
Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers?


Abstract
1. Introduction
2. Glucocorticoids and Cardiovascular Risk: More Than an Association?
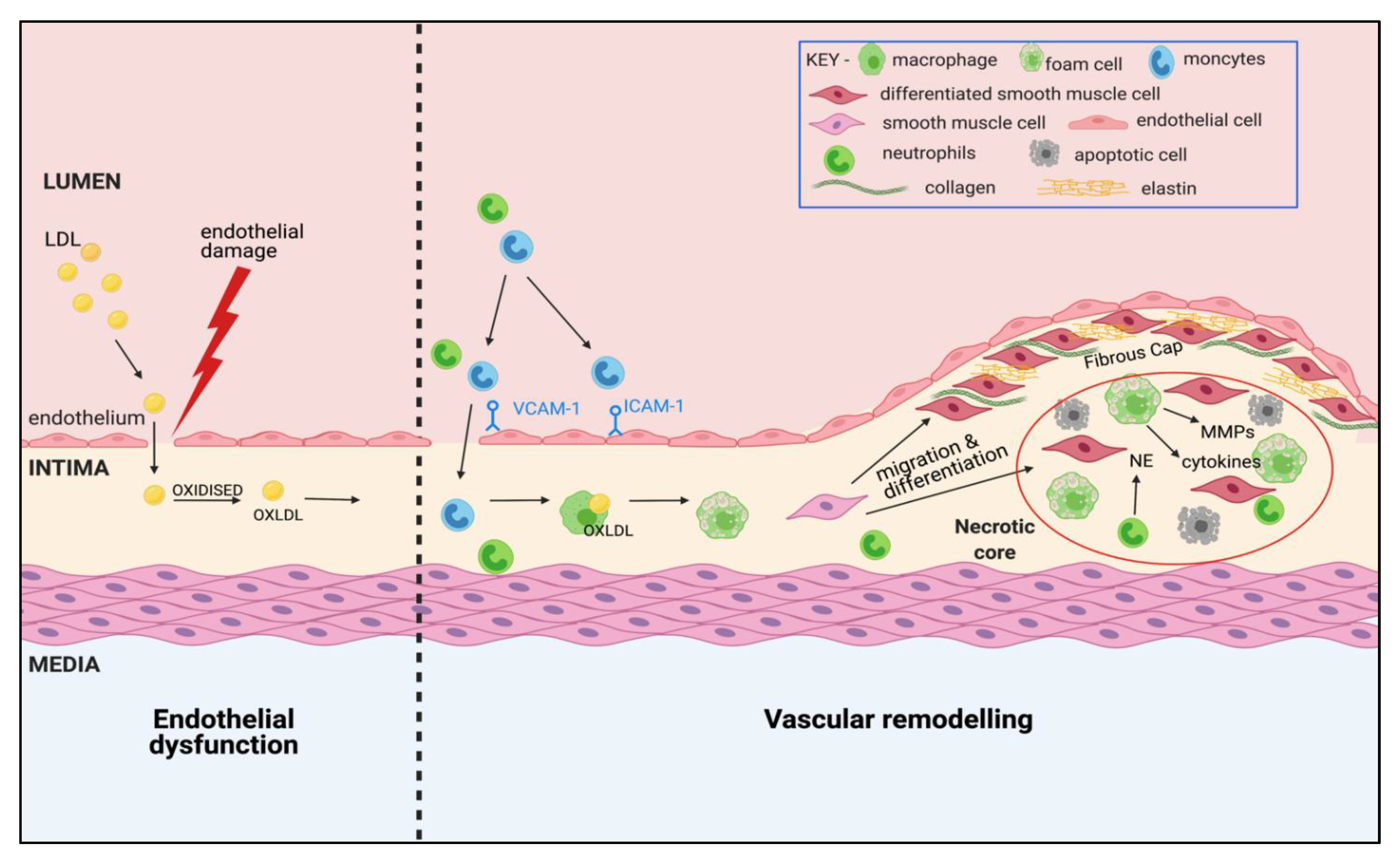
3. Atherosclerosis: A Complex, Multi-Cellular Pathophysiology
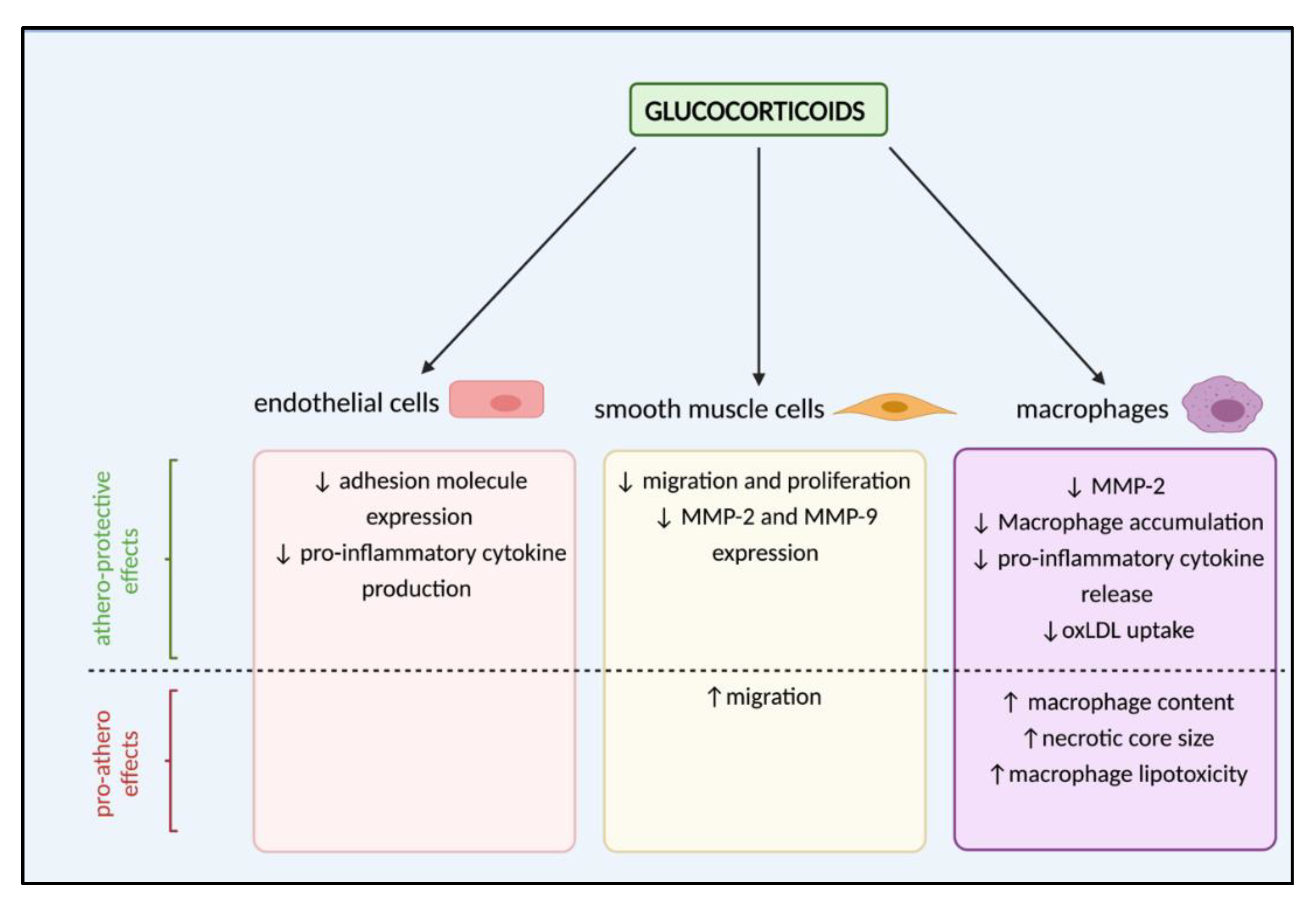
4. Are Glucocorticoids Athero-Protective or Drivers of Lesion Development?
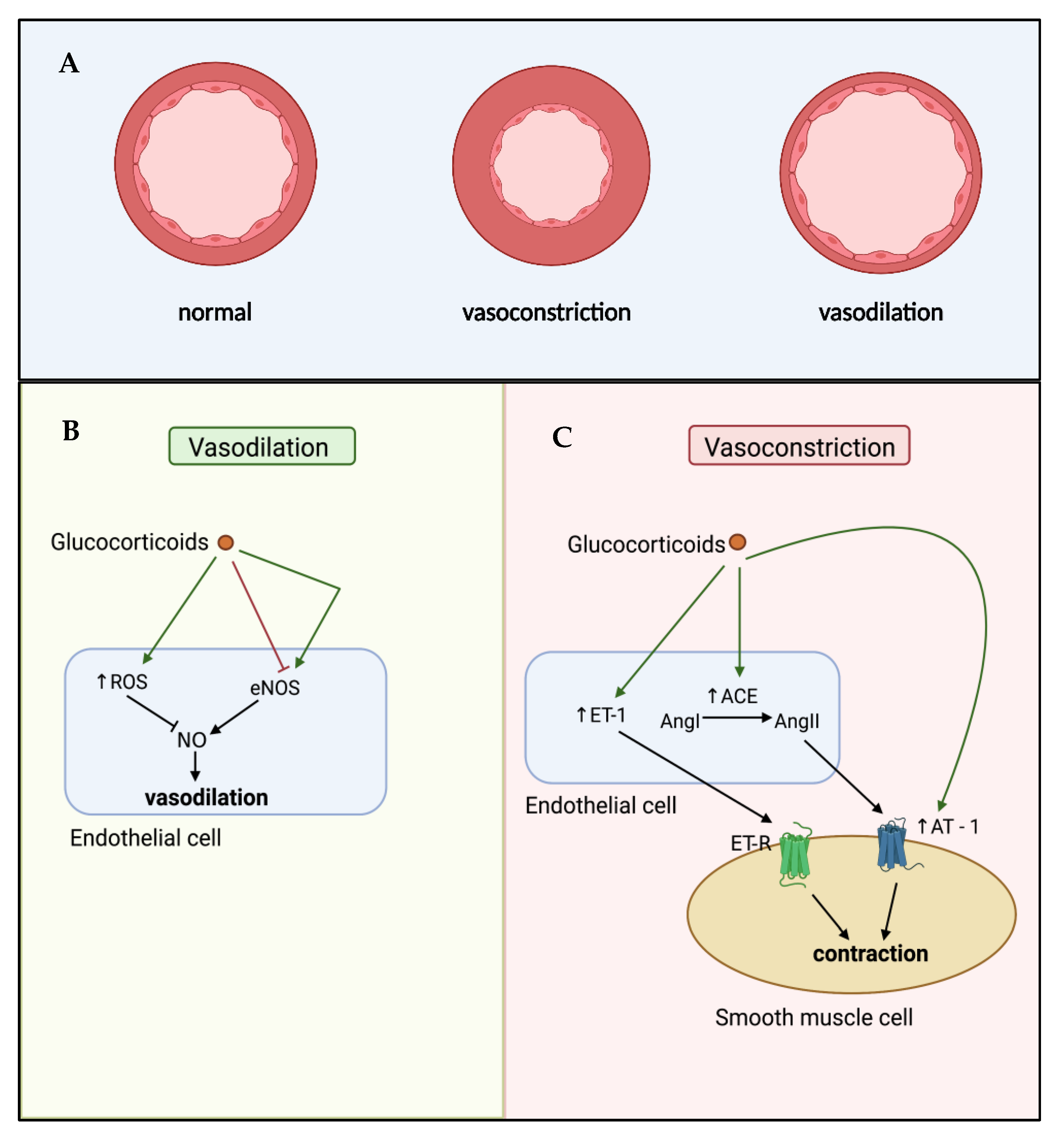
5. Glucocorticoid-Mediated Regulation of Endothelial Cell Dysfunction
6. Effects of Glucocorticoids on Vascular Remodelling
7. Mechanisms Controlling Glucocorticoid Bioavailability at the Vasculature
7.1. Pre-Receptor Metabolism of Glucocorticoids
7.2. Cognate Receptor Distribution and Regulation
7.3. Control of Glucocorticoid Delivery to the Vasculature: A Role for CBG?
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids. Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. North Am. 2016, 42, 15–31. [Google Scholar] [CrossRef]
- Rakova, N.; Jüttner, K.; Dahlmann, A.; Schröder, A.; Linz, P.; Kopp, C.; Rauh, M.; Goller, U.; Beck, L.; Agureev, A.; et al. Long-term space flight simulation reveals infradian rhythmicity in human Na+ balance. Cell Metab. 2013, 17, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, R.J.; Hoekstra, M. Glucocorticoids are active players and therapeutic targets in atherosclerotic cardiovascular disease. Mol. Cell. Endocrinol. 2020, 504, 110728. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.L. Plasma steroid-binding proteins: Primary gatekeepers of steroid hormone action. J. Endocrinol. 2016, 230, R13–R25. [Google Scholar] [CrossRef]
- Breuner, C.W.; Beyl, H.E.; Malisch, J.L. Corticosteroid-binding globulins: Lessons from biomedical research. Mol. Cell. Endocrinol. 2020, 514, 110857. [Google Scholar] [CrossRef]
- Cizza, G.; Rother, K.I. Cortisol binding globulin: More than just a carrier? J. Clin. Endocrinol. Metab. 2012, 97, 77–80. [Google Scholar] [CrossRef]
- Gustafsson, J.Å.; Carlstedt-Duke, J.; Poellinger, L.; Okret, S.; Wikström, A.C.; Brönnegård, M.; Gillner, M.; Dong, Y.; Fuxe, K.; Cintra, A.; et al. Biochemistry, molecular biology, and physiology of the glucocorticoid receptor. Endocr. Rev. 1987, 8, 185–234. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- McKay, L.I.; Cidlowski, J.A. Physiologic and Pharmacologic Effects of Corticosteroids. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, Canada, 2003. Available online: https://www.ncbi.nlm.nih.gov/books/NBK13780/ (accessed on 29 June 2021).
- Di Dalmazi, G.; Pagotto, U.; Pasquali, R.; Vicennati, V. Glucocorticoids and type 2 diabetes: From physiology to pathology. J. Nutr. Metab. 2012, 2012, 525093. [Google Scholar] [CrossRef]
- Akalestou, E.; Genser, L.; Rutter, G.A. Glucocorticoid Metabolism in Obesity and Following Weight Loss. Front. Endocrinol. 2020, 11, 59. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, T.N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells 2019, 8, 1227. [Google Scholar] [CrossRef]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology 2010, 92, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ozsoylu, S.; Strauss, H.S.; Diamond, L.K. Effects of corticosteroids on coagulation of the blood. Nature 1962, 195, 1214–1215. [Google Scholar] [CrossRef]
- Coelho, M.C.A.; Santos, C.V.; Neto, L.V.; Gadelha, M.R. Adverse effects of glucocorticoids: Coagulopathy. Eur. J. Endocrinol. 2015, 173, M11–M21. [Google Scholar] [CrossRef]
- De Leo, M.; Pivonello, R.; Auriemma, R.S.; Cozzolino, A.; Vitale, P.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Cardiovascular disease in Cushing’s syndrome: Heart versus vasculature. Neuroendocrinology 2010, 92, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Hakami, O.A.; Ahmed, S.; Karavitaki, N. Epidemiology and mortality of Cushing’s syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101521. [Google Scholar] [CrossRef]
- Li, D.; El Kawkgi, O.M.; Henriquez, A.F.; Bancos, I. Cardiovascular risk and mortality in patients with active and treated hypercortisolism. Gland. Surg. 2020, 9, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, C.; Bak, A.M.; Zøylner Rubeck, K.; Jørgensen, J.O.L. Epidemiology of Cushing’s syndrome. Neuroendocrinology 2010, 92, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tabarin, A. Do the diagnostic criteria for subclinical hypercortisolism exist? Ann. Endocrinol. 2018, 79, 146–148. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Pasquali, R.; Beuschlein, F.; Reincke, M. Subclinical hypercortisolism: A state, a syndrome, or a disease? Eur. J. Endocrinol. 2015, 173, M61–M71. [Google Scholar] [CrossRef]
- Chiodini, I. Clinical review: Diagnosis and treatment of subclinical hypercortisolism. J. Clin. Endocrinol. Metab. 2011, 96, 1223–1236. [Google Scholar] [CrossRef]
- Morelli, V.; Arosio, M.; Chiodini, I. Cardiovascular mortality in patients with subclinical Cushing. Ann. Endocrinol. 2018, 79, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Young, W.F. Management approaches to adrenal incidentalomas. A view from Rochester, Minnesota. Endocrinol. Metab. Clin. N. Am. 2000, 29, 159–185. [Google Scholar] [CrossRef]
- Walker, B.R. Cortisol—Cause and cure for metabolic syndrome? Diabet. Med. 2006, 23, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M. Tissue-specific Cushing’s syndrome, 11beta-hydroxysteroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur. J. Endocrinol. 2003, 149, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kotelevtsev, Y.; Holmes, M.C.; Burchell, A.; Houston, P.M.; Schmoll, D.; Jamieson, P.; Best, R.; Brown, R.; Edwards, C.R.W.; Seckl, J.R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. USA 1997, 94, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef]
- Ingawale, D.K.; Mandlik, S.K. New insights into the novel anti-inflammatory mode of action of glucocorticoids. Immunopharmacol. Immunotoxicol. 2020, 42, 59–73. [Google Scholar] [CrossRef]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef]
- Pujades-Rodriguez, M.; Morgan, A.W.; Cubbon, R.M.; Wu, J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immunemediated inflammatory diseases: A population-based cohort study. PLoS Med. 2020, 17, e1003432. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; MacDonald, T.M.; Walker, B.R. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann. Intern. Med. 2004, 141, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Berard, A.; Van Staa, T.P.; Cooper, C.; Egberts, A.C.G.; Leufkens, H.G.M.; Walker, B.R. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 2004, 90, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.A.; Bankier, S.; Altmaier, E.; Barnes, C.L.K.; Clark, D.W.; Ermel, R.; Friedrich, N.; van der Harst, P.; Joshi, P.K.; Karhunen, V.; et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J. Hum. Genet. 2021, 66, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Baroncini, L.A.V.; Sylvestre, L.C.; Filho, R.P. Assessment of intima-media thickness in healthy children aged 1 to 15 years. Arq. Bras. Cardiol. 2016, 106, 327–332. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.; Harker, L. Response to injury and atherogenesis. Am. J. Pathol. 1977, 86, 675–684. [Google Scholar]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef]
- Van Leeuwen, M.; Gijbels, M.J.J.; Duijvestijn, A.; Smook, M.; Van De Gaar, M.J.; Heeringa, P.; De Winther, M.P.J.; Tervaert, J.W.C. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 84–89. [Google Scholar] [CrossRef]
- Drechsler, M.; Megens, R.T.A.; Van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dollery, C.M.; Owen, C.A.; Sukhova, G.K.; Krettek, A.; Shapiro, S.D.; Libby, P. Neutrophil elastase in human atherosclerotic plaques production by macrophages. Circulation 2003, 107, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis: From mice to man. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef]
- Wen, G.; An, W.; Chen, J.; Maguire, E.M.; Chen, Q.; Yang, F.; Pearce, S.W.A.; Kyriakides, M.; Zhang, L.; Ye, S.; et al. Genetic and pharmacologic inhibition of the neutrophil elastase inhibits experimental atherosclerosis. J. Am. Heart Assoc. 2018, 7, e0081–e0087. [Google Scholar] [CrossRef] [PubMed]
- Glinzer, A.; Ma, X.; Prakash, J.; Kimm, M.A.; Lohöfer, F.; Kosanke, K.; Pelisek, J.; Thon, M.P.; Vorlova, S.; Heinze, K.G.; et al. Targeting Elastase for Molecular Imaging of Early Atherosclerotic Lesions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 525–533. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakano, M.; Narula, J.; Kolodgie, F.D.; Virmani, R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef]
- Farb, A.; Burke, A.P.; Tang, A.L.; Liang, Y.; Mannan, P.; Smialek, J.; Virmani, R. Coronary plaque erosion without rupture into a lipid core: A frequent cause of coronary thrombosis in sudden coronary death. Circulation 1996, 93, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A.; Pivonello, R.; Spiezia, S.; De Martino, M.C.; Filippella, M.; Di Somma, C.; Lombardi, G.; Colao, A. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing’s disease during active disease and 1 year after disease remission. J. Clin. Endocrinol. Metab. 2003, 88, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, R.; Ambrosino, P.; Tortora, A.; Barba, L.; Lupoli, G.A.; Di Minno, M.N.D. Markers of atherosclerosis in patients with Cushing’s syndrome: A meta-analysis of literature studies. Ann. Med. 2017, 49, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Petramala, L.; Lorenzo, D.; Iannucci, G.; Concistré, A.; Zinnamosca, L.; Marinelli, C.; De Vincentis, G.; Ciardi, A.; De Toma, G.; Letizia, C. Subclinical Atherosclerosis in Patients with Cushing Syndrome: Evaluation with Carotid Intima-Media Thickness and Ankle-Brachial Index. Endocrinol. Metab. 2015, 30, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Barahona, M.J.; Resmini, E.; Viladés, D.; Pons-Lladó, G.; Leta, R.; Puig, T.; Webb, S.M. Coronary artery disease detected by multislice computed tomography in patients after long-term cure of cushing’s syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Willenberg, T.; Diehm, N.; Zwahlen, M.; Kalka, C.; Do, D.D.; Gretener, S.; Ortmann, J.; Baumgartner, I. Impact of Long-term Corticosteroid Therapy on the Distribution Pattern of Lower Limb Atherosclerosis. Eur. J. Vasc. Endovasc. Surg. 2010, 39, 441–446. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reiss, A.B.; Jacob, B.; Ahmed, S.; Carsons, S.E.; DeLeon, J. Understanding Accelerated Atherosclerosis in Systemic Lupus Erythematosus: Toward Better Treatment and Prevention. Inflammation 2021. [Google Scholar] [CrossRef]
- Meyer, P.W.A.; Anderson, R.; Ker, J.A.; Ally, M.T.M. Rheumatoid arthritis and risk of cardiovascular disease. Cardiovasc. J. Afr. 2018, 29, 317–321. [Google Scholar] [CrossRef]
- Lewandowski, L.B.; Kaplan, M.J. Update on cardiovascular disease in lupus. Curr. Opin. Rheumatol. 2016, 28, 468–476. [Google Scholar] [CrossRef]
- Liao, K.P. Cardiovascular disease in patients with rheumatoid arthritis. Trends Cardiovasc. Med. 2017, 27, 136–140. [Google Scholar] [CrossRef]
- Crowson, C.S.; Liao, K.P.; Davis, J.M.; Solomon, D.H.; Matteson, E.L.; Knutson, K.L.; Hlatky, M.A.; Gabriel, S.E. Rheumatoid arthritis and cardiovascular disease. Am. Heart J. 2013, 166, 622–628.e1. [Google Scholar] [CrossRef]
- Naito, M.; Yasue, M.; Asai, K.; Yamada, K.; Hayashi, T.; Kuzuya, M.; Funaki, C.; Yoshimine, N.; Kuzuya, F. Effects of Dexamethasone on Experimental Atherosclerosis in Cholesterol-fed Rabbits. J. Nutr. Sci. Vitaminol. 1992, 38, 255–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Asai, K.; Funaki, C.; Hayashi, T.; Yamada, K.; Naito, M.; Kuzuya, M.; Yoshida, F.; Yoshimine, N.; Kuzuya, F. Dexamethasone-induced suppression of aortic atherosclerosis in cholesterol-fed rabbits: Possible mechanisms. Arterioscler. Thromb. 1993, 13, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, H.E.; Wang, Y.; Princen, H.; Romijn, J.A.; Havekes, L.M.; Smit, J.W.A.; Meijer, O.C.; Biermasz, N.R.; Rensen, P.C.N.; Pereira, A.M. Both Transient and Continuous Corticosterone Excess Inhibit Atherosclerotic Plaque Formation in APOE*3-Leiden.CETP Mice. PLoS ONE 2013, 8, e63882. [Google Scholar] [CrossRef]
- Jeries, H.; Volkova, N.; Grajeda-Iglesias, C.; Najjar, M.; Rosenblat, M.; Aviram, M.; Hayek, T. Prednisone and Its Active Metabolite Prednisolone Attenuate Lipid Accumulation in Macrophages. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Grahame-Clarke, C.; Shanks, N.; Marmot, M.; Lightman, S.; Vallance, P. Chronic Stress Accelerates Atherosclerosis in the Apolipoprotein E Deficient Mouse. Stress 2003, 6, 297–299. [Google Scholar] [CrossRef]
- Deuchar, G.A.; McLean, D.; Hadoke, P.W.F.; Brownstein, D.G.; Webb, D.J.; Mullins, J.J.; Chapman, K.; Seckl, J.R.; Kotelevtsev, Y.V. 11β-Hydroxysteroid dehydrogenase type 2 deficiency accelerates atherogenesis and causes proinflammatory changes in the endothelium in apoe −/− mice. Endocrinology 2011, 152, 236–246. [Google Scholar] [CrossRef]
- García, R.A.; Search, D.J.; Lupisella, J.A.; Ostrowski, J.; Guan, B.; Chen, J.; Yang, W.P.; Truong, A.; He, A.; Zhang, R.; et al. 11β-Hydroxysteroid Dehydrogenase Type 1 Gene Knockout Attenuates Atherosclerosis and In Vivo Foam Cell Formation in Hyperlipidemic apoE−/− Mice. PLoS ONE 2013, 8, e53192. [Google Scholar] [CrossRef] [PubMed]
- Kipari, T.; Hadoke, P.W.F.; Iqbal, J.; Man, T.Y.; Miller, E.; Coutinho, A.E.; Zhang, Z.; Sullivan, K.M.; Mitic, T.; Livingstone, D.E.W.; et al. 11β-hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis. FASEB J. 2013, 27, 1519–1531. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L. Glucocorticoids and Vascular Reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef]
- Suzuki, T.; Nakamura, Y.; Moriya, T.; Sasano, H. Effects of steroid hormones on vascular functions. Microsc. Res. Tech. 2003, 60, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, J.A.; Williamson, P.M.; Mangos, G.; Kelly, J.J. Cardiovascular consequences of cortisol excess. Vasc. Health Risk Manag. 2005, 1, 291–299. [Google Scholar] [CrossRef]
- Sutton, G.; Pugh, D.; Dhaun, N. Developments in the role of endothelin-1 in atherosclerosis: A potential therapeutic target? Am. J. Hypertens. 2019, 32, 813–815. [Google Scholar] [CrossRef]
- Winkles, J.A.; Alberts, G.F.; Brogi, E.; Libby, P. Endothelin-1 and endothelin receptor mRNA expression in normal and atherosclerotic human arteries. Biochem. Biophys. Res. Commun. 1993, 191, 1081–1088. [Google Scholar] [CrossRef]
- Lerman, A.; Edwards, B.S.; Hallett, J.W.; Heublein, D.M.; Sandberg, S.M.; Burnett, J.C. Circulating and Tissue Endothelin Immunoreactivity in Advanced Atherosclerosis. N. Engl. J. Med. 1991, 325, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kanse, S.M.; Takahashi, K.; Warren, J.B.; Ghatei, M.; Bloom, S.R. Glucocorticoids induce endothelin release from vascular smooth muscle cells but not endothelial cells. Eur. J. Pharmacol. 1991, 199, 99–101. [Google Scholar] [CrossRef]
- Roubert, P.; Viossat, I.; Lonchampt, M.O.; Chapelat, M.; Schulz, J.; Plas, P.; Gillard-Roubert, V.; Chabrier, P.E.; Braquet, P. Endothelin receptor regulation by endothelin synthesis in vascular smooth muscle cells: Effects of dexamethasone and phosphoramidon. J. Vasc. Res. 1993, 30, 139–144. [Google Scholar] [CrossRef]
- Morin, C.; Asselin, C.; Boudreau, F.; Provencher, P.H. Transcriptional regulation of pre-pro-endothelin-1 gene by glucocorticoids in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998, 244, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H.J.; Admiraal, P.J.J.; Derkx, F.H.M.; Schalekamp, M.A.D.H. Angiotensin I-to-II conversion in the human renal vascular bed. J. Hypertens. 1998, 16, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R.S.; Eisenberg, S.; Shai, S.Y.; Redden, R.A.; Bernstein, K.E.; Berk, B.C. Glucocorticoids induce angiotensin-converting enzyme expression in vascular smooth muscle. Hypertension 1995, 25, 343–349. [Google Scholar] [CrossRef]
- Coulet, F.; Gonzalez, W.; Boixel, C.; Meilhac, O.; Pueyo, M.E.; Michel, J. Endothelium-independent conversion of angiotensin I by vascular smooth muscle cells. Cell Tissue Res. 2001, 303, 227–234. [Google Scholar] [CrossRef]
- Sato, A.; Suzuki, H.; Nakazato, Y.; Shi Bata, H.; Inaga Mi, T.; Saruta, T. Increased expression of vascular angiotensin ii type 1a receptor gene in glucocorticoid-induced hypertension. J. Hypertens. 1994, 12, 511–516. [Google Scholar] [CrossRef]
- Rogers, K.M.; Bonar, C.A.; Estrella, J.L.; Yang, S. Inhibitory effect of glucocorticoid on coronary artery endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1922–H1928. [Google Scholar] [CrossRef]
- Iuchi, T.; Akaike, M.; Mitsui, T.; Ohshima, Y.; Shintani, Y.; Azuma, H.; Matsumoto, T. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ. Res. 2003, 92, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wallerath, T.; Witte, K.; Schafer, S.C.; Schwarz, P.M.; Prellwitz, W.; Wohlfart, P.; Kleinert, H.; Lehr, H.A.; Lemmer, B.; Forstermann, U. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc. Natl. Acad. Sci. USA 1999, 96, 13357–13362. [Google Scholar] [CrossRef] [PubMed]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef]
- Dufour, A.; Corsini, E.; Gelati, M.; Ciusani, E.; Zaffaroni, M.; Giombini, S.; Massa, G.; Salmaggi, A. Modulation of ICAM-1, VCAM-1 and HLA-DR by cytokines and steroids on HUVECs and human brain endothelial cells. J. Neurol. Sci. 1998, 157, 117–121. [Google Scholar] [CrossRef]
- Wheller, S.K.; Perretti, M. Dexamethasone inhibits cytokine-induced intercellular adhesion molecule-1 up-regulation on endothelial cell lines. Eur. J. Pharmacol. 1997, 331, 65–71. [Google Scholar] [CrossRef]
- Simoncini, T.; Maffei, S.; Basta, G.; Barsacchi, G.; Genazzani, A.R.; Liao, J.K.; De Caterina, R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 2000, 87, 19–25. [Google Scholar] [CrossRef]
- Zakkar, M.; Luong, L.A.; Chaudhury, H.; Ruud, O.; Punjabi, P.P.; Anderson, J.R.; Mullholand, J.W.; Clements, A.T.; Krams, R.; Foin, N.; et al. Dexamethasone arterializes venous endothelial cells by inducing mitogen-activated protein kinase phosphatase-1: A novel antiinflammatory treatment for vein grafts? Circulation 2011, 123, 524–532. [Google Scholar] [CrossRef]
- Shi, J.X.; Li, J.S.; Hu, R.; Shi, Y.; Su, X.; Guo, X.J.; Li, X.M. Tristetraprolin is involved in the glucocorticoid-mediated interleukin 8 repression. Int. Immunopharmacol. 2014, 22, 480–485. [Google Scholar] [CrossRef]
- Van Put, D.J.M.; Van Hove, C.E.; De Meyer, G.R.Y.; Wuyts, F.; Herman, A.G.; Bult, H. Dexamethasone influences intimal thickening and vascular reactivity in the rabbit collared carotid artery. Eur. J. Pharmacol. 1995, 294, 753–761. [Google Scholar] [CrossRef]
- Reil, T.D.; Sarkar, R.; Kashyap, V.S.; Sarkar, M.; Gelabert, H.A. Dexamethasone suppresses vascular smooth muscle cell proliferation. J. Surg. Res. 1999, 85, 109–114. [Google Scholar] [CrossRef]
- Reil, T.D.; Kashyap, V.S.; Sarkar, R.; Freishlag, J.; Gelabert, H.A. Dexamethasone inhibits the phosphorylation of retinoblastoma protein in the suppression of human vascular smooth muscle cell proliferation. J. Surg. Res. 2000, 92, 108–113. [Google Scholar] [CrossRef]
- Xu, W.; Guo, T.; Zhang, Y.; Jiang, X.; Zhang, Y.; Zen, K.; Yu, B.; Zhang, C.Y. The inhibitory effect of dexamethasone on platelet-derived growth factor-induced vascular smooth muscle cell migration through up-regulating PGC-1α expression. Exp. Cell Res. 2011, 317, 1083–1092. [Google Scholar] [CrossRef]
- Voisard, R.; Seitzer, U.; Baur, R.; Dartsch, P.C.; Osterhues, H.; Höher, M.; Hombach, V. Corticosteroid agents inhibit proliferation of smooth muscle cells from human atherosclerotic arteries in vitro. Int. J. Cardiol. 1994, 43, 257–267. [Google Scholar] [CrossRef]
- Haug, C.; Voisard, R.; Lenich, A.; Baur, R.; Höher, M.; Osterhues, H.; Hannekum, A.; Vogel, U.; Mattfeldt, T.; Hombach, V.; et al. Increased endothelin release by cultured human smooth muscle cells from atherosclerotic coronary arteries. Cardiovasc. Res. 1996, 31, 807–813. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Zhao, W.-S.; Xu, L.; Wang, X.; Li, X.-L.; Yang, X.-C. Endothelium-specific endothelin-1 expression promotes pro-inflammatory macrophage activation by regulating miR-33/NR4A axis. Exp. Cell Res. 2021, 399, 112443. [Google Scholar] [CrossRef]
- Pross, C.; Farooq, M.M.; Angle, N.; Lane, J.S.; Cerveira, J.J.; Xavier, A.E.; Freischlag, J.A.; Law, R.E.; Gelabert, H.A. Dexamethasone inhibits vascular smooth muscle cell migration via modulation of matrix metalloproteinase activity. J. Surg. Res. 2002, 102, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, J.; Jing, Z.; Xiao, Y.; Sun, Y.; Wu, Y.; Sun, H. Glucocorticoids Regulate the Vascular Remodeling of Aortic Dissection Via the p38 MAPK-HSP27 Pathway Mediated by Soluble TNF-RII. EBioMedicine 2018, 27, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.; Gertz, S.D.; Fallon, J.T.; Wiegman, P.; Berman, J.W.; Sarembock, I.J.; Taubman, M.B. Dexamethasone inhibits macrophage accumulation after balloon arterial injury in cholesterol fed rabbits. Atherosclerosis 2001, 155, 371–380. [Google Scholar] [CrossRef]
- Liberman, A.C.; Budziñski, M.L.; Sokn, C.; Gobbini, R.P.; Steininger, A.; Arzt, E. Regulatory and Mechanistic Actions of Glucocorticoids on T and Inflammatory Cells. Front. Endocrinol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Van der Valk, F.M.; Schulte, D.M.; Meiler, S.; Tang, J.; Zheng, K.H.; Van den Bossche, J.; Seijkens, T.; Laudes, M.; de Winther, M.; Lutgens, E.; et al. Liposomal prednisolone promotes macrophage lipotoxicity in experimental atherosclerosis. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1463–1470. [Google Scholar] [CrossRef]
- Van der Valk, F.M.; Van Wijk, D.F.; Lobatto, M.E.; Verberne, H.J.; Storm, G.; Willems, M.C.M.; Legemate, D.A.; Nederveen, A.J.; Calcagno, C.; Mani, V.; et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1039–1046. [Google Scholar] [CrossRef]
- Cheng, W.; Kvilekval, K.V.; Abumrad, N.A. Dexamethasone enhances accumulation of cholesteryl esters by human macrophages. Am. J. Physiol. Endocrinol. Metab. 1995, 269, E642–E648. [Google Scholar] [CrossRef]
- Petrichenko, I.E.; Daret, D.; Kolpakova, G.V.; Shakhov, Y.A.; Larrue, J. Glucocorticoids stimulate cholesteryl ester formation in human smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1143–1151. [Google Scholar] [CrossRef]
- Ledda, A.; González, M.; Gulfo, J.; Díaz Ludovico, I.; Ramella, N.; Toledo, J.; Garda, H.; Grasa, M.; Esteve, M. Decreased OxLDL uptake and cholesterol efflux in THP1 cells elicited by cortisol and by cortisone through 11β-hydroxysteroid dehydrogenase type 1. Atherosclerosis 2016, 250, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Hadoke, P.W.F.; Kipari, T.; Seckl, J.R.; Chapman, K.E. Modulation of 11β-hydroxysteroid dehydrogenase as a strategy to reduce vascular inflammation. Curr. Atheroscler. Rep. 2013, 15, 320. [Google Scholar] [CrossRef]
- Hadoke, P.W.F.; Iqbal, J.; Walker, B.R. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br. J. Pharmacol. 2009, 156, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Draper, N.; Stewart, P.M. 11β-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J. Endocrinol. 2005, 186, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R.; Yau, J.L.; Brett, L.P.; Seckl, J.R.; Monder, C.; Williams, B.C.; Edwards, C.R.W. 1lβ-hydroxysteroid dehydrogenase in vascular smooth muscle and heart: Implications for cardiovascular responses to glucocorticoids. Endocrinology 1991, 129, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Dover, A.R.; Hadoke, P.W.F.; Macdonald, L.J.; Miller, E.; Newby, D.E.; Walker, B.R. Intravascular glucocorticoid metabolism during inflammation and injury in mice. Endocrinology 2007, 148, 166–172. [Google Scholar] [CrossRef]
- Ayari, H.; Legedz, L.; Lantelme, P.; Feugier, P.; Randon, J.; Cerutti, C.; Lohez, O.; Scoazec, J.Y.; Li, J.Y.; Gharbi-Chihi, J.; et al. Auto-amplification of cortisol actions in human carotid atheroma is linked to arterial remodeling and stroke. Fundam. Clin. Pharmacol. 2014, 28, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Atalar, F.; Gormez, S.; Caynak, B.; Akan, G.; Tanriverdi, G.; Bilgic-Gazioglu, S.; Gunay, D.; Duran, C.; Akpinar, B.; Ozbek, U.; et al. The role of mediastinal adipose tissue 11β-hydroxysteroid d ehydrogenase type 1 and glucocorticoid expression in the development of coronary atherosclerosis in obese patients with ischemic heart disease. Cardiovasc. Diabetol. 2012, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Macdonald, L.J.; Low, L.; Seckl, J.R.; Yau, C.W.; Walker, B.R.; Hadoke, P.W.F. Contribution of endogenous glucocorticoids and their intravascular metabolism by 11β-HSDs to postangioplasty neointimal proliferation in mice. Endocrinology 2012, 153, 5896–5905. [Google Scholar] [CrossRef]
- Hermanowski-Vosatka, A.; Balkovec, J.M.; Cheng, K.; Chen, H.Y.; Hernandez, M.; Koo, G.C.; Le Grand, C.B.; Li, Z.; Metzger, J.M.; Mundt, S.S.; et al. 11β-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J. Exp. Med. 2005, 202, 517–527. [Google Scholar] [CrossRef]
- Anderson, A.; Walker, B.R. 11β-HSD1 inhibitors for the treatment of type 2 diabetes and cardiovascular disease. Drugs 2013, 73, 1385–1393. [Google Scholar] [CrossRef]
- De Kloet, E.R. From receptor balance to rational glucocorticoid therapy. Endocrinology 2014, 155, 2754–2769. [Google Scholar] [CrossRef]
- Moss, M.E.; Lu, Q.; Iyer, S.L.; Engelbertsen, D.; Marzolla, V.; Caprio, M.; Lichtman, A.H.; Jaffe, I.Z. Endothelial Mineralocorticoid Receptors Contribute to Vascular Inflammation in Atherosclerosis in a Sex-Specific Manner. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1588–1601. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernández-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis—Brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Preusch, M.R.; Rattazzi, M.; Albrecht, C.; Merle, U.; Tuckermann, J.; Schütz, G.; Blessing, E.; Zoppellaro, G.; Pauletto, P.; Krempien, R.; et al. Critical role of macrophages in glucocorticoid driven vascular calcification in a mouse-model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Hayward, C.; Direk, N.; Lewis, J.G.; Hammond, G.L.; Hill, L.A.; Anderson, A.; Huffman, J.; Wilson, J.F.; Campbell, H.; et al. Genome Wide Association Identifies Common Variants at the SERPINA6/SERPINA1 Locus Influencing Plasma Cortisol and Corticosteroid Binding Globulin. PLoS Genet. 2014, 10, e1004474. [Google Scholar] [CrossRef]
- Ho, J.T.; Al-Musalhi, H.; Chapman, M.J.; Quach, T.; Thomas, P.D.; Bagley, C.J.; Lewis, J.G.; Torpy, D.J. Septic shock and sepsis: A comparison of total and free plasma cortisol levels. J. Clin. Endocrinol. Metab. 2006, 91, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.H.; Andreassen, T.K.; Breiderhoff, T.; Bräsen, J.H.; Schulz, H.; Gross, V.; Gröne, H.-J.; Nykjaer, A.; Willnow, T.E. Hyporesponsiveness to Glucocorticoids in Mice Genetically Deficient for the Corticosteroid Binding Globulin. Mol. Cell. Biol. 2006, 26, 7236–7245. [Google Scholar] [CrossRef]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.F.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef]
- Bartalena, L.; Hammond, G.L.; Farsetti, A.; Flink, I.L.; Robbins, J. Interleukin-6 inhibits corticosteroid-binding globulin synthesis by human hepatoblastoma-derived (Hep g2) cells. Endocrinology 1993, 133, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Tsigos, C.; Kyrou, I. Prolonged suppression of corticosteroid-binding globulin by recombinant human interleukin-6 in man. J. Clin. Endocrinol. Metab. 1998, 83, 3379. [Google Scholar] [CrossRef]
- Hammond, G.L. Molecular properties of corticosteroid binding globulin and the sex-steroid binding proteins. Endocr. Rev. 1990, 11, 65–79. [Google Scholar] [CrossRef]
- Lin, H.Y.; Underhill, C.; Gardill, B.R.; Muller, Y.A.; Hammond, G.L. Residues in the human corticosteroid-binding globulin reactive center loop that influence steroid binding before and after elastase cleavage. J. Biol. Chem. 2009, 284, 884–896. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Acute, Adaptive Physiological Roles | Chronic, Maladaptive Effects | Pathological Outcomes | References |
|---|---|---|---|
| Regulation of glucose homeostasis | Hyperglycaemia | Diabetes mellitus | [11] |
| Insulin resistance | |||
| Maintenance of energy homeostasis | Visceral adiposity | Obesity | [12] |
| General weight gain | |||
| Maintenance of vascular tone and blood pressure | Impaired vasodilation | Hypertension | [13] |
| Increased contractility | |||
| Plasma volume expansion | |||
| Regulation of lipid metabolism | Elevated cholesterol | Dyslipidaemia | [14] |
| Elevated triglycerides | |||
| Heart development and function | Bradycardia Cardiac hypertrophy | Heart failure | [13] |
| Regulation of clotting factors and fibrinogen | Hypercoagulability | Thrombosis | [15,16] |
| Potential Impact | Target | Effect | Reference |
|---|---|---|---|
| Pro-Atherosclerotic | Endothelium | ↓ generation of EDRFs | [83] |
| Endothelium | ↑ ROS generation | [84] | |
| Endothelium | ↓ eNOS | [85] | |
| vSMC | ↑ vasoconstrictor (ET-1) generation | [78] | |
| vSMC | ↑ vasoconstrictor (AngII) generation | [80,81] | |
| vSMC | ↑ AT-1 receptor expression | [82] | |
| vSMC | ↑ accumulation of cholesterol esters | [106] | |
| Macrophages | ↑ lipotoxicity | [104] | |
| Macrophages | ↑ accumulation of cholesterol esters | [105] | |
| Anti-Atherosclerotic | Endothelium | ↑ eNOS (non-genomic) | [86] |
| Endothelium | ↓ adhesion molecule expression | [87,88,89] | |
| Endothelium | ↓ expression of pro-inflammatory cytokines | [90,91] | |
| Macrophages | ↓ accumulation | [101] | |
| Macrophages | ↓ expression of pro-inflammatory cytokines | [102] | |
| Macrophages | ↓ oxLDL uptake and cholesterol efflux | [107] | |
| Unclear | vSMCs | ↓ proliferation and migration | [92,93,94,95,96,99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacLeod, C.; Hadoke, P.W.F.; Nixon, M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? Int. J. Mol. Sci. 2021, 22, 7622. https://doi.org/10.3390/ijms22147622
MacLeod C, Hadoke PWF, Nixon M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? International Journal of Molecular Sciences. 2021; 22(14):7622. https://doi.org/10.3390/ijms22147622
Chicago/Turabian StyleMacLeod, Clare, Patrick W. F. Hadoke, and Mark Nixon. 2021. "Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers?" International Journal of Molecular Sciences 22, no. 14: 7622. https://doi.org/10.3390/ijms22147622
APA StyleMacLeod, C., Hadoke, P. W. F., & Nixon, M. (2021). Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? International Journal of Molecular Sciences, 22(14), 7622. https://doi.org/10.3390/ijms22147622