role for the -secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. USA 2002, 99, 4697–4702. [Google Scholar] [CrossRef]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R. Efficacy and Safety of Memantine in Moderate-to-Severe Alzheimer Disease: The Evidence to Date. Alzheimer Dis. Assoc. Disord. 2006, 20, 23–29. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ Oligomers Induce Neuronal Oxidative Stress through an N-Methyl-D-aspartate Receptor-dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qiu, X.; Mak, S.; Guo, B.; Hu, S.; Wang, J.; Luo, F.; Xu, D.; Sun, Y.; Zhang, G.; et al. Multifunctional memantine nitrate significantly protects against glutamate-induced excitotoxicity via inhibiting calcium influx and attenuating PI3K/Akt/GSK3beta pathway. Chem. Interact. 2020, 325, 109020. [Google Scholar] [CrossRef]
- Kelly, B.L.; Ferreira, A. β-Amyloid-induced Dynamin 1 Degradation Is Mediated by N-Methyl-D-Aspartate Receptors in Hippocampal Neurons. J. Biol. Chem. 2006, 281, 28079–28089. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; E Shepardson, N.; Smith, I.; Brett, F.M.; A Farrell, M.; Rowan, M.J.; A Lemere, C.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Dewachter, I.; Filipkowski, R.; Priller, C.; Ris, L.; Neyton, J.; Croes, S.; Terwel, D.; Gysemans, M.; Devijver, H.; Borghgraef, P.; et al. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol. Aging 2009, 30, 241–256. [Google Scholar] [CrossRef]
- Chappell, A.S.; Gonzales, C.; Williams, J.; Witte, M.M.; Mohs, R.C.; Sperling, R. AMPA potentiator treatment of cognitive deficits in Alzheimer disease. Neurology 2007, 68, 1008–1012. [Google Scholar] [CrossRef]
- Trzepacz, P.T.; Cummings, J.; Konechnik, T.; Forrester, T.D.; Chang, C.; Dennehy, E.B.; Willis, B.A.; Shuler, C.; Tabas, L.B.; Lyketsos, C. Mibampator (LY451395) randomized clinical trial for agitation/aggression in Alzheimer’s disease. Int. Psychogeriatrics 2013, 25, 707–719. [Google Scholar] [CrossRef]
- Bloss, E.B.; Hunter, R.G.; Waters, E.M.; Muñoz, C.; Bernard, K.; McEwen, B.S. Behavioral and biological effects of chronic S18986, a positive AMPA receptor modulator, during aging. Exp. Neurol. 2008, 210, 109–117. [Google Scholar] [CrossRef]
- Jhee, S.S.; Chappell, A.S.; Zarotsky, V.; Moran, S.V.; Rosenthal, M.; Kim, E.; Chalon, S.; Toublanc, N.; Brandt, J.; Coutant, D.E.; et al. Multiple-Dose Plasma Pharmacokinetic and Safety Study of LY450108 and LY451395 (AMPA Receptor Potentiators) and Their Concentration in Cerebrospinal Fluid in Healthy Human Subjects. J. Clin. Pharmacol. 2006, 46, 424–432. [Google Scholar] [CrossRef]
- Nimmrich, V.; Grimm, C.; Draguhn, A.; Barghorn, S.; Lehmann, A.; Schoemaker, H.; Hillen, H.; Gross, G.; Ebert, U.; Bruehl, C. Amyloid Oligomers (A 1-42 Globulomer) Suppress Spontaneous Synaptic Activity by Inhibition of P/Q-Type Calcium Currents. J. Neurosci. 2008, 28, 788–797. [Google Scholar] [CrossRef]
- Rovira, C.; Arbez, N.; Mariani, J. Aβ(25–35) and Aβ(1–40) act on different calcium channels in CA1 hippocampal neurons. Biochem. Biophys. Res. Commun. 2002, 296, 1317–1321. [Google Scholar] [CrossRef]
- Hermann, D.; Mezler, M.; Müller, M.K.; Wicke, K.; Gross, G.; Draguhn, A.; Bruehl, C.; Nimmrich, V. Synthetic Aβ oligomers (Aβ1–42 globulomer) modulate presynaptic calcium currents: Prevention of Aβ-induced synaptic deficits by calcium channel blockers. Eur. J. Pharmacol. 2013, 702, 44–55. [Google Scholar] [CrossRef]
- Mark, R.J.; Hensley, K.; A Butterfield, D.; Mattson, M.P. Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J. Neurosci. 1995, 15, 6239–6249. [Google Scholar] [CrossRef]
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538. [Google Scholar] [CrossRef]
- Tanis, J.E.; Ma, Z.; Krajacic, P.; He, L.; Foskett, J.K.; Lamitina, T. CLHM-1 is a Functionally Conserved and Conditionally Toxic Ca2+-Permeable Ion Channel in Caenorhabditis elegans. J. Neurosci. 2013, 33, 12275–12286. [Google Scholar] [CrossRef]
- Wang, X.S.; Gruenstein, E. Rapid elevation of neuronal cytoplasmic calcium by apolipoprotein E peptide. J. Cell Physiol. 1997, 173, 73–83. [Google Scholar] [CrossRef]
- Müller, W.; Meske, V.; Berlin, K.; Scharnagl, H.; Marz, W.; Ohm, T. Apolipoprotein E Isoforms Increase Intracellular Ca2+Differentially Through a ω-Agatoxin IVa-Sensitive Ca2+-Channel. Brain Pathol. 1998, 8, 641–653. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, S.; Zhao, H.; Wang, Y.; Chen, X.; Sun, X. Effects of apolipoprotein E gene polymorphism on the intracellular Ca2+ concentration of astrocytes in the early stages post injury. Exp. Ther. Med. 2017, 15, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-? peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-β-(1-42) Increases Ryanodine Receptor-3 Expression and Function in Neurons of TgCRND8 Mice. J. Biol. Chem. 2006, 281, 38440–38447. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ozawa, H.; Saito, T.; Takahata, N.; Takemura, H. Calcium mobilization evoked by amyloid β-protein involves inositol 1,4,5-trisphosphate production in human platelets. Life Sci. 1998, 62, 705–713. [Google Scholar] [CrossRef]
- Schapansky, J.; Olson, K.; Van Der Ploeg, R.; Glazner, G. NF-κB activated by ER calcium release inhibits Aβ-mediated expression of CHOP protein: Enhancement by AD-linked mutant presenilin 1. Exp. Neurol. 2007, 208, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Shtifman, A.; Ward, C.W.; Laver, D.R.; Bannister, M.L.; Lopez, J.R.; Kitazawa, M.; LaFerla, F.M.; Ikemoto, N.; Querfurth, H.W. Amyloid-β protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging 2010, 31, 2080–2090. [Google Scholar] [CrossRef]
- Müller, M.; Cárdenas, C.; Mei, L.; Cheung, K.-H.; Foskett, J.K. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 13293–13298. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, A.; Cerullo, M.S.; Buxeda, P.; Tomagra, G.; Giustetto, M.; Chiantia, G.; Carabelli, V.; Carbone, E. Amyloid Beta42 oligomers up-regulate the excitatory synapses by potentiating presynaptic release while impairing postsynaptic NMDA receptors. J. Physiol. 2020, 598, 2183–2197. [Google Scholar] [CrossRef]
- Stutzmann, G.E. Calcium Dysregulation, IP3 Signaling, and Alzheimer’s Disease. Neuroscience 2005, 11, 110–115. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.-Y.; Foskett, J.K. Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Rybalchenko, V.; Hwang, S.-Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Hayrapetyan, V.; Rybalchenko, V.; Rybalchenko, N.; Koulen, P. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein–protein interaction. Cell Calcium 2008, 44, 507–518. [Google Scholar] [CrossRef]
- Green, K.N.; DeMuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Cedazo-Mínguez, A.; Popescu, B.O.; Ankarcrona, M.; Nishimura, T.; Cowburn, R.F. The Presenilin 1 ΔE9 Mutation Gives Enhanced Basal Phospholipase C Activity and a Resultant Increase in Intracellular Calcium Concentrations. J. Biol. Chem. 2002, 277, 36646–36655. [Google Scholar] [CrossRef]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; A Leissring, M.; Shepel, P.; Geiger, J.D. Calcium signaling in the ER: Its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Mei, L.; Mak, D.-O.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-Function Enhancement of IP3 Receptor Modal Gating by Familial Alzheimer’s Disease-Linked Presenilin Mutants in Human Cells and Mouse Neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef]
- Ohkubo, N.; Mitsuda, N.; Tamatani, M.; Yamaguchi, A.; Lee, Y.-D.; Ogihara, T.; Vitek, M.P.; Tohyama, M. Apolipoprotein E4 Stimulates cAMP Response Element-binding Protein Transcriptional Activity through the Extracellular Signal-regulated Kinase Pathway. J. Biol. Chem. 2001, 276, 3046–3053. [Google Scholar] [CrossRef]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Stutzmann, G.E. The Pathogenesis of Alzheimers Disease—Is It a Lifelong “Calciumopathy”? Neuroscientist 2007, 13, 546–559. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; Oliveira, C.R. ER stress is involved in Aβ-induced GSK-3β activation and tau phosphorylation. J. Neurosci. Res. 2008, 86, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai 1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, L.; Herms, J.; Kuznicki, J. Calcium dysregulation in Alzheimer’s disease. Neurochem. Int. 2008, 52, 621–633. [Google Scholar] [CrossRef]
- Zeiger, W.; Vetrivel, K.S.; Buggia-Prévot, V.; Nguyen, P.D.; Wagner, S.L.; Villereal, M.L.; Thinakaran, G. Ca2+ Influx through Store-operated Ca2+ Channels Reduces Alzheimer Disease β-Amyloid Peptide Secretion. J. Biol. Chem. 2013, 288, 26955–26966. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.-J.; Feske, S.; White, C.L.; Bezprozvanny, I. Reduced Synaptic STIM2 Expression and Impaired Store-Operated Calcium Entry Cause Destabilization of Mature Spines in Mutant Presenilin Mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef]
- Ryazantseva, M.; Goncharova, A.; Skobeleva, K.; Erokhin, M.; Methner, A.; Georgiev, P.; Kaznacheyeva, E. Presenilin-1 Delta E9 Mutant Induces STIM1-Driven Store-Operated Calcium Channel Hyperactivation in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 4667–4680. [Google Scholar] [CrossRef]
- Li, H.-S.; Xu, X.-Z.S.; Montell, C. Activation of a TRPC3-Dependent Cation Current through the Neurotrophin BDNF. Neuron 1999, 24, 261–273. [Google Scholar] [CrossRef]
- Lessard, C.B.; Lussier, M.P.; Cayouette, S.; Bourque, G.; Boulay, G. The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell. Signal. 2005, 17, 437–445. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, Q.; Zhou, P.; Li, S.; Zhu, F. HERV-W env regulates calcium influx via activating TRPC3 channel together with depressing DISC1 in human neuroblastoma cells. J. Neuro Virol. 2019, 25, 101–113. [Google Scholar] [CrossRef]
- Keller, J.N.; Guo, Q.; Holtsberg, F.W.; Bruce-Keller, A.J.; Mattson, M.P. Increased Sensitivity to Mitochondrial Toxin-Induced Apoptosis in Neural Cells Expressing Mutant Presenilin-1 Is Linked to Perturbed Calcium Homeostasis and Enhanced Oxyradical Production. J. Neurosci. 1998, 18, 4439–4450. [Google Scholar] [CrossRef]
- Kruman, I.; Guo, Q.; Mattson, M.P. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J. Neurosci. Res. 1998, 51, 293–308. [Google Scholar] [CrossRef]
- Toglia, P.; Ullah, G. The gain-of-function enhancement of IP3-receptor channel gating by familial Alzheimer’s disease-linked presenilin mutants increases the open probability of mitochondrial permeability transition pore. Cell Calcium 2016, 60, 13–24. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced Expression of the Voltage-Dependent Anion Channel 1 (VDAC1) in Alzheimer’s Disease Transgenic Mice: An Insight into the Pathogenic Effects of Amyloid-β. J. Alzheimer’s Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef]
- Williams, G.S.B.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nat. Cell Biol. 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Gunter, T.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: Mechanisms and functions. Cell Calcium 2000, 28, 285–296. [Google Scholar] [CrossRef]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-Calcium Exchange Is Mediated by a Distinct Potassium-independent Sodium-Calcium Exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef]
- Lytton, J. Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+ transport. Biochem. J. 2007, 406, 365–382. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y.; Anraku, Y. Calcium transport driven by a proton motive force in vacuolar membrane vesicles of Saccharomyces cerevisiae. J. Biol. Chem. 1983, 258, 5614–5617. [Google Scholar] [CrossRef]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef]
- McBrayer, M.; Nixon, R.A. Lysosome and calcium dysregulation in Alzheimer’s disease: Partners in crime. Biochem. Soc. Trans. 2013, 41, 1495–1502. [Google Scholar] [CrossRef]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef]
- Fox, A.P.; Nowycky, M.C.; Tsien, R.W. Single-channel recordings of three types of calcium channels in chick sensory neurones. J. Physiol. 1987, 394, 173–200. [Google Scholar] [CrossRef]
- Sun, L.; Wei, H. Ryanodine Receptors: A Potential Treatment Target in Various Neurodegenerative Disease. Cell. Mol. Neurobiol. 2020, 1–12. [Google Scholar] [CrossRef]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 Mutations Increase Levels of Ryanodine Receptors and Calcium Release in PC12 Cells and Cortical Neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Liang, G.; Xu, Z.; Chu, C.T.; Wei, H. Alzheimer’s Disease Presenilin-1 Mutation Sensitizes Neurons to Impaired Autophagy Flux and Propofol Neurotoxicity: Role of Calcium Dysregulation. J. Alzheimer’s Dis. 2019, 67, 137–147. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP Mobilizes Ca2+ from Reserve Granules, Lysosome-Related Organelles, in Sea Urchin Eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef]
- Querfurth, H.W.; Selkoe, D.J. Calcium Ionophore Increases Amyloid.beta. Peptide Production by Cultured Cells. Biochemistry 1994, 33, 4550–4561. [Google Scholar] [CrossRef]
- Querfurth, H.W.; Jiang, J.; Geiger, J.D.; Selkoe, D.J. Caffeine Stimulates Amyloid β-Peptide Release from β-Amyloid Precursor Protein-Transfected HEK293 Cells. J. Neurochem. 1997, 69, 1580–1591. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Kim, J.; Ham, S.; Park, J.H.Y.; Han, S.; Jung, Y.-K.; Shim, I.; Han, J.-S.; Lee, K.W.; et al. Ca2+-permeable TRPV1 pain receptor knockout rescues memory deficits and reduces amyloid-β and tau in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2019, 29, 228–237. [Google Scholar] [CrossRef]
- Itkin, A.; Dupres, V.; Dufrêne, Y.F.; Bechinger, B.; Ruysschaert, J.-M.; Raussens, V. Calcium Ions Promote Formation of Amyloid β-Peptide (1–40) Oligomers Causally Implicated in Neuronal Toxicity of Alzheimer’s Disease. PLoS ONE 2011, 6, e18250. [Google Scholar] [CrossRef]
- Green, K.N.; LaFerla, F.M. Linking Calcium to Aβ and Alzheimer’s Disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef]
- Ahmad, A.; Muzaffar, M.; Ingram, V.M. Ca2+, within the physiological concentrations, selectively accelerates Aβ42 fibril formation and not Aβ40 in vitro. Biochim. Biophys. Acta BBA Proteins Proteom. 2009, 1794, 1537–1548. [Google Scholar] [CrossRef]
- Guo, Q.; Fu, W.; Sopher, B.L.; Miller, M.W.; Ware, C.B.; Martin, G.M.; Mattson, M.P. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat. Med. 1999, 5, 101–106. [Google Scholar] [CrossRef]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Ueda, M.; Masuda, T.; Misumi, Y.; Yamashita, T.; Ando, Y. Memantine, a Noncompetitive N-Methyl-d-Aspartate Receptor Antagonist, Attenuates Cerebral Amyloid Angiopathy by Increasing Insulin-Degrading Enzyme Expression. Mol. Neurobiol. 2019, 56, 8573–8588. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.-Z.; Li, B.; Li, Y.-C.; Yang, X.-L.; Zhang, W.; Zhong, L.; Tang, S.-J. Activation of NMDA Receptors Upregulates A Disintegrin and Metalloproteinase 10 via a Wnt/MAPK Signaling Pathway. J. Neurosci. 2012, 32, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Hoey, S.E.; Buonocore, F.; Cox, C.J.; Hammond, V.J.; Perkinton, M.S.; Williams, R.J. AMPA Receptor Activation Promotes Non-Amyloidogenic Amyloid Precursor Protein Processing and Suppresses Neuronal Amyloid-β Production. PLoS ONE 2013, 8, e78155. [Google Scholar] [CrossRef] [PubMed]
- Dreses-Werringloer, U.; Lambert, J.-C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A Polymorphism in CALHM1 Influences Ca2+ Homeostasis, Aβ Levels, and Alzheimer’s Disease Risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE Isoforms Differentially Regulate Brain Amyloid- Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef]
- Kelliher, M.; Fastbom, J.; Cowburn, R.; Bonkale, W.; Ohm, T.; Ravid, R.; Sorrentino, V.; O’Neill, C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and β-amyloid pathologies. Neuroscience 1999, 92, 499–513. [Google Scholar] [CrossRef]
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ Channel Function as an Early Preventative Strategy for Alzheimer’s Disease. PLoS ONE 2012, 7, e52056. [Google Scholar] [CrossRef]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine Receptor Blockade Reduces Amyloid- Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Supnet, C.; Sun, S.; Zhang, H.; Good, L.; Popugaeva, E.; Bezprozvanny, I. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels 2014, 8, 230–242. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Ruefli, A.A.; Parker, C.A.; Cypess, A.M.; Greengard, P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc. Natl. Acad. Sci. USA 1994, 91, 4489–4493. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.D.; Saito, T.; Saido, T.C.; Bezprozvanny, I. Neuronal Store-Operated Calcium Entry and Mushroom Spine Loss in Amyloid Precursor Protein Knock-In Mouse Model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef]
- Kyung, T.; Lee, S.; Kim, J.E.; Cho, T.; Park, H.; Jeong, Y.-M.; Kim, D.; Shin, A.; Kim, S.; Baek, J.; et al. Optogenetic control of endogenous Ca2+ channels in vivo. Nat. Biotechnol. 2015, 33, 1092–1096. [Google Scholar] [CrossRef]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-Mediated Modulation of Capacitative Calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nuñez, L.; Villalobos, C. Amyloid β Oligomers Increase ER-Mitochondria Ca2+ Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca2+ Remodeling. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Culwell, A.R.; Esch, F.S.; Lieberburg, I.; Rydel, R.E. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 1993, 10, 243–254. [Google Scholar] [CrossRef]
- Scremin, E.; Agostini, M.; Leparulo, A.; Pozzan, T.; Greotti, E.; Fasolato, C. ORAI2 Down-Regulation Potentiates SOCE and Decreases Aβ42 Accumulation in Human Neuroglioma Cells. Int. J. Mol. Sci. 2020, 21, 5288. [Google Scholar] [CrossRef]
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 Protects Against Alzheimer’s Disease, Mitochondria, and Synaptic Deficiencies. J. Alzheimer’s Dis. 2013, 37, 679–690. [Google Scholar] [CrossRef]
- Šoškić, V.; Klemm, M.; Proikas-Cezanne, T.; Schwall, G.P.; Poznanović, S.; Stegmann, W.; Groebe, K.; Zengerling, H.; Schoepf, R.; Burnet, M.; et al. A Connection between the Mitochondrial Permeability Transition Pore, Autophagy, and Cerebral Amyloidogenesis. J. Proteome Res. 2008, 7, 2262–2269. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Johnson, G.V. Transient Increases in Intracellular Calcium Result in Prolonged Site-selective Increases in Tau Phosphorylation through a Glycogen Synthase Kinase 3β-dependent Pathway. J. Biol. Chem. 1999, 274, 21395–21401. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hiragami, Y.; Murayama, M.; Ishizuka, K.; Kawahara, M.; Takashima, A. Phosphorylation of tau at serine 416 by Ca2+/calmodulin-dependent protein kinase II in neuronal soma in brain. J. Neurochem. 2005, 94, 1438–1447. [Google Scholar] [CrossRef]
- Avila, J.; Pérez, M.; Lim, F.; Gómez-Ramos, A.; Hernández, F.; Lucas, J.J. Tau in neurodegenerative diseases: Tau phosphorylation and assembly. Neurotox. Res. 2004, 6, 477–482. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.-H.; Kim, E.; George-Hyslop, P.S.; et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 2015, 5, 10934. [Google Scholar] [CrossRef]
- Pierrot, N.; Ghisdal, P.; Caumont, A.-S.; Octave, J.-N. Intraneuronal amyloid-β1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 2004, 88, 1140–1150. [Google Scholar] [CrossRef]
- Bruno, A.M.; Huang, J.Y.; Bennett, D.A.; Marr, R.A.; Hastings, M.L.; Stutzmann, G.E. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1001.e1–1001.e6. [Google Scholar] [CrossRef]
- Antonell, A.; Lladó, A.; Altirriba, J.; Botta-Orfila, T.; Balasa, M.; Fernández, M.; Ferrer, I.; Sánchez-Valle, R.; Molinuevo, J.L. A preliminary study of the whole-genome expression profile of sporadic and monogenic early-onset Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1772–1778. [Google Scholar] [CrossRef]
- Chami, M.; Checler, F. Ryanodine receptors. Channels 2014, 8, 168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Zhang, Y.; Li, B.; Wu, Y.; Li, B. Effect of β-amyloid (25-35) on mitochondrial function and expression of mitochondrial permeability transition pore proteins in rat hippocampal neurons. J. Cell. Biochem. 2011, 112, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid β-Peptide Promotes Permeability Transition Pore in Brain Mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Toescu, E.C.; Verkhratsky, A. The importance of being subtle: Small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Reijo, T.; Mikko, B.; Antti, S.; Timo, S.; Mikko, B. Serum Calcium And Prediction Of Cognitive Decline In Old Age. J. Am. Geriatr. Soc. 2008, 56, 1573–1574. [Google Scholar] [CrossRef]
- Heck, A.; Fastenrath, M.; Coynel, D.; Auschra, B.; Bickel, H.; Freytag, V.; Gschwind, L.; Hartmann, F.; Jessen, F.; Kaduszkiewicz, H.; et al. Genetic Analysis of Association Between Calcium Signaling and Hippocampal Activation, Memory Performance in the Young and Old, and Risk for Sporadic Alzheimer Disease. JAMA Psychiatry 2015, 72, 1029–1036. [Google Scholar] [CrossRef]
- Walsh, D.M.; Townsend, M.; Podlisny, M.B.; Shankar, G.M.; Fadeeva, J.V.; El Agnaf, O.; Hartley, D.M.; Selkoe, D.J. Certain Inhibitors of Synthetic Amyloid -Peptide (A ) Fibrillogenesis Block Oligomerization of Natural A and Thereby Rescue Long-Term Potentiation. J. Neurosci. 2005, 25, 2455–2462. [Google Scholar] [CrossRef]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nat. Cell Biol. 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Hu, W.-Y.; He, Z.-Y.; Yang, L.-J.; Zhang, M.; Xing, D.; Xiao, Z.-C. The Ca2+channel inhibitor 2-APB reverses β-amyloid-induced LTP deficit in hippocampus by blocking BAX and caspase-3 hyperactivation. Br. J. Pharmacol. 2015, 172, 2273–2285. [Google Scholar] [CrossRef]
- Trinchese, F.; Fa’, M.; Liu, S.; Zhang, H.; Hidalgo, A.; Schmidt, S.D.; Yamaguchi, H.; Yoshii, N.; Mathews, P.M.; Nixon, R.A.; et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 2796–2807. [Google Scholar] [CrossRef]
- Dineley, K.T.; Hogan, D.; Zhang, W.-R.; Taglialatela, G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol. Learn. Mem. 2007, 88, 217–224. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflügers Arch. Eur. J. Physiol. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Dudek, S.M.; Bear, M.F. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. USA 1992, 89, 4363–4367. [Google Scholar] [CrossRef]
- Morris, R.G.M.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nat. Cell Biol. 1986, 319, 774–776. [Google Scholar] [CrossRef]
- Hsiung, G.-Y.R.; Feldman, H.H. Pharmacological treatment in moderate-to-severe Alzheimer’s disease. Expert Opin. Pharmacother. 2008, 9, 2575–2582. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Banerjee, P.; Tanila, H. Memantine Improves Spatial Learning in a Transgenic Mouse Model of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2004, 311, 677–682. [Google Scholar] [CrossRef]
- Palop, J.J. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef]
- Wang, Y.; Mattson, M.P. L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol. Aging 2014, 35, 88–95. [Google Scholar] [CrossRef]
- Tsukuda, K.; Mogi, M.; Li, J.-M.; Iwanami, J.; Min, L.-J.; Sakata, A.; Fujita, T.; Iwai, M.; Horiuchi, M. Diabetes-Associated Cognitive Impairment Is Improved by a Calcium Channel Blocker, Nifedipine. Hypertension 2008, 51, 528–533. [Google Scholar] [CrossRef]
- Doody, R.S.; I Gavrilova, S.; Sano, M.; Thomas, R.G.; Aisen, P.S.; O Bachurin, S.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Moriguchi, S.; Shioda, N.; Yamamoto, Y.; Tagashira, H.; Fukunaga, K. The T-type voltage-gated calcium channel as a molecular target of the novel cognitive enhancer ST101: Enhancement of long-term potentiation and CaMKII autophosphorylation in rat cortical slices. J. Neurochem. 2012, 121, 44–53. [Google Scholar] [CrossRef]
- Deshpande, L.S.; Sun, D.A.; Sombati, S.; Baranova, A.; Wilson, M.S.; Attkisson, E.; Hamm, R.J.; DeLorenzo, R.J. Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 2008, 441, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Mallmann, R.T.; Elgueta, C.; Sleman, F.; Castonguay, J.; Wilmes, T.; Maagdenberg, A.V.D.; Klugbauer, N. Ablation of CaV2.1 Voltage-Gated Ca2+ Channels in Mouse Forebrain Generates Multiple Cognitive Impairments. PLoS ONE 2013, 8, e78598. [Google Scholar] [CrossRef]
- Micale, V.; Cristino, L.; Tamburella, A.; Petrosino, S.; Leggio, G.M.; Di Marzo, V.; Drago, F. Enhanced cognitive performance of dopamine D3 receptor “knock-out” mice in the step-through passive-avoidance test: Assessing the role of the endocannabinoid/endovanilloid systems. Pharmacol. Res. 2010, 61, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, P.; Oleksik, A.M.; Mooijaart, S.P.; De Craen, A.; Westendorp, R.G. APOE genotype modulates the effect of serum calcium levels on cognitive function in old age. Neurology 2009, 72, 821–828. [Google Scholar] [CrossRef]
- Cui, P.-J.; Zheng, L.; Cao, L.; Wang, Y.; Deng, Y.-L.; Wang, G.; Xu, W.; Tang, H.-D.; Ma, J.-F.; Zhang, T.; et al. CALHM1 P86L Polymorphism is a Risk Factor for Alzheimer’s Disease in the Chinese Population. J. Alzheimer’s Dis. 2010, 19, 31–35. [Google Scholar] [CrossRef]
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.-O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 Receptor-Mediated Ca2+ Signaling Alleviates Mutant Presenilin-Linked Familial Alzheimer’s Disease Pathogenesis. J. Neurosci. 2014, 34, 6910–6923. [Google Scholar] [CrossRef]
- Jaworska, A.; Dzbek, J.; Styczynska, M.; Kuznicki, J. Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer’s disease or mild cognitive impairment. Biochim. Biophys. Acta BBA Bioenerg. 2013, 1833, 1692–1699. [Google Scholar] [CrossRef][Green Version]
- Lü, N.; Lü, B.-C.; Cheng, L.-Z.; Zhang, Y.-Q.; Li, Y.-Q.; Zhao, Z.-Q. Involvement of ryanodine receptors in tetanic sciatic stimulation-induced long-term potentiation of spinal dorsal horn and persistent pain in rats. J. Neurosci. Res. 2012, 90, 1096–1104. [Google Scholar] [CrossRef]
- Matsuo, N.; Tanda, K.; Nakanishi, K.; Yamasaki, N.; Toyama, K.; Takao, K.; Takeshima, H.; Miyakawa, T. Comprehensive behavioral phenotyping of ryanodine receptor type3 (RyR3) knockout mice: Decreased social contact duration in two social interaction tests. Front. Behav. Neurosci. 2009, 3, 3. [Google Scholar] [CrossRef]
- Alkon, D.L.; Nelson, T.J.; Zhao, W.; Cavallaro, S. Time domains of neuronal Ca2+ signaling and associative memory: Steps through a calexcitin, ryanodine receptor, K+ channel cascade. Trends Neurosci. 1998, 21, 529–537. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Lee, C.S.-K.; Cheng, W.-H.; Lai, K.-O.; Foskett, J.K.; Cheung, K.-H. Familial Alzheimer’s disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef]
- Emptage, N.J.; Reid, C.; Fine, A. Calcium Stores in Hippocampal Synaptic Boutons Mediate Short-Term Plasticity, Store-Operated Ca2+ Entry, and Spontaneous Transmitter Release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Kon, N.; Satoh, A.; Miyoshi, N. A small-molecule DS44170716 inhibits Ca2+-induced mitochondrial permeability transition. Sci. Rep. 2017, 7, 3864. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.-C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Birks, J.; López-Arrieta, J.; López-Arrieta, J.M. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst. Rev. 2002, 2002, CD000147. [Google Scholar] [CrossRef]
- Hopp, S.C.; D’Angelo, H.M.; E Royer, S.; Kaercher, R.M.; Crockett, A.M.; Adzovic, L.; Wenk, G.L. Calcium dysregulation via L-type voltage-dependent calcium channels and ryanodine receptors underlies memory deficits and synaptic dysfunction during chronic neuroinflammation. J. Neuroinflamm. 2015, 12, 56. [Google Scholar] [CrossRef]
- Luengo, E.; Buendia, I.; Fernández-Mendívil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernández-García, B.; Sánchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef]
- Mattson, M.P.; Partin, J.; Begley, J. Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998, 807, 167–176. [Google Scholar] [CrossRef]
- Jin, Y. Synaptogenesis: Insights from worm and fly. Curr. Opin. Neurobiol. 2002, 12, 71–79. [Google Scholar] [CrossRef]
- Mattson, M.P.; Chan, S.L. Dysregulation of Cellular Calcium Homeostasis in Alzheimer’s Disease: Bad Genes and Bad Habits. J. Mol. Neurosci. 2001, 17, 205–224. [Google Scholar] [CrossRef]
- Chan, S.L.; Furukawa, K.; Mattson, M.P. Presenilins and APP in Neuritic and Synaptic Plasticity: Implications for the Pathogenesis of Alzheimer’s Disease. NeuroMol. Med. 2002, 2, 167–196. [Google Scholar] [CrossRef]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Goncalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.-P.; et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nat. Cell Biol. 1997, 387, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Nelson, O.; Supnet, C.; Liu, H.; Bezprozvanny, I. Familial Alzheimer’s Disease Mutations in Presenilins: Effects on Endoplasmic Reticulum Calcium Homeostasis and Correlation with Clinical Phenotypes. J. Alzheimer’s Dis. 2010, 21, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Tonkikh, A.; Janus, C.; El-Beheiry, H.; Pennefather, P.S.; Samoilova, M.; McDonald, P.; Ouanounou, A.; Carlen, P.L. Calcium chelation improves spatial learning and synaptic plasticity in aged rats. Exp. Neurol. 2006, 197, 291–300. [Google Scholar] [CrossRef]
- Winder, D.G.; Mansuy, I.M.; Osman, M.; Moallem, T.M.; Kandel, E.R. Genetic and Pharmacological Evidence for a Novel, Intermediate Phase of Long-Term Potentiation Suppressed by Calcineurin. Cell 1998, 92, 25–37. [Google Scholar] [CrossRef]
- Foster, T.C.; Sharrow, K.M.; Masse, J.R.; Norris, C.M.; Kumar, A. Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 2001, 21, 4066–4073. [Google Scholar] [CrossRef]
- Yamin, G. NMDA receptor-dependent signaling pathways that underlie amyloid β-protein disruption of LTP in the hippocampus. J. Neurosci. Res. 2009, 87, 1729–1736. [Google Scholar] [CrossRef]
- Raymond, C.R.; Redman, S.J. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J. Physiol. 2006, 570, 97–111. [Google Scholar] [CrossRef]
- Sanderson, J.L.; Gorski, J.A.; Dell’Acqua, M.L. NMDA Receptor-Dependent LTD Requires Transient Synaptic Incorporation of Ca 2+ -Permeable AMPARs Mediated by AKAP150-Anchored PKA and Calcineurin. Neuron 2016, 89, 1000–1015. [Google Scholar] [CrossRef]
- Jia, Z.; Agopyan, N.; Miu, P.; Xiong, Z.; Henderson, J.; Gerlai, R.; A Taverna, F.; Velumian, A.; MacDonald, J.; Carlen, P.; et al. Enhanced LTP in Mice Deficient in the AMPA Receptor GluR2. Neuron 1996, 17, 945–956. [Google Scholar] [CrossRef]
- Plant, K.; A Pelkey, K.; A Bortolotto, Z.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef]
- Fortin, D.A.; Davare, M.A.; Srivastava, T.; Brady, J.D.; Nygaard, S.; Derkach, V.A.; Soderling, T.R. Long-Term Potentiation-Dependent Spine Enlargement Requires Synaptic Ca2+-Permeable AMPA Receptors Recruited by CaM-Kinase I. J. Neurosci. 2010, 30, 11565–11575. [Google Scholar] [CrossRef]
- Chowdhury, D.; Hell, J.W. Ca2+/Calmodulin Binding to PSD-95 Downregulates Its Palmitoylation and AMPARs in Long-Term Depression. Front. Synaptic Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Huber, K.M.; Mauk, M.D.; Kelly, P.T. LTP induced by activation of voltage-dependent Ca2+ channels requires protein kinase activity. NeuroReport 1995, 6, 1281–1284. [Google Scholar] [CrossRef][Green Version]
- White, J.A.; McKinney, B.C.; John, M.C.; Powers, P.A.; Kamp, T.J.; Murphy, G.G. Conditional forebrain deletion of the L-type calcium channel CaV1.2 disrupts remote spatial memories in mice. Learn. Mem. 2008, 15, 1–5. [Google Scholar] [CrossRef]
- Li, H.-B.; Mao, R.-R.; Zhang, J.-C.; Yang, Y.; Cao, J.; Xu, L. Antistress Effect of TRPV1 Channel on Synaptic Plasticity and Spatial Memory. Biol. Psychiatry 2008, 64, 286–292. [Google Scholar] [CrossRef]
- Gerdeman, G.L.; Ronesi, J.; Lovinger, D.M. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat. Neurosci. 2002, 5, 446–451. [Google Scholar] [CrossRef]
- Chávez, A.E.; Chiu, C.Q.; Castillo, P.E. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat. Neurosci. 2010, 13, 1511–1518. [Google Scholar] [CrossRef]
- James, E.O.; Clevenger, A.C.; Dietz, R.M.; Patsos, O.P. A Novel Peptide Inhibitor Of Trpm2 Channels Reduces Memory Deficits And Improves Ltp Following Traumatic Brain Injury In Mice. J. Neurotrauma 2018. Available online: https://www.google.com/search?tbm=bks&q=104.+232.+Orfila%2C+James%2C+E.%2C+Clevenger%2C+Amy%2C+C.%2C+Dietz%2C+Robert%2C+M.%2C+Patsos+%282018%29+A+NOVEL+PEPTIDE+INHIBITOR+OF+TRPM2+CHANNELS+REDUCES+MEMORY+DEFICITS+AND+IMPROVES+LTP+FOLLOWING+TRAUMATIC+BRAIN+INJURY+IN+MICE.+Journal+of+Neurotrauma (accessed on 13 March 2021).
- Menigoz, A.; Ahmed, T.; Sabanov, V.; Philippaert, K.; Pinto, S.; Kerselaers, S.; Segal, A.; Freichel, M.; Voets, T.; Nilius, B.; et al. TRPM4-dependent post-synaptic depolarization is essential for the induction of NMDA receptor-dependent LTP in CA1 hippocampal neurons. Pflügers Arch. Eur. J. Physiol. 2016, 468, 593–607. [Google Scholar] [CrossRef]
- Miyata, M.; Finch, E.A.; Khiroug, L.; Hashimoto, K.; Hayasaka, S.; Oda, S.-I.; Inouye, M.; Takagishi, Y.; Augustine, G.J.; Kano, M. Local Calcium Release in Dendritic Spines Required for Long-Term Synaptic Depression. Neuron 2000, 28, 233–244. [Google Scholar] [CrossRef]
- A Cummings, J.; Mulkey, R.M.; A Nicoll, R.; Malenka, R.C. Ca2+ Signaling Requirements for Long-Term Depression in the Hippocampus. Neuron 1996, 16, 825–833. [Google Scholar] [CrossRef]
- Kato, H.K.; Kassai, H.; Watabe, A.M.; Aiba, A.; Manabe, T. Functional coupling of the metabotropic glutamate receptor, InsP3receptor and L-type Ca2+channel in mouse CA1 pyramidal cells. J. Physiol. 2012, 590, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- Shimuta, M.; Yoshikawa, M.; Fukaya, M.; Watanabe, M.; Takeshima, H.; Manabe, T. Postsynaptic Modulation of AMPA Receptor-Mediated Synaptic Responses and LTP by the Type 3 Ryanodine Receptor. Mol. Cell. Neurosci. 2001, 17, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Kim, J.; Schneider, C.; Jacobson, C.; Molgó, J.; Stutzmann, G.E. Early Presynaptic and Postsynaptic Calcium Signaling Abnormalities Mask Underlying Synaptic Depression in Presymptomatic Alzheimer’s Disease Mice. J. Neurosci. 2012, 32, 8341–8353. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, B.; Beglopoulos, V.; Wines-Samuelson, M.; Zhang, D.; Dragatsis, I.; Südhof, T.C.; Shen, J. Presenilins are essential for regulating neurotransmitter release. Nat. Cell Biol. 2009, 460, 632–636. [Google Scholar] [CrossRef]
- Majewski, Ł.; Maciąg, F.; Boguszewski, P.M.; Wasilewska, I.; Wiera, G.; Wójtowicz, T.; Mozrzymas, J.; Kuznicki, J. Overexpression of STIM1 in neurons in mouse brain improves contextual learning and impairs long-term depression. Biochim. Biophys. Acta BBA Bioenerg. 2017, 1864, 1071–1087. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Levy, M.; Sampson, M.J.; Anflous, K.; Armstrong, D.L.; Brown, S.E.; Sweatt, J.D.; Craigen, W.J. The Role of Mitochondrial Porins and the Permeability Transition Pore in Learning and Synaptic Plasticity. J. Biol. Chem. 2002, 277, 18891–18897. [Google Scholar] [CrossRef]
- Mulkey, R.M.; Malenka, R.C. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 1992, 9, 967–975. [Google Scholar] [CrossRef]
- Obenaus, A.; Mody, I.; Baimbridge, K.G. Dantrolene-Na (Dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci. Lett. 1989, 98, 172–178. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cat. | Stimulator or Mediator | Mechanism | Experimental Model | Reference |
|---|---|---|---|---|
| Aβ | Aβ1–40 | Aβ1–40→IL-1β→Ca2+ influx | Rat cortical synaptosomes and cultured cortical neurons | [29] |
| Aβ25–35 | Aβ25–35→L-/T-VGCC→Ca2+ influx | Rat CA1 pyramidal neurons | [31] | |
| Aβ | Aβ→Ca2+ influx | APP/PS1 Tg mice | [30] | |
| Aβ→PKA∪L-VGCC→Ca2+ influx | Neurons | [34] | ||
| APs | Ca2+ in the spines and dendrites of cortical pyramidal neurons of APs → Ca2+ in the adjacent resting neurons. | The spines and dendrites of cortical pyramidal neurons in 3 × Tg AD animals | [22] | |
| APs→Ca2+ influx | The astrocytes of 6-month-old APP/PS1 mice | [35] | ||
| Aβ→Formation of cation channels→Ca2+ passage | Artificial lipid membranes | [39] | ||
| Oligomeric Aβ→Ca2+ influx and leakage from intracellular Ca2+ stores | SH-SY5Y cells | [35] | ||
| Aβ→Formation of pores in the cell membrane of post-mortem→Ca2+ influx | Post-mortem of AD brains | [36] | ||
| sAPP | sAPP→cGMP→K+ channel┤Ca2+ | Hippocampal neurons | [56] | |
| γ-secretase | γ-secretase→ER-Ca2+ | SH-SY5Y cells (control and PSEN2T122R-expressing) | [61] | |
| CM | NMDAR | memantine nitrate-06 (MN-06)┤NMDAR→Ca2+ influx | Primary rat cerebellar granule hippocampal neurons | [64] |
| Aβ∪endogenous Ca2+ channels→ NMDAR→Ca2+ influx | Mature hippocampal neurons | [63] | ||
| AMPAR | LY451395, LY450108 and S18986┤AMPAR→Ca2+ influx | AD animal models | [69,70,71,72] | |
| P/Q-VGCC | Aβ┤P/Q-VGCC→Ca2+ influx | Hippocampal neurons | [73] | |
| N/T/L-VGCC | Aβ1–40→N/T/L-VGCC→postsynaptic Ca2+ response | Cortical neurons | [29,74,128] | |
| Na+/K+-ATPase | Aβ┤ion-motive ATPases┤NMDAR and VGCCs→Ca2+ influx Aβ┤Ca2+-ATPase┤Ca2+ efflux | Primary neurons and synaptosomes of adult post-mortem hippocampus | [76] | |
| CALHM1 | Voltage∪extracellular Ca2+→CALHM1 | hippocampal slices from wild-type Calhm1+/+, Calhm1+/−, and Calhm1−/− mice | [78] | |
| APOE | APOE→G-protein-linked PLC→Ca2+ influx and mobilization | Neurons | [79] | |
| APOE4>E3>E2→P/Q type Ca2+-channels→ intracellular free Ca2+ | Rat hippocampal astrocytes and neurons | [80] | ||
| APOEε4→ intracellular Ca2+ | Primary cultured astrocytes of APOE−/− mice | [81] | ||
| ER | Aβ/InsP3R | Aβ→InsP3R→Ca2+ response | Cultured neurons | [87] |
| Aβ1–42/RyR | Aβ1–42→RyRs→Ca2+ flux | primary cultured hippocampal neurons | [88] | |
| Aβ aggregates/InsP3R/RyR | Aβ aggregates→InsP3R and RyR→Ca2+ flux from ER | Human brain tissues and cells, hippocampal CA1 pyramidal neurons | [82,129] | |
| PS1/InsP3R/RyR/SERCA | PS1∪InsP3R, RyR and SERCA→Ca2+ signaling cascade | Primary rat cortical neurons | [89,90,91,93,94] | |
| PS/InsP3R | PS∪InsP3R→Ca2+ flux | Primary cortical neurons | [90] | |
| PS1mut/InsP3 | PS1mut→PLC→InsP3→Ca2+ flux from ER | SH-SY5Y cell | [95] | |
| PSmut/RyR | PSmut→InsP3R and RyR→Ca2+ release from ER | PC12 cells, mouse neurons and lipid bilayers | [93,96,130,131] | |
| PSmut/SERCA | PSmut∪SERCA→Ca2+ influx | SH-SY5Y cells and patient-derived fibroblasts | [132] | |
| APOE4/RyR | APOE4→RyR→Ca2+ release from ER→APs and NFTs | Rat primary hippocampal neurons | [98,99,100] | |
| Stim1D76A | Stim1D76A mutation┤SOCE→Ca2+ influx | Primary neurons from the PS1mut mice | [104,105] | |
| Stim2 | PS1M146V mutation┤STIM2→SOCE→Ca2+ influx | PS1M146V mice | [106] | |
| Stim1 | PS1 ΔE9 mutation→Stim1→SOCE→Ca2+ influx | mouse hippocampal neurons | [107] | |
| TRPC3 | BDNF→TRPC3→Ca2+ influx. | Pontine neurons and SH-SY5Y cells | [108,110] | |
| TRPC6 | PS2→TRPC6┤Ca2+ influx | HEK293 cells | [109] | |
| MT | PS1L286V and PS1M146L | PS1L286V mutation┤Mitochondria→Ca2+ flux | PS1L286V mutated PC12 cells and PS1M146L lymphoblasts | [111,112,113] |
| VDAC | hAPPSwe→VDAC1→Ca2+ flux to the mitochondria | Tg2576 mice | [114] | |
| MCU | MCU→Ca2+ flux to the mitochondrial matrix | COS-7 cell | [115,116] | |
| Na+/Ca2+ exchanger | Na+/Ca2+ exchanger→Ca2+ across IMM | HEK293T cells | [117,118,119] | |
| mPTP | mPTP→Efflux of Ca2+ from mitochondria | SH-SY5Y cells | [120] | |
| LM | v-ATPase/CAX | V-ATPase and CAX→Ca2+ influx to lysosomes | Rat kidney fibroblasts | [122,123,133] |
| TRPML/TPC | TRPML and TPC→Ca2+ efflux from lysosomes | HEK293 cells | [124] | |
| VGCC | VGCC→Ca2+ release┤autophagic fusion and/or autophagy flux. | Cacna1a−/− and Cacna2d2−/− mice | [125] | |
| PS1mut/− | Mutation or deletion of PS1┤v-ATPase →Ca2+ uptake by lysosomes | APP/PS1 mice | [126] | |
| PS1/2−/− | PS1 and 2 knockout┤Ca2+ uptake by lysosomes→autophagy process | PS1/2−/− neurons | [127] |
| Cat. | Stimulator or Mediator | Mechanism | Experimental Model | Reference |
|---|---|---|---|---|
| Ca2+ | Aβ | Ca2+ ionophore, A23187→free Ca2+→Aβ production | hAPP overexpressed HEK293 cells, Primary cultured neurons from 3 × Tg AD mice | [134,135,136] |
| Ca2+→Aβ | SH-SY5Y cells | [36] | ||
| Ca2+→Aβ1–40 oligomers | Neurons | [137] | ||
| Ca2+→Aβ fibrils | AD mice and in vitro Aβ peptides | [138,139] | ||
| CM | NMDAR | Memantine┤NMDAR→Aβ | SH-SY5Y cells | [138] |
| AMPAR | AMPAR→Ca2+→tau phosphorylation | PS1mut mice | [140,141] | |
| Memantine┤NMDAR→Aβ1–40 | APP23 mice | [142] | ||
| NMDAR→ADAM10 | Primary mouse cortical neurons | [143] | ||
| AMPAR | AMPAR→α-secretase→sAPPα┤Aβ | Cortical neurons | [144] | |
| CALMH1 | CALHM1P86L→sAPPβ→Aβ | APP Tg mice | [138,145] | |
| L-VGCC | L-VGCC→Ca2+→Aβ | Rat cortical neurons | [134,138] | |
| Cav1.2 | Isradipine┤Cav1.2→Aβ | 3 × Tg mice | [34] | |
| APOE4 | APOE4→Aβ42 in CSF | AD patients | [34] | |
| APOE | APOE1-3┤Aβ | hAPOE isoforms (PDAPP/TRE) expressing Aβ-amyloidosis mice | [146] | |
| ER | InsP3R | InsP3R−/− receptor┤Aβ | InsP3R−/− Sf9 and DT40 cells | [90] |
| RyR | RyR→NFTs | AD patients, Primary cultured rat neurons | [101,147] | |
| RyR→Ca2+→Aβ | βAPP expressed HEK293 cells | [134,135] | ||
| Dantrolene→RyR→β-/γ-secretase→phosphorylation of APP and formation of APs | Dantrolene treated AD mice | [148,149] | ||
| RyR2 | APP mutation→RyR2PTM→Ca2+ leaky┤Aβ | SH-SY5Y cells | [150] | |
| FKBP12.6∪RyR2→Ca2+ leaky┤APs | 3 × Tg mice | [150] | ||
| RyR3 | RyR3−/−┤APs | APP/PS1 mice | [151] | |
| SERCA | Thapsigargin or siRNA┤SERCA→Aβ | PS1−/− and PS2−/− fibroblasts | [94] | |
| Thapsigargin┤SERCA→Ca2+→Aβ | APP overexpressed HEK293 cells | [135] | ||
| 10 nM thapsigargin→Aβ 20 nM thapsigargin┤Aβ | APP overexpressed CHO cells | [152] | ||
| Stim1/Orail | Stim1/Orai1→SOCE→Ca2+→Aβ/APs | APP expressed HEK293 cells | [105] | |
| SOCE | SOCE→mushroom spines ┤Aβ┤memory functions | PS1M146V knockin hippocampal neurons | [153,154] | |
| SOCE→Ca2+ influx ┤Aβ→AD | Human neuroblastoma cells, Primary cultured hippocampal neurons | [155,156] | ||
| SOCE inhibition→Aβ1–42 | SH-SY5Y cells, Human neuroglioma H4 cells | [157,158] | ||
| MT | VDAC1 | Reduced expression of VDAC1┤βAPP, Tau, PS1, PS2, and BACE1 | VDAC1+/− vs VDAC1+/+ mice | [159] |
| mPTP | APPKM670/671NL/PS1L166P∪dutasteride┤mPTP→APs | Primary neurons and APP/PS1 Tg mice | [160] | |
| Ca2+ | p-tau | Ca2+→p-tau | SH-SY5Y cells | [100] |
| Ca2+→GSK3β→p-tau | SH-SY5Y cells | [161] | ||
| Ca2+→p-tau | Primary hippocampal neurons and the immortalized GnRH neurons (GT1-7 cells). | [162] | ||
| Ca2+→mPGES-1/PGE2/EPs/CDK5/p35/p25→p-tau | N2a and APP/PS1 Tg mice | [19] | ||
| NFTs | Ca2+→Ca2+-activated kinases→p-tau→NFTs | SH-SY5Y, N2a and AD mice models | [100,163] |
| Cat. | Stimulator or Mediator | Mechanism | Experimental Model | Reference |
|---|---|---|---|---|
| Ca2+ | Serum Ca2+→cognitive decline | Aging people | [174] | |
| Ca2+→dementia | AD patients | [175] | ||
| Aβ oligomes | Aβ oligomers→Ca2+ influx┤LTP→synaptic plasticity→learning and memory | AD models, Hippocampal slices and APP/PS1 Tg mice | [176,177,178] | |
| Calpain | Inhibitor┤calpain→Aβ┤learning and memory | APP/PS1 mice | [179] | |
| Calcineurin | Inhibitor┤calcineurin┤learning and memory | Tg2576 mice | [180] | |
| CM | NMDAR | Calcineurin→removing NMDAR/AMPAR by endocytosis┤cognition of AD | APP/PS1 mice | [181] |
| Antagonist┤NMDAR┤synaptic plasticity┤cognitive decline | Rats | [182,183] | ||
| Blocking NMDAR┤Ca2+┤cognition | AD patients and AD mouse models | [184,185] | ||
| CP-AMPAR→Ca2+ influx→neuronal network dysfunction/excitotoxicity→cognitive decline | APP/PS1 mice | [186] | ||
| L-VGCC | L-VGCC→Ca2+ currents→cognitive decline | CA1 synapses of 3 × Tg AD mice | [187] | |
| Nifedipine┤Ca2+ channel→cognitive impairment | KK-A(y) mice | [188] | ||
| Nimodipine┤L-VGCC┤learning ability | Mild-to-moderate AD patients | [189] | ||
| T-VGCC | ST101┤T-VGCC┤LTP/p-CaMKII →cognitive decline | Rat cortical slices | [190] | |
| NMDAR | MK-801┤NMDAR→Ca2+→cognitive decline | Traumatic brain injury (TBI) mice | [191] | |
| Cav 2.1 | Cav 2.1−/−┤Ca2+┤learninig ability | Cav 2.1 knocking out mice | [192] | |
| TRPV1 | SB366791┤TRPV1┤cognitive performance | Dopamine D3 receptor (D3R)−/− mice | [193] | |
| APOE4 | APOE4→serum Ca2+┤cognitive function | Aging people | [194] | |
| CALHM1 | CALHM1P86L polymorphism→AD | Chinese populations | [195] | |
| ER | InsP3 | PS1M146V┤InsP3→InsP3R1→Ca2+ →memory loss | PS1M146V mice | [196] |
| InsP3R | SOCE∪InsP3R→Ca2+┤cognitive impairment | Sporadic or mild AD patients | [197] | |
| RyR | Dantrolene┤RyR┤synaptic plasticity→cognitive ability | AD mouse model | [198] | |
| RyR2/RyR3 | RyR3−/−/RyR2+/+┤social behavior and memory | RyR3−/−/RyR2+/+ mice | [199,200] | |
| RyRPTM→ER→Ca2+ leaky →cognitive deficits | 3 × Tg mice | [150] | ||
| Stim2/SOCE | STIM2−∪SOCE−┤mushroom spines→LTP→memory | PSmut mice | [106,201] | |
| SOCE−→cognitive decline→AD | Hippocampal slice cultures | [202] | ||
| MT | VDAC1 | VDAC1∪p-tau, Aβ, and γ-secretase→neurotoxicity→cell death→dementia→AD | APP, APP/PS1 and 3 × Tg mice | [203] |
| mPTP | DS16570511, DS44170716┤MCU→Ca2+ influx to mitochondria→mPTP→apoptotic cell death | HEK293 cells | [204,205] | |
| LM | TPC | Tetrandrine, NED-19┤TPCE2┤re-acidify lysosome→autophagy | MEFs cells | [206] |
| Beclin1−/−→Aβ | hAPP mice | [207] |
| Cat. | Stimulator or Mediator | Mechanism | Experimental Model | Reference |
|---|---|---|---|---|
| Ca2+ | Aβ→Ca2+ influx→LTD┤memory┤AD | Tg2576 mice | [28] | |
| Aβ oligomers→Ca2+┤LTP | Hippocampal slices | [176] | ||
| PS1−/−┤LTP | PS1−/− mice | [216] | ||
| BAPTA-AM┤Ca2+┤LTP. | Aged rat hippocampal slices | [217] | ||
| CM | CaN | CaN+┤LTP CaN−→synpatic strength →LTP | CaN+ mice | [218] |
| Ca2+→CaN→LTD | Aged or APP mice | [180,219] | ||
| Inhibitors┤CaN┤LTP | APP mice | [180] | ||
| Aβ┤CaN┤synaptic plasticity | Tg2576 mice | [28] | ||
| NMDAR | Aβ oligomers→NMDAR→Ca2+┤LTP | Hippocampal CA1 and DG regions | [220] | |
| NMDAR→Ca2+┤LTP | Rat hippocampus | [221] | ||
| AMPAR | AMPAR→Ca2+→LTP∪LTD | CA1 pyramidal cells | [242] | |
| GluR2−/−→LTP | GluR2−/− mice | [223] | ||
| CP-AMPAR→LTP | CA1 hippocampal neurons | [224] | ||
| Glycine→CP-AMPAR→CaMKI→LTP | Mature hippocampal neurons | [225] | ||
| Ca2+/Calmodulin∪PSD-95┤PSD-95∪AMPAR┤LTD | Rat hippocampal neurons | [226] | ||
| VGCC | VGCC→CaMKII→LTP | Hippocampus slides | [227] | |
| Cav1.2 | Cav1.2+→LTP, synaptic plasticity, and the memory | Ca(V)1.2 (cKO) mice | [228] | |
| TRPV1 | Capsaicin and resiniferatoxin→TRPV1→LTP | Hippocampus slides | [229] | |
| Capsaicin→TRPV1→Ca2+ influx→LTP | Hippocampus slides | [229] | ||
| TRPV1/4 | Endocannabinoid anandamide (AEA) →TRPV1/4→LTP | CB1−/− mice TRPV1−/− mice | [230,231] | |
| TRPM2 | Inhibitor┤TRPM2┤LTP | Traumatic injured brain of mice | [232] | |
| TRPM4 | TRPM4−┤NMDAR→LTP | CA1 hippocampal neurons | [233] | |
| ER | IP3 | IP3→Ca2+ efflux from ER→LTD | Myosin-Va mutation mice or rats | [234] |
| Metabotropic glutamatergic receptors→InsP3→Ca2+ efflux from ER→LTD | Hippocampal slices | [235] | ||
| InsP3R/RyR | InsP3R−→LTP∪┤LTD RyR−┤LTP∪LTD | Rat hippocampus slides, 3 × Tg AD mice | [196,243] | |
| RyR | RyR3−/−→LTP∪┤LTD | RyR3−/− mice, 3 × Tg mice | [237,238] | |
| PS−→RyR→Ca2+ release from ER ┤LTP | PS conditioned neurons from CA1 and CA3 | [239] | ||
| SOCE | SOCE−┤Ca2+ influx→CaMKII→LTP→memory | FVB/NJ mice | [240] | |
| STIM1+┤LTD┤contextual learning | FVB/NJ mice | [240] | ||
| MT | VDAC | VDAC1−/−┤synaptic plasticity | VDAC1−/− mice | [159] |
| mPTP | Cyclosporine A┤mPTP→long and short term synaptic plasticity | Porin-deficient or cyclosporin A-treated mice | [241] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. https://doi.org/10.3390/ijms22115900
Guan P-P, Cao L-L, Wang P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. International Journal of Molecular Sciences. 2021; 22(11):5900. https://doi.org/10.3390/ijms22115900
Chicago/Turabian StyleGuan, Pei-Pei, Long-Long Cao, and Pu Wang. 2021. "Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau" International Journal of Molecular Sciences 22, no. 11: 5900. https://doi.org/10.3390/ijms22115900
APA StyleGuan, P.-P., Cao, L.-L., & Wang, P. (2021). Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. International Journal of Molecular Sciences, 22(11), 5900. https://doi.org/10.3390/ijms22115900