
Acute Lung Injury in Cholinergic-Deficient Mice Supports Anti-Inflammatory Role of α7 Nicotinic Acetylcholine Receptor

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of VAChT Deficiency and PNU 282987 Treatment on LPS-Induced Lung Inflammation
2.2. Effect of VAChT Deficiency, LPS-Instillation, and PNU 282987 Treatment on the Level of Lung Cholinergic Receptors
2.3. Effect of Vagotomy in LPS-Induced Lung Inflammation and Cholinergic Receptors in WT Mice
3. Discussion
4. Materials and Methods
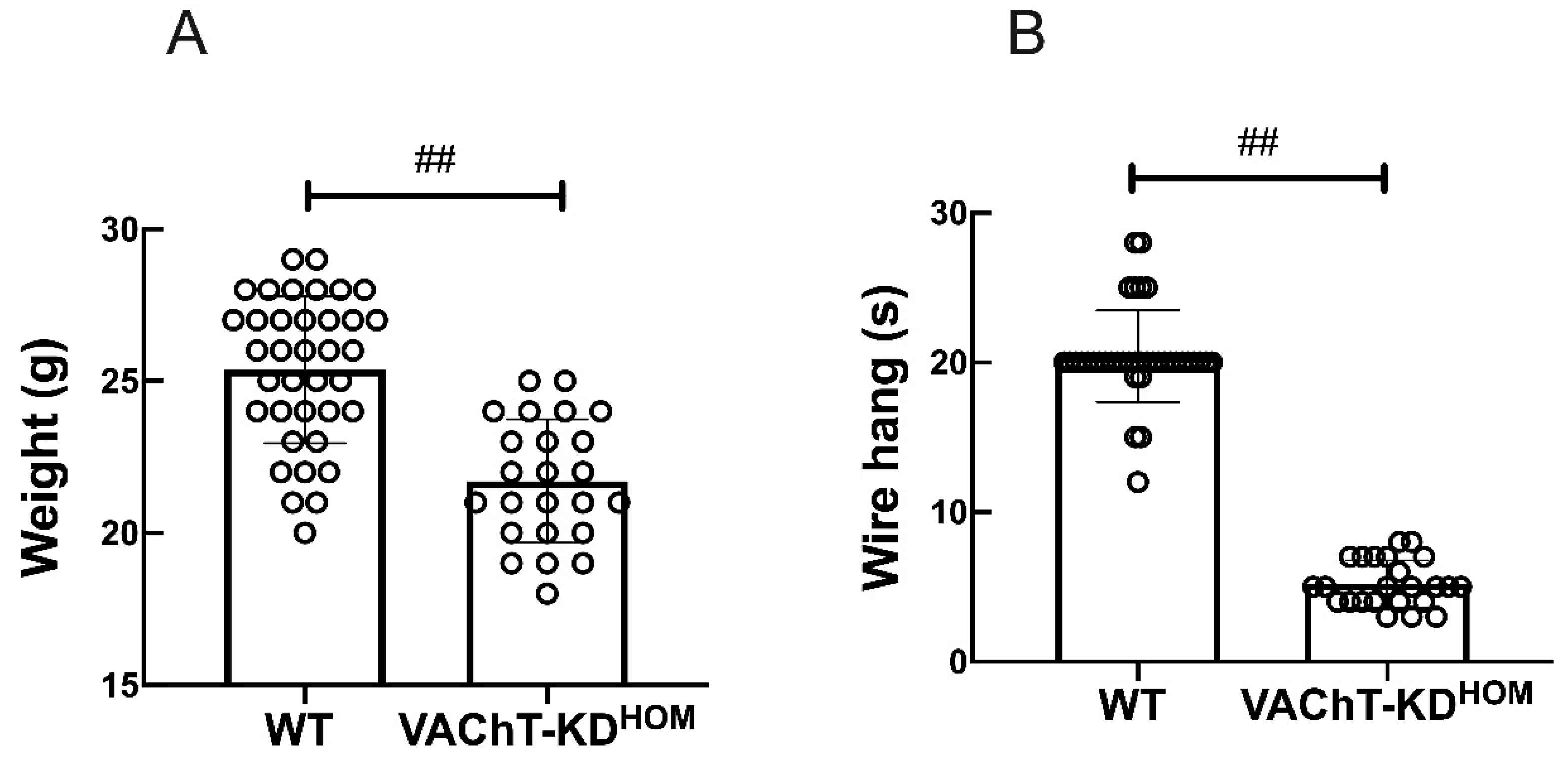
4.1. Weight and Wire-Hang Test
4.2. LPS Instillation Protocol
4.3. Treatment with PNU 282987
4.4. Vagotomy
4.5. Bronchoalveolar Lavage Fluid (BALF)
4.6. Immunoenzymatic Assay (ELISA) for Cytokine Detection in BALF
4.7. Evaluation of nAChR Nicotinic Receptors and Identification of Receptor Subunits in Lung Tissue
4.7.1. Preparation of Lung Membrane Fractions
4.7.2. Radioligand Binding Assay
4.8. Data Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferguson, N.D.; Fan, E.; Camporota, L.; Antonelli, M.; Anzueto, A.; Beale, R.; Brochard, L.; Brower, R.; Esteban, A.; Gattinoni, L.; et al. The Berlin definition of ARDS: An expanded rationale, justification, and supplementary material. Intensive Care Med. 2012, 38, 1573–1582. [Google Scholar] [CrossRef]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef]
- Máca, J.; Jor, O.; Holub, M.; Sklienka, P.; Burša, F.; Burda, M.; Janout, V.; Ševčík, P. Past and Present ARDS Mortality Rates: A Systematic Review. Respir. Care 2017, 62, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Salton, F.; Fabiano, F. Acute respiratory distress syndrome. Eur. Respir. Rev. 2017, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Rocco, P.R.; Dos Santos, C.; Pelosi, P. Lung parenchyma remodeling in acute respiratory distress syndrome. Minerva Anestesiol. 2009, 75, 730–740. [Google Scholar]
- Santos, F.B.; Nagato, L.K.; Boechem, N.M.; Negri, E.M.; Guimarães, A.; Capelozzi, V.L.; Faffe, D.S.; Zin, W.A.; Rocco, P.R. Time course of lung parenchyma remodeling in pulmonary and extrapulmonary acute lung injury. J. Appl. Physiol. 2006, 100, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; José, R.J.; Mercer, P.F.; Brealey, D.; Parekh, D.; Thickett, D.R.; O’Kane, C.; McAuley, D.F.; Chambers, R.C. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax 2017, 72, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Santamaria, L.B. Cholinesterase inhibitors improve survival in experimental sepsis: A new way to activate the cholinergic anti-inflammatory pathway. Crit. Care Med. 2008, 36, 622–623. [Google Scholar] [CrossRef]
- Metzen, J.; Bittinger, F.; Kirkpatrick, C.J.; Kilbinger, H.; Wessler, I. Proliferative effect of acetylcholine on rat trachea epithelial cells is mediated by nicotinic receptors and muscarinic receptors of the M1-subtype. Life Sci. 2003, 72, 2075–2080. [Google Scholar] [CrossRef]
- Coulson, F.R.; Fryer, A.D. Muscarinic acetylcholine receptors and airway diseases. Pharmacol. Ther. 2003, 98, 59–69. [Google Scholar] [CrossRef]
- Fryer, A.D.; Jacoby, D.B. Muscarinic receptors and control of airway smooth muscle. Am. J. Respir. Crit. Care Med. 1998, 158, S154–S160. [Google Scholar] [CrossRef] [PubMed]
- Giebelen, I.A.; van Westerloo, D.J.; LaRosa, G.J.; de Vos, A.F.; van der Poll, T. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock 2007, 27, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Yang, K.; Xu, G.N.; Zhu, L.; Hou, L.N.; Zhang, W.H.; Chen, H.Z.; Cui, Y.Y. Role of M3 mAChR in in vivo and in vitro models of LPS-induced inflammatory response. Int. Immunopharmacol. 2012, 14, 320–327. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Lv, Z.; Meng, L.; Xu, J.; Yuan, S.; Fu, Z. Activation of Alpha-7 Nicotinic Acetylcholine Receptors (α7nAchR) Promotes the Protective Autophagy in LPS-Induced Acute Lung Injury (ALI) In Vitro and In Vivo. Inflammation 2019, 42, 2236–2245. [Google Scholar] [CrossRef]
- Pinheiro, N.M.; Santana, F.P.; Almeida, R.R.; Guerreiro, M.; Martins, M.A.; Caperuto, L.C.; Camara, N.O.; Wensing, L.A.; Prado, V.F.; Tiberio, I.F.; et al. Acute lung injury is reduced by the alpha7nAChR agonist PNU-282987 through changes in the macrophage profile. FASEB J. 2017, 31, 320–332. [Google Scholar] [CrossRef]
- Pinheiro, N.M.; Miranda, C.J.; Perini, A.; Camara, N.O.; Costa, S.K.; Alonso-Vale, M.I.; Caperuto, L.C.; Tiberio, I.F.; Prado, M.A.; Martins, M.A.; et al. Pulmonary inflammation is regulated by the levels of the vesicular acetylcholine transporter. PLoS ONE 2015, 10, e0120441. [Google Scholar] [CrossRef]
- Bagdas, D.; Gurun, M.S.; Flood, P.; Papke, R.L.; Damaj, M.I. New Insights on Neuronal Nicotinic Acetylcholine Receptors as Targets for Pain and Inflammation: A Focus on alpha7 nAChRs. Curr. Neuropharmacol. 2018, 16, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Hone, A.J.; McIntosh, J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018, 592, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt-Mernak, M.I.; Pinheiro, N.M.; Santana, F.P.; Guerreiro, M.P.; Saraiva-Romanholo, B.M.; Grecco, S.S.; Caperuto, L.C.; Felizardo, R.J.; Câmara, N.O.; