
N-3-Hydroxy Dodecanoyl-DL-homoserine Lactone (OH-dDHL) Triggers Apoptosis of Bone Marrow-Derived Macrophages through the ER- and Mitochondria-Mediated Pathways


Abstract
1. Introduction
2. Results
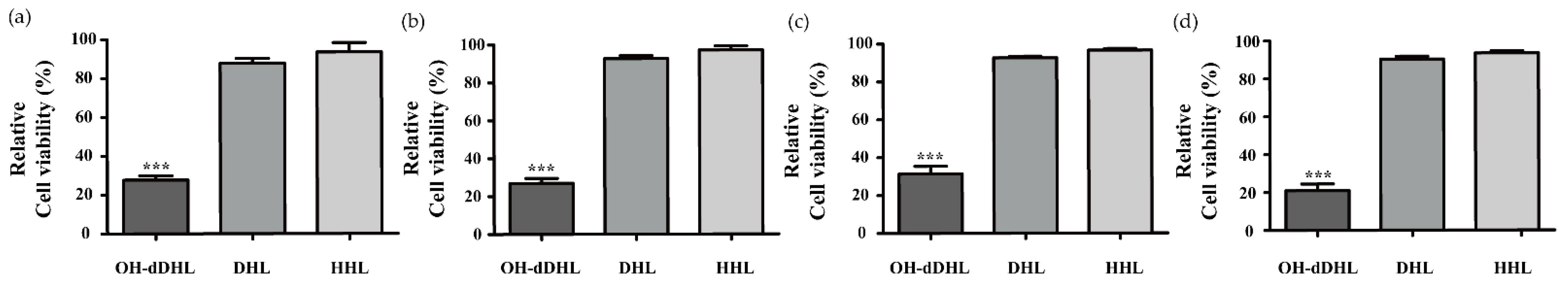
2.1. Effect of AHLs on Cell Viability of Host Cells
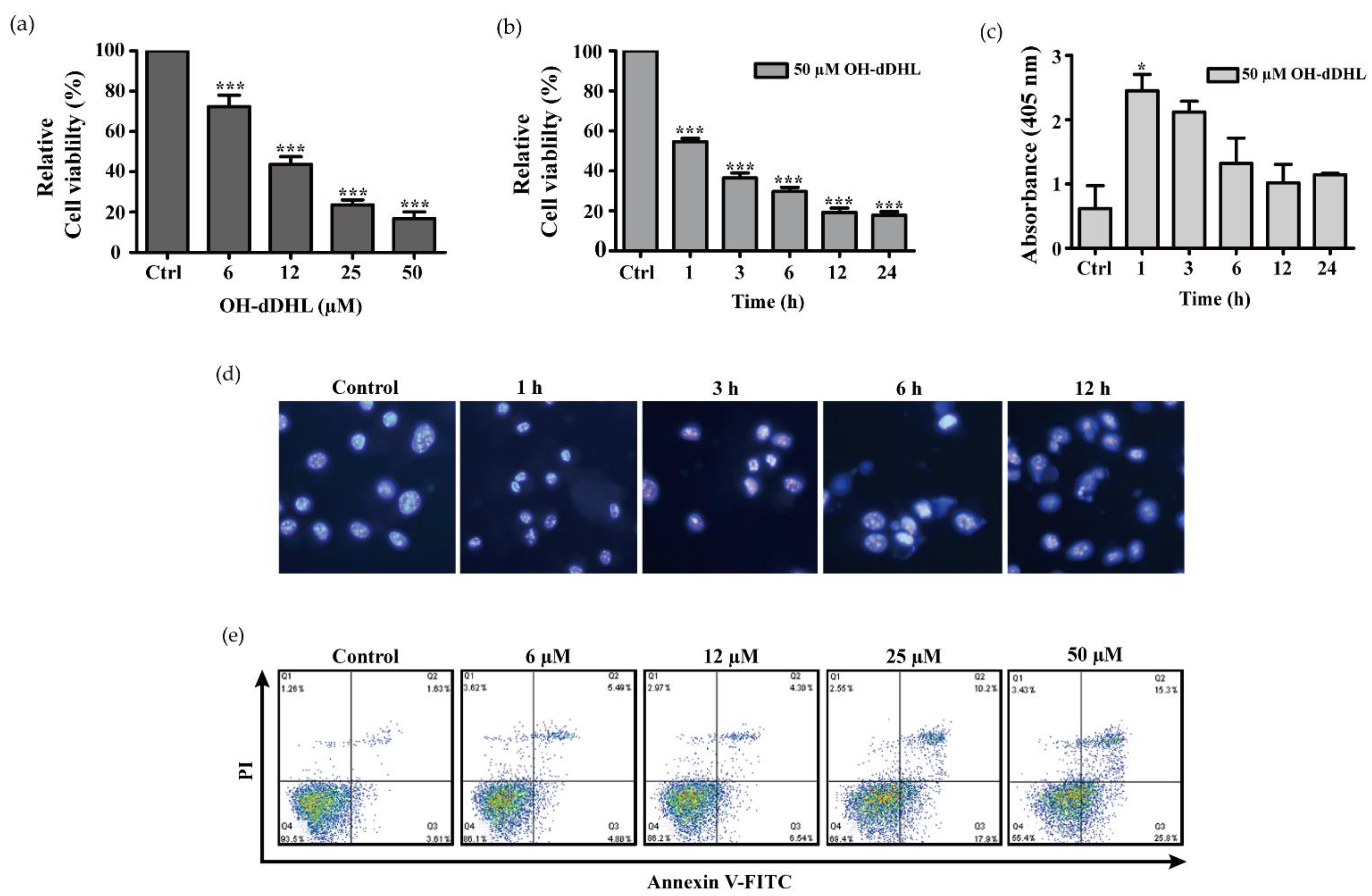
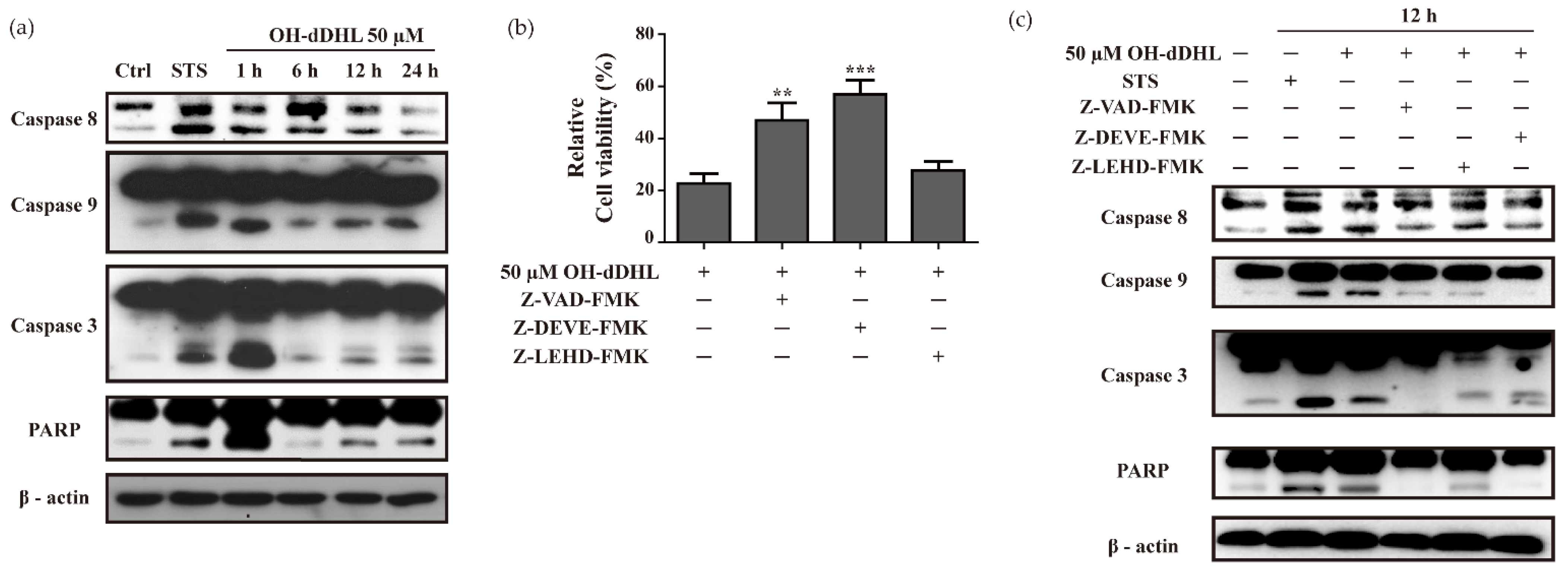
2.2. OH-dDHL Induced Cell Death by Caspase Activation
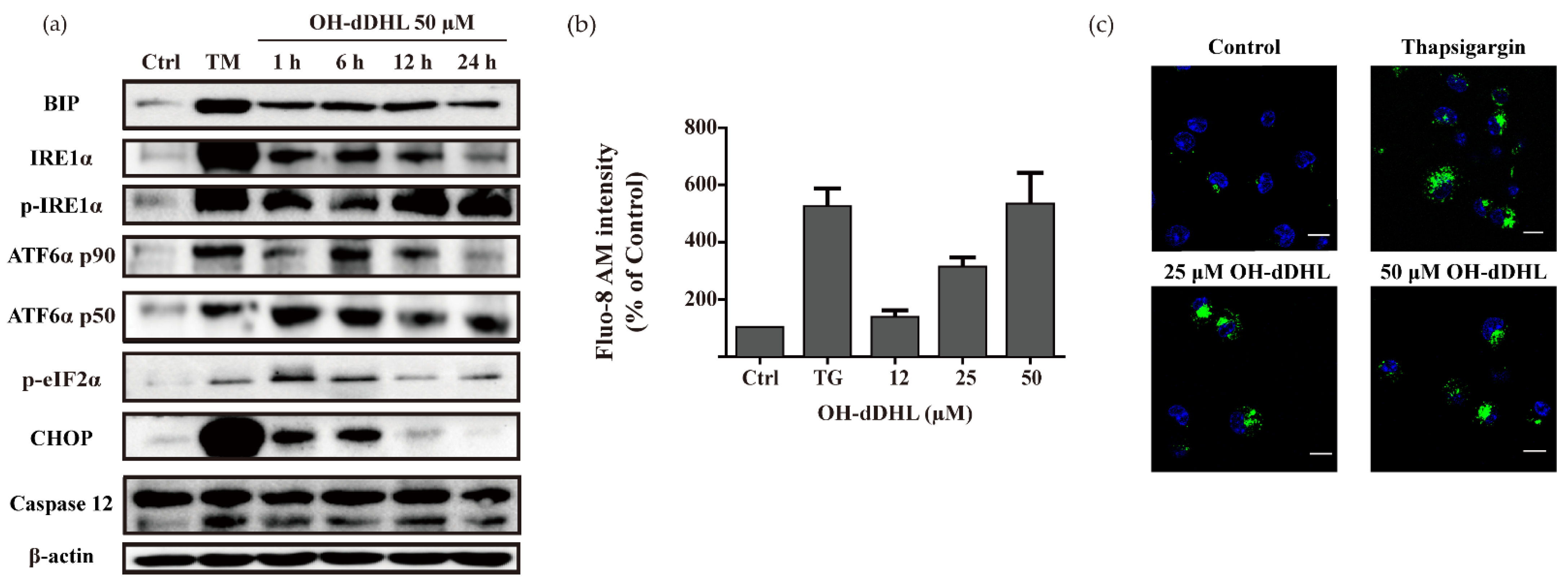
2.3. OH-dDHL Induced ER Stress Response in BMDM
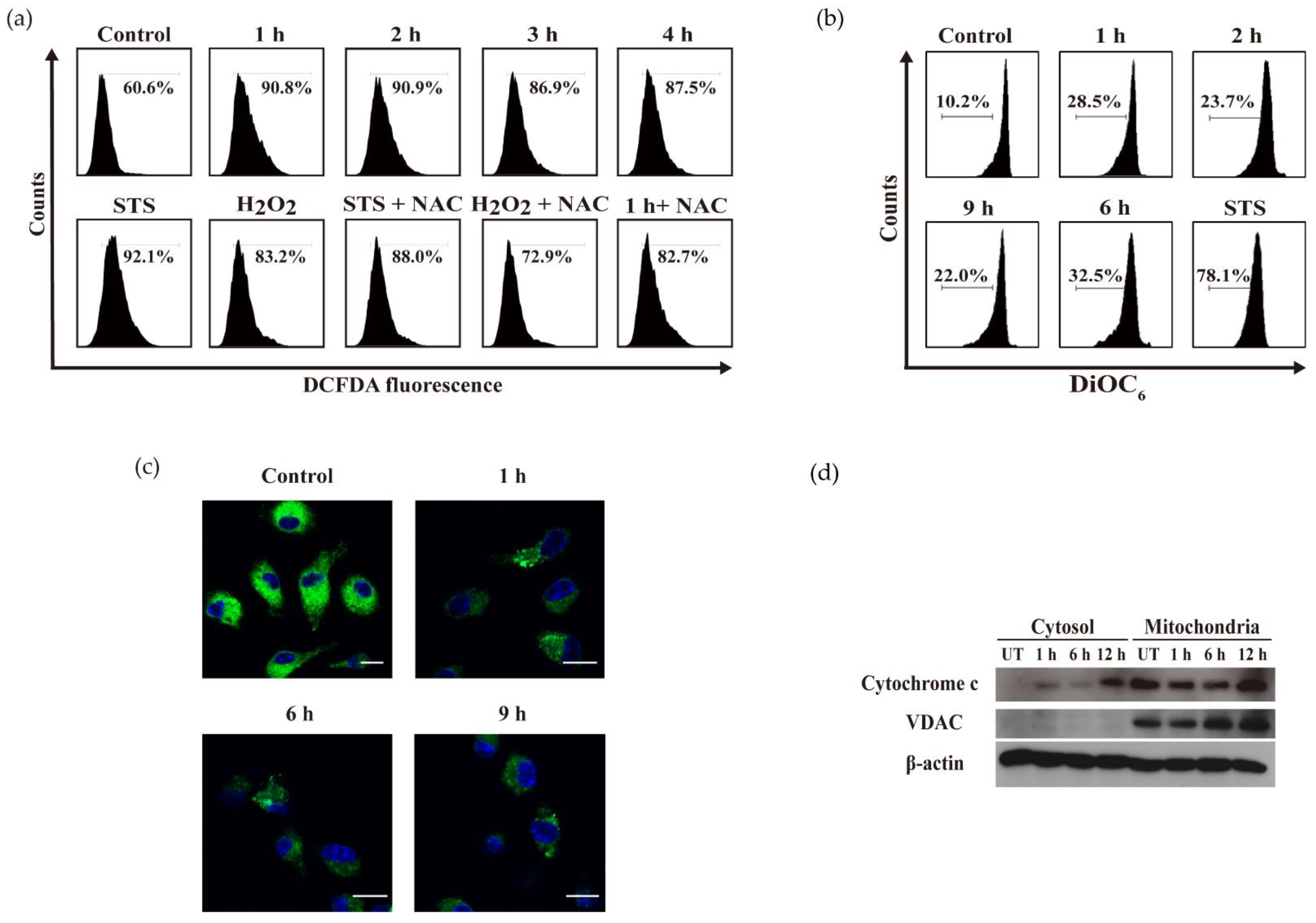
2.4. OH-dDHL Induced ROS Production and Mitochondrial Dysfunction in BMDM
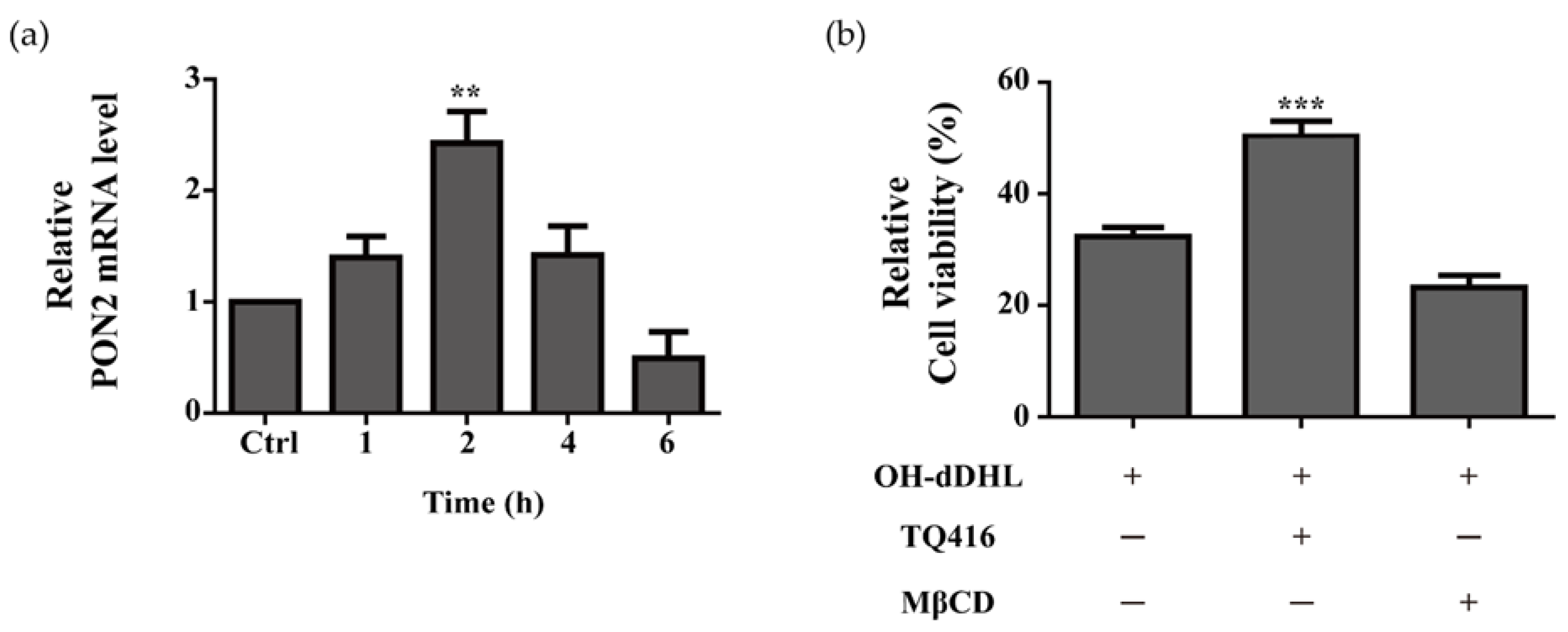
2.5. Effect of Lipid Raft and PON2 on Cell Viability of BMDMs
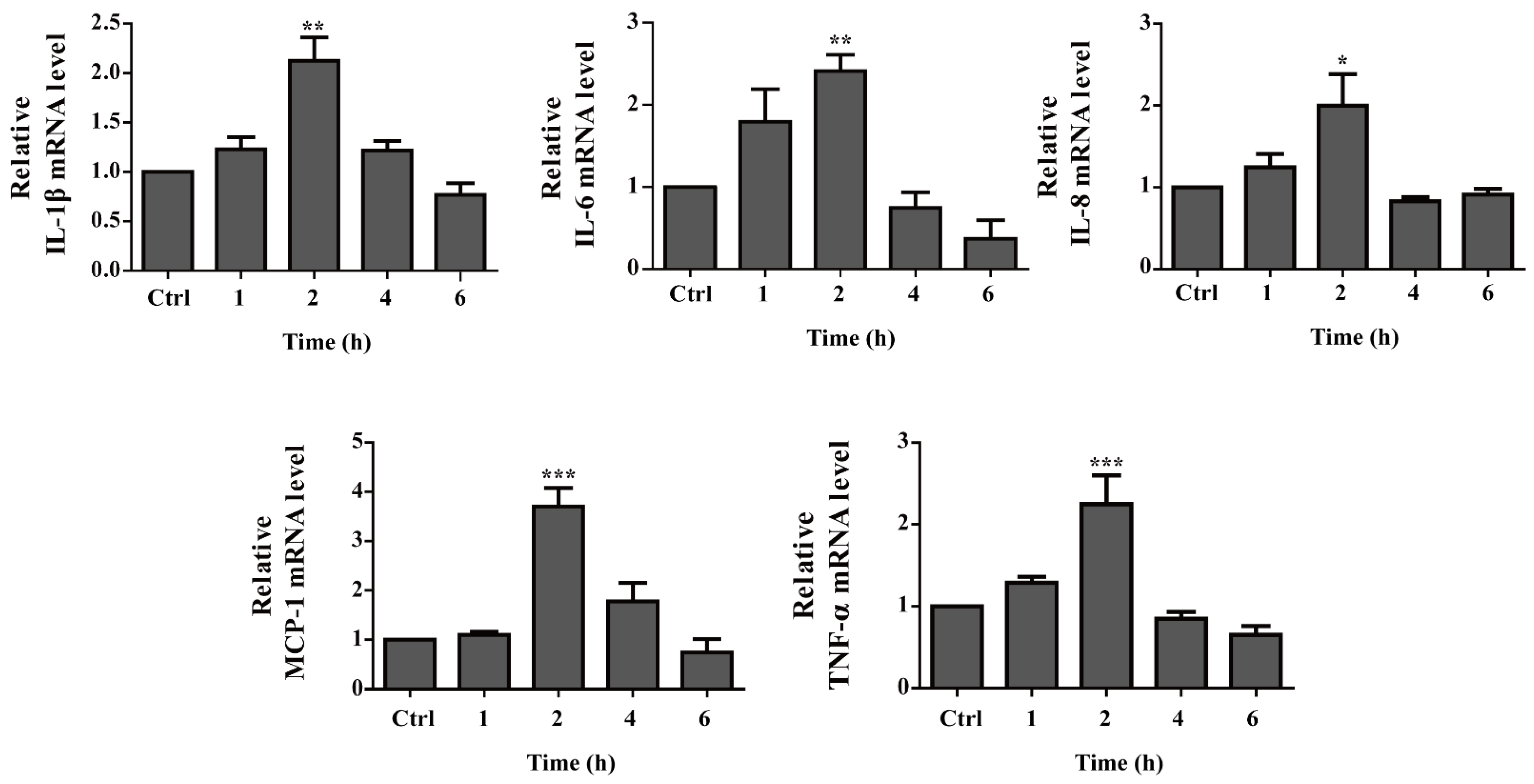
2.6. Pro-Inflammatory Cytokine Gene Expression Is up Regulated by OH-dDHL on BMDMs
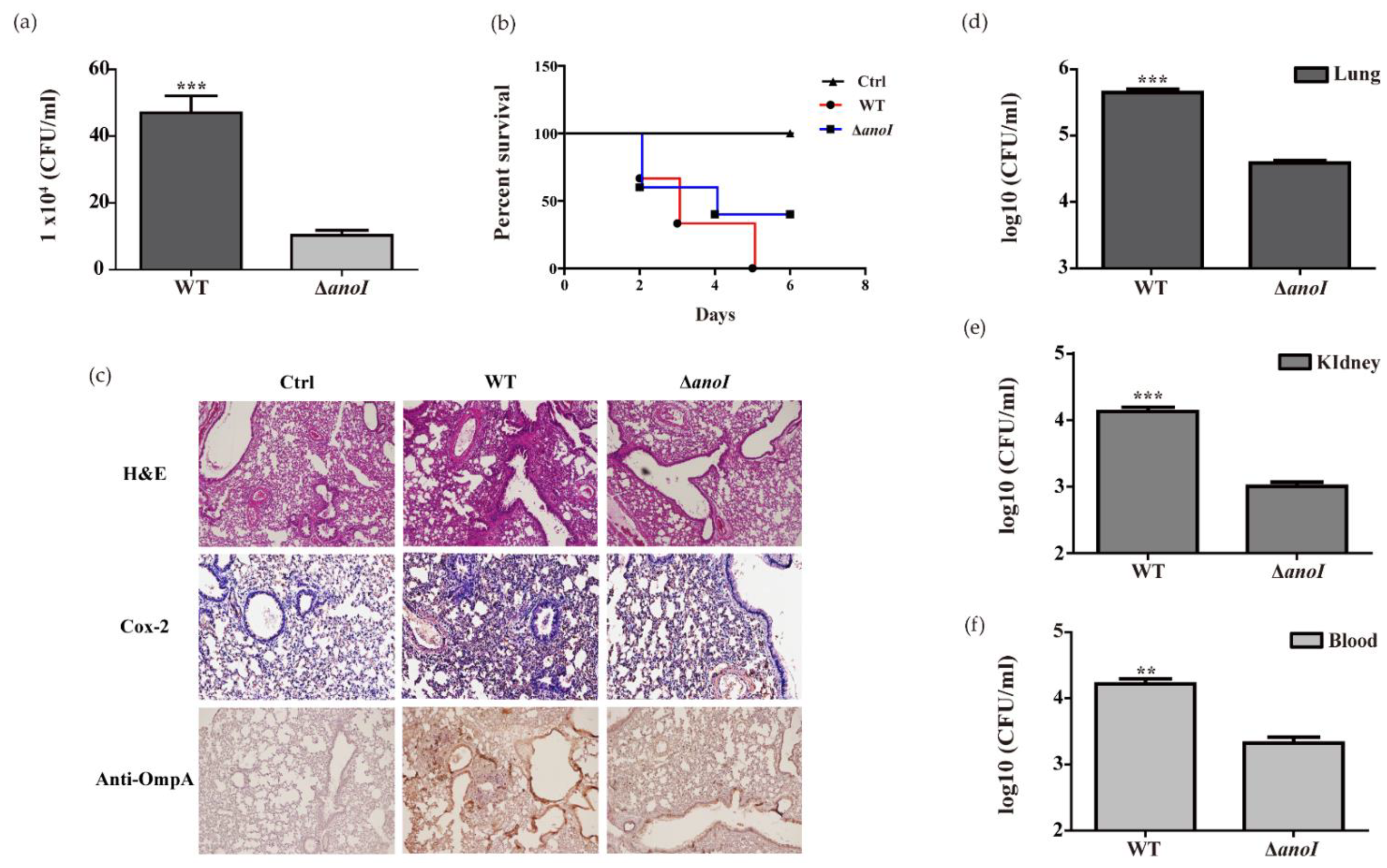
2.7. Lung Colonization and Virulence by A. nosocomialis
3. Discussion
4. Materials and Methods
4.1. The Bacterial Strains, Culture Conditions
4.2. Reagent and Antibody
4.3. Cell Culture
4.4. Determination of Cell Viability
4.5. Apoptosis Analysis
4.6. Immunofluorescence Microscopic Analysis
4.7. Measurement of DNA Fragmentation by Cell Death Detection ELISAplus Kit
4.8. Western Blot Analysis
4.9. Measurement of ROS Production
4.10. Measurement of Mitochondria Membrane Potential (ΔΨm) and Confocal Microscopic Analysis
4.11. Mitochondria Fractionation
4.12. Measurement of Intracellular Calcium (Ca2+)
4.13. RNA Extraction, Revers Transcription, and Real-Time Quantitative PCR (RT-qPCR)
4.14. Bacterial Invasion Assay
4.15. Animal Experiments
4.16. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peleg, A.Y.; De Breij, A.; Adams, M.D.; Cerqueira, G.; Mocali, S.; Galardini, M.; Nibbering, P.H.; Earl, A.; Ward, D.V.; Paterson, D.; et al. The Success of Acinetobacter Species; Genetic, Metabolic and Virulence Attributes. PLoS ONE 2012, 7, e46984. [Google Scholar] [CrossRef]
