
Wild-Type and Mutant FUS Expression Reduce Proliferation and Neuronal Differentiation Properties of Neural Stem Progenitor Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
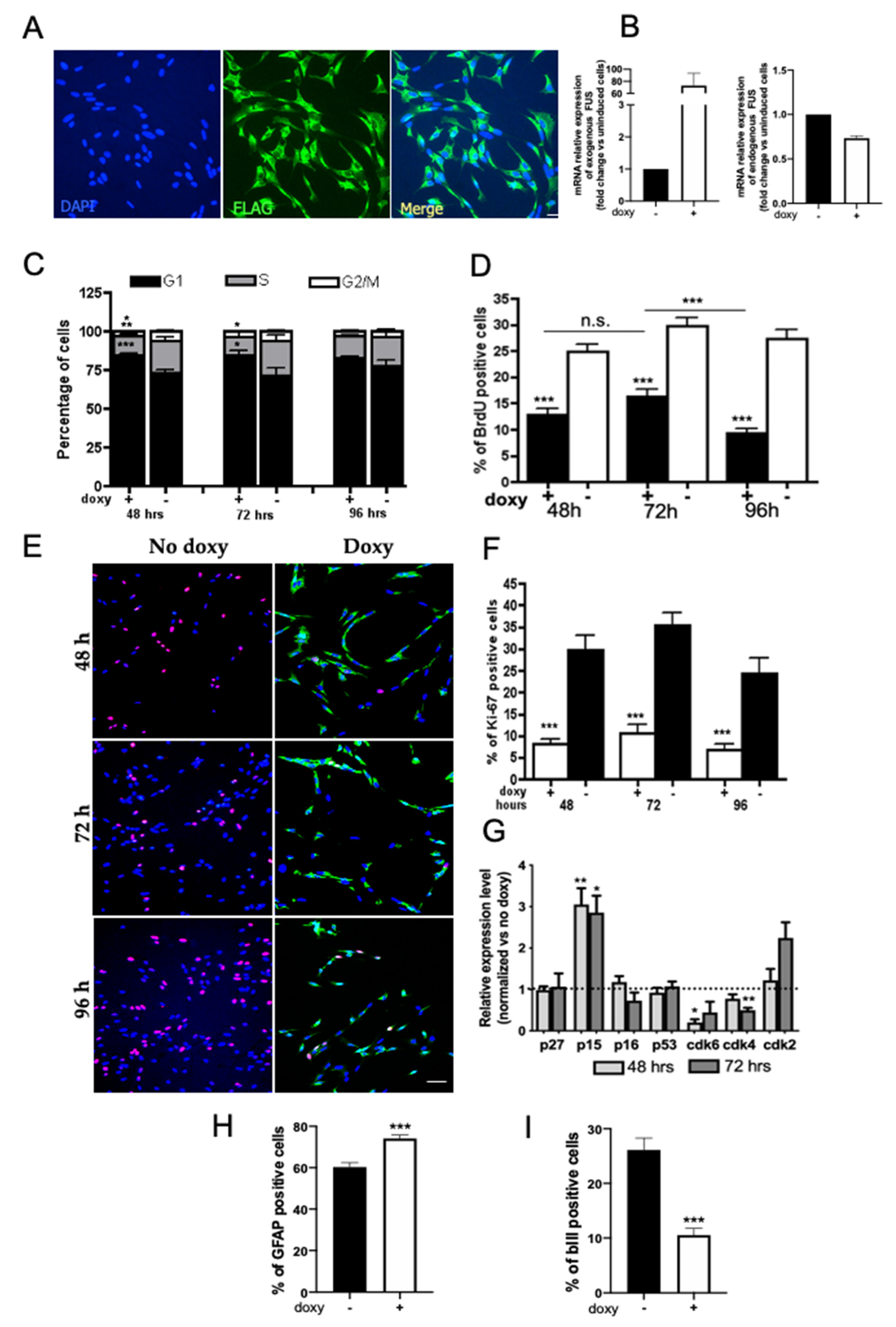{kind=link}
Abstract
1. Introduction
2. Results
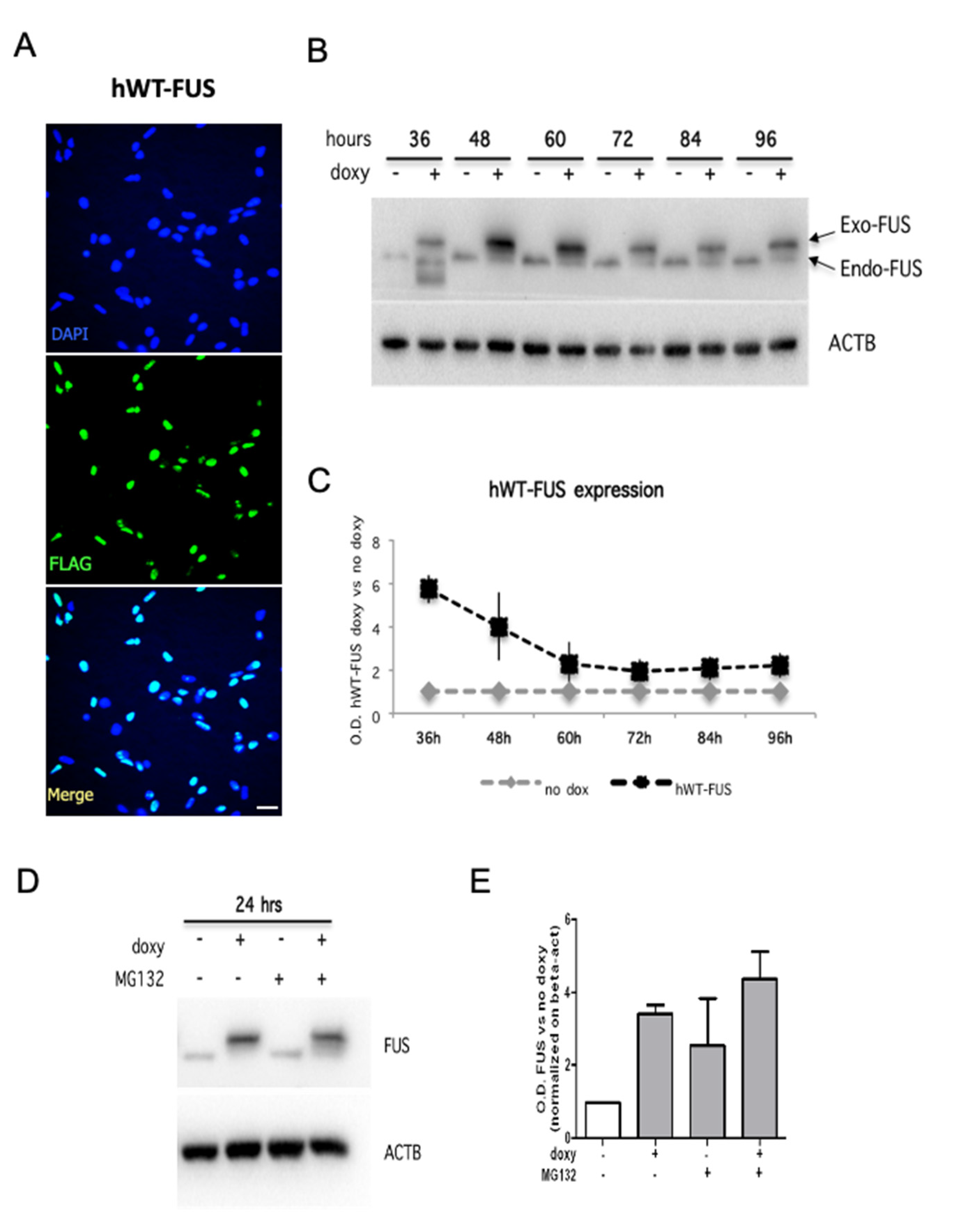
2.1. Expression and Localization of HumanWT-FUS in NSPCs
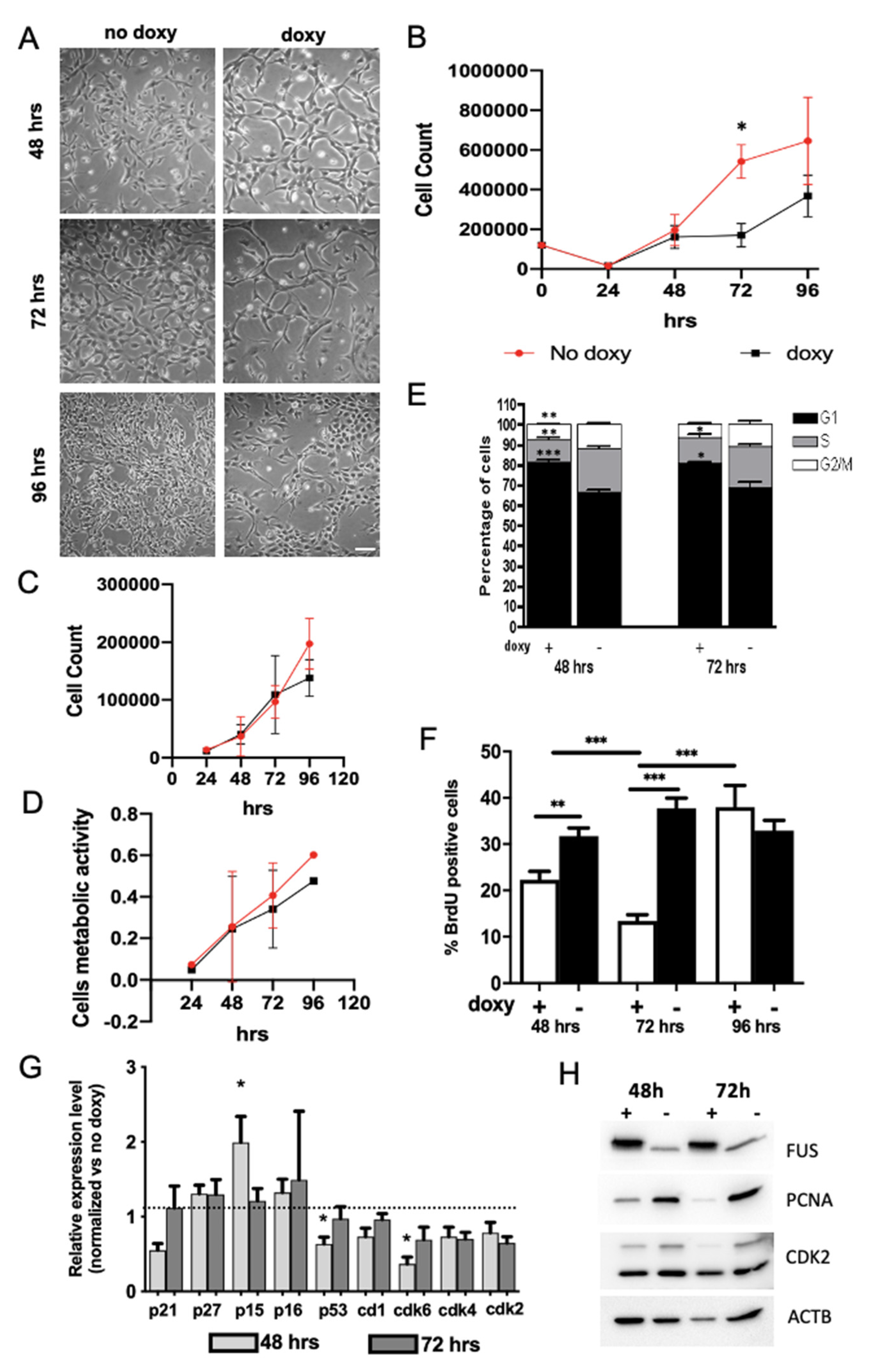
2.2. Expression of hWT-FUS Protein Markedly Decreases Cell Growth
2.3. FUS Regulates the Expression of Cell Cycle Factors
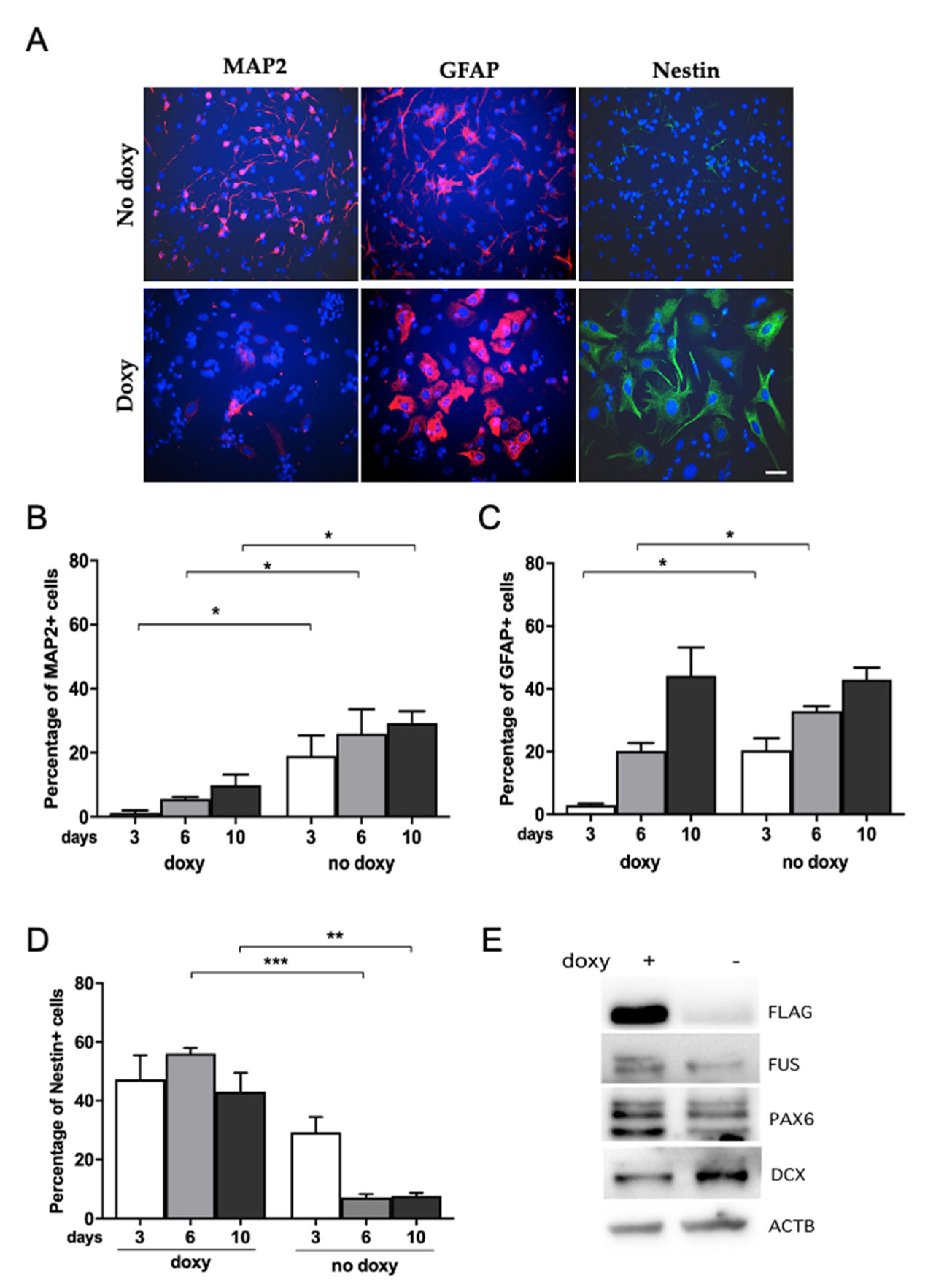
2.4. Human WT-FUS Expression Has an Anti-Neurogenic Effect
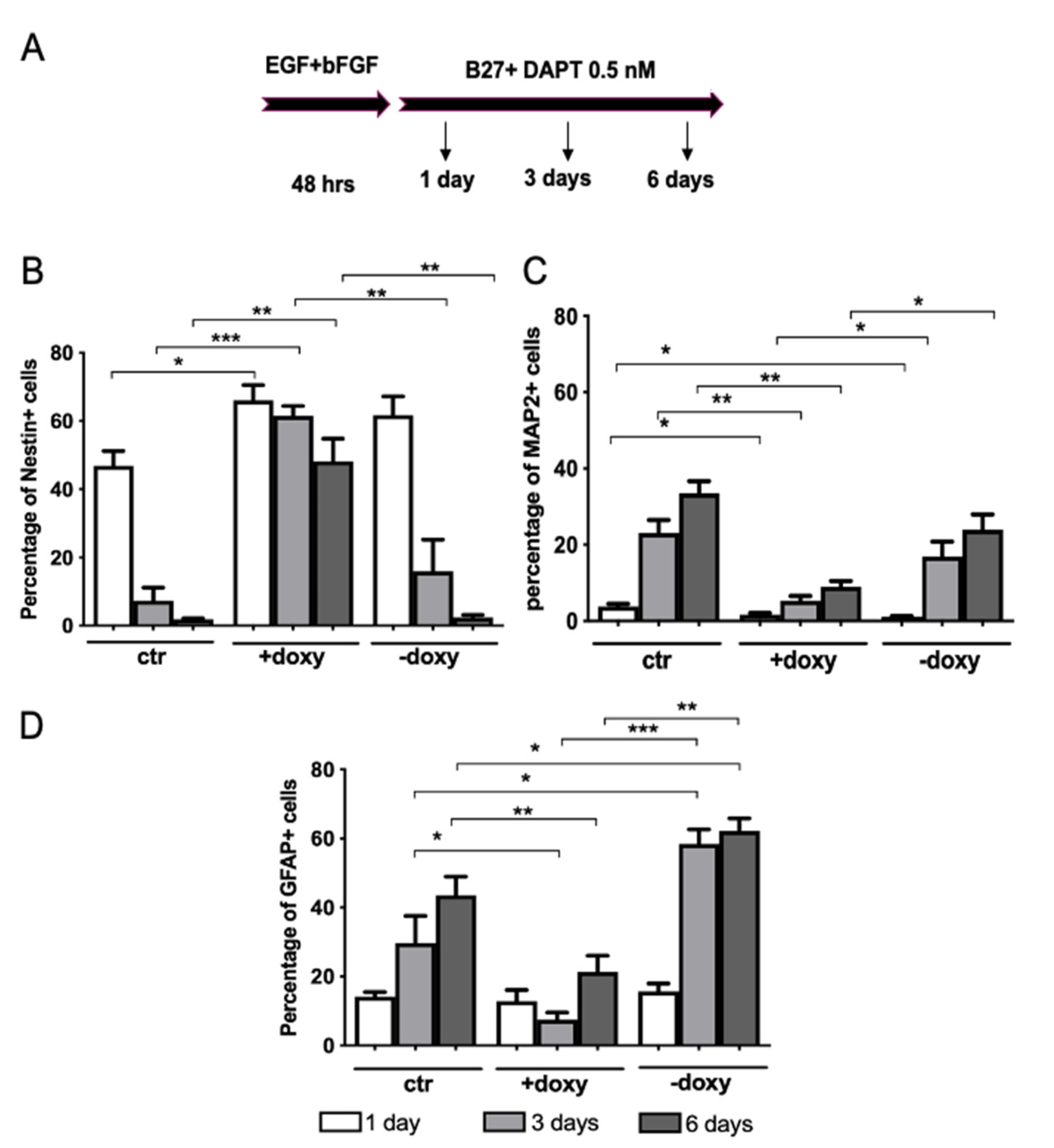
2.5. Expression of Mutant Human FUS Arrests Proliferation and Promotes Glial Differentiation
3. Discussion
Conclusions and Perspectives
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Electroporation of NSPCs
4.4. Immunocytochemistry
4.5. RNA Extraction
4.6. Real-Time RT-PCR and Quantification of Gene Expression
| FUS human | F: GCT GCC ATC ACA AGC ATA GC R: CAG CCT GGA TGA CAG AGC AA |
| FUS mouse | F: CTG GAA CTT TGT TGC TTG GCG R: CCC CGT AGC TTT GAG TTG CTT G |
| beta-actin | F: CCC AGG CAT TGC TGA CAG G R: GCT GGA AGG TGG ACA GTG AG |
| p27 | F: GGG TTC CAG CTT GTT GTG TT R: GGC CAT TTT CCA TCT CTG AA |
| CD1 | F: CAG CTT GCT AGG GAA CTT GC R: GAG TCT CCG GTG CAT CAT TT |
| p15 | F: CCT TCC CCT GTG AAC TGA AA R: GTG AAT CCC CAC ACA TGA CA |
| Cdk4 | F: GGC CCT CAA GAG TGT GAG A R: CAT CAG CCG TAC AAC ATT G |
| Cdk2 | F: AGA CTC CTG CTG CCA CTG TT R: CCC TCT TGC TTG GTT CTG AG |
| p16 | F: CAT CTG GAG CAG CAT GGA GTC R: GGG TAC GAC CGA AAG AGT TCG |
| p57 | F: TTG AGT AAA AGC CCC CAC A R: TGG GCA GTA CAG GAA CCA T |
| Cdk6 | F: ACT CTC ACC TGC CAT CCA A R: GAG GCT TGC TTA GGG GAC T |
4.7. Cell Growth
4.8. MTT Assay
4.9. Cell Cycle Analysis
4.10. BrdU Incorporation
4.11. Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Rabbitts, T.H.; Forster, A.; Larson, R.; Nathan, P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993, 4, 175–180. [Google Scholar] [CrossRef]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Soraru, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2010, 47, 190–194. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Andersson, M.K.; Stahlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Aman, P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef]
- Hoell, J.I.; Larsson, E.; Runge, S.; Nusbaum, J.D.; Duggimpudi, S.; Farazi, T.A.; Hafner, M.; Borkhardt, A.; Sander, C.; Tuschl, T. RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 2011, 18, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Batra, R.; Lagier-Tourenne, C.; Yeo, G.W. RNA-binding proteins in neurodegeneration: Seq and you shall receive. Trends Neurosci. 2015, 38, 226–236. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef]
- Ling, S.C.; Dastidar, S.G.; Tokunaga, S.; Ho, W.Y.; Lim, K.; Ilieva, H.; Parone, P.A.; Tyan, S.H.; Tse, T.M.; Chang, J.C.; et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife 2019, 8, e40811. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Moncada, A.; Conte, A.; Lattante, S.; Marangi, G.; Luigetti, M.; Lucchini, M.; Mirabella, M.; Romano, A.; Del Grande, A.; et al. Mutations in the 3’ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4748–4755. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Onori, A.; Toselli, C.; Stronati, E.; Morlando, M.; Bozzoni, I.; Monni, E.; Kokaia, Z.; Lupo, G.; Minghetti, L.; et al. Increased FUS levels in astrocytes leads to astrocyte and microglia activation and neuronal death. Sci. Rep. 2019, 9, 4572. [Google Scholar] [CrossRef]
- Copani, A.; Uberti, D.; Sortino, M.A.; Bruno, V.; Nicoletti, F.; Memo, M. Activation of cell-cycle-associated proteins in neuronal death: A mandatory or dispensable path? Trends Neurosci. 2001, 24, 25–31. [Google Scholar] [CrossRef]
- Greene, L.A.; Liu, D.X.; Troy, C.M.; Biswas, S.C. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 2007, 1772, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Boudreau, M.; Kriz, J.; Couillard-Despres, S.; Kaplan, D.R.; Julien, J.P. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J. Neurosci. 2003, 23, 2131–2140. [Google Scholar] [CrossRef]
- Ranganathan, S.; Bowser, R. Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am. J. Pathol. 2003, 162, 823–835. [Google Scholar] [CrossRef]
- Ranganathan, S.; Bowser, R. p53 and Cell Cycle Proteins Participate in Spinal Motor Neuron Cell Death in ALS. Open Pathol. J. 2010, 4, 11–22. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Ward, C.L.; Boggio, K.J.; Johnson, B.N.; Boyd, J.B.; Douthwright, S.; Shaffer, S.A.; Landers, J.E.; Glicksman, M.A.; Bosco, D.A. A loss of FUS/TLS function leads to impaired cellular proliferation. Cell Death Dis. 2014, 5, e1572. [Google Scholar] [CrossRef]
- Endo, K.; Ishigaki, S.; Masamizu, Y.; Fujioka, Y.; Watakabe, A.; Yamamori, T.; Hatanaka, N.; Nambu, A.; Okado, H.; Katsuno, M.; et al. Silencing of FUS in the common marmoset (Callithrix jacchus) brain via stereotaxic injection of an adeno-associated virus encoding shRNA. Neurosci. Res. 2018, 130, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Brooke, G.N.; Culley, R.L.; Dart, D.A.; Mann, D.J.; Gaughan, L.; McCracken, S.R.; Robson, C.N.; Spencer-Dene, B.; Gamble, S.C.; Powell, S.M.; et al. FUS/TLS is a novel mediator of androgen-dependent cell-cycle progression and prostate cancer growth. Cancer Res. 2011, 71, 914–924. [Google Scholar] [CrossRef]
- Ke, H.; Zhao, L.; Feng, X.; Xu, H.; Zou, L.; Yang, Q.; Su, X.; Peng, L.; Jiao, B. NEAT1 is Required for Survival of Breast Cancer Cells Through FUS and miR-548. Gene Regul. Syst. Biol. 2016, 10 (Suppl 1), 11–17. [Google Scholar] [CrossRef]
- Wang, Z.; Lei, H.; Sun, Q. MicroRNA-141 and its associated gene FUS modulate proliferation, migration and cisplatin chemosensitivity in neuroblastoma cell lines. Oncol. Rep. 2016, 35, 2943–2951. [Google Scholar] [CrossRef]
- Soldati, C.; Bithell, A.; Johnston, C.; Wong, K.Y.; Stanton, L.W.; Buckley, N.J. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 2013, 124, 418–430. [Google Scholar] [CrossRef]
- Carucci, N.; Cacci, E.; Nisi, P.S.; Licursi, V.; Paul, Y.L.; Biagioni, S.; Negri, R.; Rugg-Gunn, P.J.; Lupo, G. Transcriptional response of Hoxb genes to retinoid signalling is regionally restricted along the neural tube rostrocaudal axis. R Soc. Open Sci. 2017, 4, 160913. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Liu, G.; Ozturk, A.; Hicks, G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.Y.; Ling, S.C. Elevated FUS levels by overriding its autoregulation produce gain-of-toxicity properties that disrupt protein and RNA homeostasis. Autophagy 2019, 15, 1665–1667. [Google Scholar] [CrossRef]
- Quinet, G.; Gonzalez-Santamarta, M.; Louche, C.; Rodriguez, M.S. Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy. Molecules 2020, 25, 2352. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, K.; Yamashita, T.; Shang, J.; Ohta, Y.; Nomura, E.; Morihara, R.; Omote, Y.; Takemoto, M.; Abe, K. Switching the Proteolytic System from the Ubiquitin-Proteasome System to Autophagy in the Spinal Cord of an Amyotrophic Lateral Sclerosis Mouse Model. Neuroscience 2021, 466, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Perez-Losada, J.; Pintado, B.; Gutierrez-Adan, A.; Flores, T.; Banares-Gonzalez, B.; del Campo, J.C.; Martin-Martin, J.F.; Battaner, E.; Sanchez-Garcia, I. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 2000, 19, 2413–2422. [Google Scholar] [CrossRef]
- Liu, S.; Yang, N.; Jiang, X.; Wang, J.; Dong, J.; Gao, Y. FUS-induced circular RNA ZNF609 promotes tumorigenesis and progression via sponging miR-142-3p in lung cancer. J. Cell Physiol. 2021, 236, 79–92. [Google Scholar] [CrossRef]
- Svetoni, F.; De Paola, E.; La Rosa, P.; Mercatelli, N.; Caporossi, D.; Sette, C.; Paronetto, M.P. Post-transcriptional regulation of FUS and EWS protein expression by miR-141 during neural differentiation. Hum. Mol. Genet. 2017, 26, 2732–2746. [Google Scholar] [CrossRef]
- Sinno, M.; Biagioni, S.; Ajmone-Cat, M.A.; Pafumi, I.; Caramanica, P.; Medda, V.; Tonti, G.; Minghetti, L.; Mannello, F.; Cacci, E. The matrix metalloproteinase inhibitor marimastat promotes neural progenitor cell differentiation into neurons by gelatinase-independent TIMP-2-dependent mechanisms. Stem. Cells Dev. 2013, 22, 345–358. [Google Scholar] [CrossRef]
- Soldati, C.; Caramanica, P.; Burney, M.J.; Toselli, C.; Bithell, A.; Augusti-Tocco, G.; Stanton, L.W.; Biagioni, S.; Buckley, N.J.; Cacci, E. RE1 silencing transcription factor/neuron-restrictive silencing factor regulates expansion of adult mouse subventricular zone-derived neural stem/progenitor cells in vitro. J. Neurosci. Res. 2015, 93, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, K.; McKay, R.D. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J. Neurosci. 1988, 8, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Dahlstrand, J.; Lardelli, M.; Lendahl, U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Brain Res. Dev. Brain Res. 1995, 84, 109–129. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein. Cell 2011, 2, 223–236. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Azuma, Y.; Yamaguchi, M. Cancer-related genes and ALS. Front. Biosci. 2019, 24, 1241–1258. [Google Scholar]
- Mirra, A.; Rossi, S.; Scaricamazza, S.; Di Salvio, M.; Salvatori, I.; Valle, C.; Rusmini, P.; Poletti, A.; Cestra, G.; Carri, M.T.; et al. Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice. Sci. Rep. 2017, 7, 2033. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Ishigaki, S.; Masuda, A.; Fujioka, Y.; Iguchi, Y.; Katsuno, M.; Shibata, A.; Urano, F.; Sobue, G.; Ohno, K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2012, 2, 529. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Alexiou, P.; Maragkakis, M.; Chang, A.; Mourelatos, Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA 2013, 19, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Orozco, D.; Tahirovic, S.; Rentzsch, K.; Schwenk, B.M.; Haass, C.; Edbauer, D. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 2012, 13, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; Wang, E.T.; Friedman, B.A.; Keagle, P.J.; Lowe, P.; Leclerc, A.L.; van den Berg, L.H.; Housman, D.E.; Veldink, J.H.; Landers, J.E. Characterization of FUS mutations in amyotrophic lateral sclerosis using RNA-Seq. PLoS ONE 2013, 8, e60788. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Chen, S.; Yu, Y.; Yan, B.; Haertlein, T.C.; Carrasco, M.A.; Tapia, J.C.; Zhai, B.; Das, R.; Lalancette-Hebert, M.; et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012, 2, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, R.; Suzuki, S.; Mashima, T.; Kondo, K.; Nagata, T.; Katahira, M.; Kurokawa, R. The binding specificity of Translocated in LipoSarcoma/FUsed in Sarcoma with lncRNA transcribed from the promoter region of cyclin D1. Cell Biosci. 2016, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Ozturk, A.; Hicks, G.G. FUS-regulated RNA metabolism and DNA damage repair: Implications for amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis. Rare Dis. 2014, 2, e29515. [Google Scholar] [CrossRef]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Raczynska, K.D.; Ruepp, M.D.; Brzek, A.; Reber, S.; Romeo, V.; Rindlisbacher, B.; Heller, M.; Szweykowska-Kulinska, Z.; Jarmolowski, A.; Schumperli, D. FUS/TLS contributes to replication-dependent histone gene expression by interaction with U7 snRNPs and histone-specific transcription factors. Nucleic. Acids Res. 2015, 43, 9711–9728. [Google Scholar] [CrossRef]
- Caputo, M.; La Bella, V.; Notaro, A. Differential subcellular expression of (P525L)FUS as a putative biomarker for ALS phenoconversion. Neurol. Genet. 2020, 6, e410. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Kundu, R.K.; Cheng, H.; Maegdefessel, L.; Toh, R.; Ahn, G.O.; Ali, Z.A.; Anderson, D.R.; et al. Loss of CDKN2B promotes p53-dependent smooth muscle cell apoptosis and aneurysm formation. Arterioscler. Thromb Vasc. Biol 2013, 33, e1–e10. [Google Scholar] [CrossRef] [PubMed]
- Nanda, V.; Downing, K.P.; Ye, J.; Xiao, S.; Kojima, Y.; Spin, J.M.; DiRenzo, D.; Nead, K.T.; Connolly, A.J.; Dandona, S.; et al. CDKN2B Regulates TGFbeta Signaling and Smooth Muscle Cell Investment of Hypoxic Neovessels. Circ. Res. 2016, 118, 230–240. [Google Scholar] [CrossRef]
- Zheng, K.; Li, H.; Zhu, Y.; Zhu, Q.; Qiu, M. MicroRNAs are essential for the developmental switch from neurogenesis to gliogenesis in the developing spinal cord. J. Neurosci. 2010, 30, 8245–8250. [Google Scholar] [CrossRef]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Lanson, N.A., Jr.; Pandey, U.B. FUS-related proteinopathies: Lessons from animal models. Brain Res. 2012, 1462, 44–60. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuoka, M. Overexpression of nuclear FUS induces neuronal cell death. Neuroscience 2015, 287, 113–124. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Licursi, V.; Anzellotti, S.; Favaro, J.; Sineri, S.; Carucci, N.; Cundari, E.; Fiore, M.; Guarguaglini, G.; Pippa, S.; Nisi, P.S.; et al. X-ray irradiated cultures of mouse cortical neural stem/progenitor cells recover cell viability and proliferation with dose-dependent kinetics. Sci. Rep. 2020, 10, 6562. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 1997, 21, A–3B. [Google Scholar] [CrossRef]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Krishan, A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol 1975, 66, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.; Darzynkiewicz, Z.; Staiano-Coico, L.; Melamed, M.R. Detection of 5-bromodeoxyuridine (BrdUrd) incorporation by monoclonal antibodies: Role of the DNA denaturation step. J. Histochem. Cytochem. 1985, 33, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Taupin, P. BrdU immunohistochemistry for studying adult neurogenesis: Paradigms, pitfalls, limitations, and validation. Brain Res. Rev. 2007, 53, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stronati, E.; Biagioni, S.; Fiore, M.; Giorgi, M.; Poiana, G.; Toselli, C.; Cacci, E. Wild-Type and Mutant FUS Expression Reduce Proliferation and Neuronal Differentiation Properties of Neural Stem Progenitor Cells. Int. J. Mol. Sci. 2021, 22, 7566. https://doi.org/10.3390/ijms22147566
Stronati E, Biagioni S, Fiore M, Giorgi M, Poiana G, Toselli C, Cacci E. Wild-Type and Mutant FUS Expression Reduce Proliferation and Neuronal Differentiation Properties of Neural Stem Progenitor Cells. International Journal of Molecular Sciences. 2021; 22(14):7566. https://doi.org/10.3390/ijms22147566
Chicago/Turabian StyleStronati, Eleonora, Stefano Biagioni, Mario Fiore, Mauro Giorgi, Giancarlo Poiana, Camilla Toselli, and Emanuele Cacci. 2021. "Wild-Type and Mutant FUS Expression Reduce Proliferation and Neuronal Differentiation Properties of Neural Stem Progenitor Cells" International Journal of Molecular Sciences 22, no. 14: 7566. https://doi.org/10.3390/ijms22147566
APA StyleStronati, E., Biagioni, S., Fiore, M., Giorgi, M., Poiana, G., Toselli, C., & Cacci, E. (2021). Wild-Type and Mutant FUS Expression Reduce Proliferation and Neuronal Differentiation Properties of Neural Stem Progenitor Cells. International Journal of Molecular Sciences, 22(14), 7566. https://doi.org/10.3390/ijms22147566