- Rocha, G.A.; Lima, D.F.; Rodrigues, E.R.; Leão, R.S.; Folescu, T.W.; Firmida, M.C.; Cohen, R.W.F.; Albano, R.M.; Marques, E.A. Species distribution, sequence types and antimicrobial resistance of Acinetobacter spp. from cystic fibrosis patients. Epidemiol. Infect. 2017, 146, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-T.; Kuo, S.-C.; Yang, S.-P.; Lin, Y.-T.; Chiang, D.-H.; Tseng, F.-C.; Chen, T.-L.; Fung, C.-P. Bacteremic nosocomial pneumonia caused by Acinetobacter baumannii and Acinetobacter nosocomialis: A single or two distinct clinical entities? Clin. Microbiol. Infect. 2013, 19, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-M.; Lee, Y.-T.; Kuo, S.-C.; Chen, T.-L.; Liu, C.-P.; Liu, C.-E. Comparison between bacteremia caused by Acinetobacter pittii and Acinetobacter nosocomialis. J. Microbiol. Immunol. Infect. 2017, 50, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Gowda, L.K.; Marie, M.A.M. Role of quorum-sensing molecules in infections caused by Gram-negative bacteria and host cell response. Rev. Med. Microbiol. 2014, 25, 66–70. [Google Scholar] [CrossRef]
- Bhargava, N.; Sharma, P.; Capalash, N. Quorum sensing in Acinetobacter: An emerging pathogen. Crit. Rev. Microbiol. 2010, 36, 349–360. [Google Scholar] [CrossRef]
- Lade, H.; Paul, D.; Kweon, J.H. N-Acyl Homoserine Lactone-Mediated Quorum Sensing with Special Reference to Use of Quorum Quenching Bacteria in Membrane Biofouling Control. BioMed Res. Int. 2014, 2014, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Muras, A.; Romero, M.; López, M.; Tomas, M.; Otero, A. Multiple Quorum Quenching Enzymes Are Active in the Nosocomial Pathogen Acinetobacter baumannii ATCC17978. Front. Cell. Infect. Microbiol. 2018, 8, 310. [Google Scholar] [CrossRef]
- Oh, M.H.; Choi, C.H. Role of LuxIR Homologue AnoIR in Acinetobacter nosocomialis and the Effect of Virstatin on the Expression of anoR Gene. J. Microbiol. Biotechnol. 2015, 25, 1390–1400. [Google Scholar] [CrossRef]
- Saipriya, K.; Swathi, C.; Ratnakar, K.; Sritharan, V.; Kamaraju, S.; Ch, S. Quorum-sensing system in Acinetobacter baumannii: A potential target for new drug development. J. Appl. Microbiol. 2019, 128, 15–27. [Google Scholar] [CrossRef]
- Erdönmez, D.; Rad, A.Y.; Aksöz, N. Quorum sensing molecules production by nosocomial and soil isolates Acinetobacter baumannii. Arch. Microbiol. 2017, 199, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Krzymińska, S.; Frąckowiak, H.; Kaznowski, A. Acinetobacter calcoaceticus–baumannii Complex Strains Induce Caspase-Dependent and Caspase-Independent Death of Human Epithelial Cells. Curr. Microbiol. 2012, 65, 319–329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shames, S.R.; Finlay, B.B. Breaking the Stereotype: Virulence Factor–Mediated Protection of Host Cells in Bacterial Pathogenesis. PLoS Pathog. 2010, 6, e1001057. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, J.H.; Park, M.; Park, K.S.; Bae, I.K.; Kim, Y.B.; Cha, C.-J.; Jeong, B.C.; Lee, S.H. Biology of Acinetobacter baumannii: Pathogenesis, Antibiotic Resistance Mechanisms, and Prospective Treatment Options. Front. Cell. Infect. Microbiol. 2017, 7, 55. [Google Scholar] [CrossRef]
- Morris, F.C.; Dexter, C.; Kostoulias, X.; Uddin, M.I.; Peleg, A.Y. The Mechanisms of Disease Caused by Acinetobacter baumannii. Front. Microbiol. 2019, 10, 1601. [Google Scholar] [CrossRef]
- Choi, C.H.; Hyun, S.H.; Lee, J.Y.; Lee, J.S.; Lee, Y.; Kim, S.A.; Chae, J.-P.; Yoo, S.M.; Lee, J.C. Acinetobacter baumannii outer membrane protein A targets the nucleus and induces cytotoxicity. Cell. Microbiol. 2007, 10, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.S.; Kwon, S.-O.; Moon, D.C.; Gurung, M.; Lee, J.H.; Kim, S.I.; Lee, J.C. Acinetobacter baumannii Secretes Cytotoxic Outer Membrane Protein A via Outer Membrane Vesicles. PLoS ONE 2011, 6, e17027. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Oh, M.H.; Jun, S.H.; Jeon, H.; Kim, S.I.; Kim, K.; Lee, Y.C.; Lee, J.C. Outer membrane Protein A plays a role in pathogenesis of Acinetobacter nosocomialis. Virulence 2016, 7, 413–426. [Google Scholar] [CrossRef]
- Savitskaya, M.A.; Onishchenko, G.E. Mechanisms of apoptosis. Biochemistry 2015, 80, 1393–1405. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, C.A.; Schiffner, F.; Henkel, M.; Waibel, M.; Stork, B.; Daubrawa, M.; Eberl, L.; Gregor, M.; Wesselborg, S. Effects of bacterial N-acyl homoserine lactones on human Jurkat T lymphocytes-OdDHL induces apoptosis via the mitochondrial pathway. Int. J. Med. Microbiol. 2009, 299, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Glucksam-Galnoy, Y.; Sananes, R.; Silberstein, N.; Krief, P.; Kravchenko, V.V.; Meijler, M.M.; Zor, T. The Bacterial Quorum-Sensing Signal Molecule N-3-Oxo-Dodecanoyl-l-Homoserine Lactone Reciprocally Modulates Pro- and Anti-Inflammatory Cytokines in Activated Macrophages. J. Immunol. 2013, 191, 337–344. [Google Scholar] [CrossRef]
- Stacy, D.M.; Welsh, M.; Rather, P.N.; Blackwell, H.E. Attenuation of Quorum Sensing in the Pathogen Acinetobacter baumannii Using Non-native N-Acyl Homoserine Lactones. ACS Chem. Biol. 2012, 7, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Kohler, T.; Bacchetta, M.; Saab, J.B.; Frieden, M.; van Delden, C.; Chanson, M. Airway Epithelial Cell Integrity Protects from Cytotoxicity of Pseudomonas aeruginosa Quorum-Sensing Signals. Am. J. Respir. Cell Mol. Biol. 2015, 53, 265–275. [Google Scholar] [CrossRef]
- Shih, D.M.; Lusis, A.J. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr. Opin. Lipidol. 2009, 20, 288–292. [Google Scholar] [CrossRef]
- Eum, S.Y.; Jaraki, D.; Bertrand, L.; András, I.E.; Toborek, M. Disruption of epithelial barrier by quorum-sensing N-3-(oxododecanoyl)-homoserine lactone is mediated by matrix metalloproteinases. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G992–G1001. [Google Scholar] [CrossRef]
- Tao, S.; Luo, Y.; He, B.; Liu, J.; Qian, X.; Ni, Y.; Zhao, R. Paraoxonase 2 modulates a proapoptotic function in LS174T cells in response to quorum sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone. Sci. Rep. 2016, 6, 28778. [Google Scholar] [CrossRef]
- Kobayashi, S.D.; Malachowa, N.; DeLeo, F.R. Neutrophils and Bacterial Immune Evasion. J. Innate Immun. 2018, 10, 432–441. [Google Scholar] [CrossRef]
- García-Patiño, M.G.; García-Contreras, R.; Licona-Limón, P. The Immune Response against Acinetobacter baumannii, an Emerging Pathogen in Nosocomial Infections. Front. Immunol. 2017, 8, 441. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; KuoLee, R.; Harris, G.; Van Rooijen, N.; Patel, G.B.; Chen, W. Role of Macrophages in Early Host Resistance to Respiratory Acinetobacter baumannii Infection. PLoS ONE 2012, 7, e40019. [Google Scholar] [CrossRef]
- Lancellotti, M.; Pereira, R.F.C.; Cury, G.G.; De Hollanda, L.M. Pathogenic and opportunistic respiratory bacteria-induced apoptosis. Braz. J. Infect. Dis. 2009, 13, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Labbe, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.H.; Deo, P.; Naderer, T. Macrophage cell death in microbial infections. Cell. Microbiol. 2016, 18, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Tateda, K.; Ishii, Y.; Horikawa, M.; Matsumoto, T.; Miyairi, S.; Pechere, J.C.; Standiford, T.J.; Ishiguro, M.; Yamaguchi, K. The Pseudomonas aeruginosa Autoinducer N -3-Oxododecanoyl Homoserine Lactone Accelerates Apoptosis in Macrophages and Neutrophils. Infect. Immun. 2003, 71, 5785–5793. [Google Scholar] [CrossRef]
- Taguchi, R.; Tanaka, S.; Joe, G.-H.; Maseda, H.; Nomura, N.; Ohnishi, J.; Ishizuka, S.; Shimizu, H.; Miyazaki, H. Mucin 3 is involved in intestinal epithelial cell apoptosis via N-(3-oxododecanoyl)-l-homoserine lactone-induced suppression of Akt phosphorylation. Am. J. Physiol. Cell Physiol. 2014, 307, C162–C168. [Google Scholar] [CrossRef]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef]
- Schwarzer, C.; Fu, Z.; Shuai, S.; Babbar, S.; Zhao, G.; Li, C.; Machen, T.E. Pseudomonas aeruginosa homoserine lactone triggers apoptosis and Bak/Bax-independent release of mitochondrial cytochrome C in fibroblasts. Cell. Microbiol. 2014, 16, 1094–1104. [Google Scholar] [CrossRef]
- Schwarzer, C.; Fu, Z.; Patanwala, M.; Hum, L.; Lopez-Guzman, M.; Illek, B.; Kong, W.; Lynch, S.V.; Machen, T.E. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cell. Microbiol. 2012, 14, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Olsson, S.; Sundler, R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol. Immunol. 2006, 43, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, C.; Fu, Z.; Morita, T.; Whitt, A.G.; Neely, A.M.; Liu, Y.; Machen, T.E.; Sun, B.; Xiao, Z.; Wang, R.; et al. Paraoxonase 2 Serves a Proapopotic Function in Mouse and Human Cells in Response to the Pseudomonas aeruginosa quorum-sensing Molecule N-(3-Oxododecanoyl)-homoserine Lactone. J. Biol. Chem. 2015, 290, 7247–7258. [Google Scholar] [CrossRef]
- Nho, J.S.; Jun, S.H.; Oh, M.H.; Park, T.I.; Choi, C.W.; Kim, S.I.; Choi, C.H.; Lee, J.C. Acinetobacter nosocomialis secretes outer membrane vesicles that induce epithelial cell death and host inflammatory responses. Microb. Pathog. 2015, 81, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Brossard, K.A.; Campagnari, A.A. The Acinetobacter baumannii Biofilm-Associated Protein Plays a Role in Adherence to Human Epithelial Cells. Infect. Immun. 2012, 80, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Zhu, J.; Feng, L.; Li, J.; Liu, X. Characterization of LuxI/LuxR and their regulation involved in biofilm formation and stress resistance in fish spoilers Pseudomonas fluorescens. Int. J. Food Microbiol. 2019, 297, 60–71. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Hou, H.M.; Zhang, G.L.; Wang, Y.F.; Hao, H.S. AHLs Regulate Biofilm Formation and Swimming Motility of Hafnia alvei H4. Front. Microbiol. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Lee, J.; Wu, J.; Deng, Y.; Wang, J.; Wang, C.; Wang, J.; Chang, C.; Dong, Y.; Williams, P.; Zhang, L.H. A cell-cell communication signal integrates quorum sensing and stress response. Nat. Chem. Biol. 2013, 9, 339–343. [Google Scholar] [CrossRef]
- Horke, S.; Xiao, J.; Schutz, E.M.; Kramer, G.L.; Wilgenbus, P.; Witte, I.; Selbach, M.; Teiber, J.F. Novel Paraoxonase 2-Dependent Mechanism Mediating the Biological Effects of the Pseudomonas aeruginosa Quorum-Sensing Molecule N-(3-Oxo-Dodecanoyl)-l-Homoserine Lactone. Infect. Immun. 2015, 83, 3369–3380. [Google Scholar] [CrossRef]
- Oh, M.H.; Lee, J.C.; Kim, J.; Choi, C.H.; Han, K. Simple Method for Markerless Gene Deletion in Multidrug-Resistant Acinetobacter baumannii. Appl. Environ. Microbiol. 2015, 81, 3357–3368. [Google Scholar] [CrossRef]
- Subhadra, B.; Surendran, S.; Kim, D.H.; Woo, K.; Oh, M.H.; Choi, C.H. The transcription factor NemR is an electrophile-sensing regulator important for the detoxification of reactive electrophiles in Acinetobacter nosocomialis. Res. Microbiol. 2019, 170, 123–130. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| TNF-α | AGGCACTCCCCCAAAAGATG | GTAGACAGAAGAGCGTGGTGG |
| IL-1β | CAACAAGAGCTTCAGGCAGG | TGCTCATGTCCTCATCCTGG |
| IL-6 | GTTGCCTTCTTGGGACTGAT | GGTATAGACAGGTCTGTTGG |
| IL-8 | GCTACGATGTCTGTGTATTC | TCACTTCCTTTCTGTTGCAG |
| MCP-1 | CCACTCACCTGCTGCTACTC | ACAGCTTCTTTGGGACACCT |
| PON2 | CTAATGGACAGAGGCTCTTC | TACACCGTTGTCACTGATGG |
| GAPDH | GTTCCAGTATGACTCCACTC | GTCTCGCTCCTGGAAGATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woo, K.; Kim, D.H.; Oh, M.H.; Park, H.S.; Choi, C.H. N-3-Hydroxy Dodecanoyl-DL-homoserine Lactone (OH-dDHL) Triggers Apoptosis of Bone Marrow-Derived Macrophages through the ER- and Mitochondria-Mediated Pathways. Int. J. Mol. Sci. 2021, 22, 7565. https://doi.org/10.3390/ijms22147565
Woo K, Kim DH, Oh MH, Park HS, Choi CH. N-3-Hydroxy Dodecanoyl-DL-homoserine Lactone (OH-dDHL) Triggers Apoptosis of Bone Marrow-Derived Macrophages through the ER- and Mitochondria-Mediated Pathways. International Journal of Molecular Sciences. 2021; 22(14):7565. https://doi.org/10.3390/ijms22147565
Chicago/Turabian StyleWoo, Kyungho, Dong Ho Kim, Man Hwan Oh, Ho Sung Park, and Chul Hee Choi. 2021. "N-3-Hydroxy Dodecanoyl-DL-homoserine Lactone (OH-dDHL) Triggers Apoptosis of Bone Marrow-Derived Macrophages through the ER- and Mitochondria-Mediated Pathways" International Journal of Molecular Sciences 22, no. 14: 7565. https://doi.org/10.3390/ijms22147565
APA StyleWoo, K., Kim, D. H., Oh, M. H., Park, H. S., & Choi, C. H. (2021). N-3-Hydroxy Dodecanoyl-DL-homoserine Lactone (OH-dDHL) Triggers Apoptosis of Bone Marrow-Derived Macrophages through the ER- and Mitochondria-Mediated Pathways. International Journal of Molecular Sciences, 22(14), 7565. https://doi.org/10.3390/ijms22147565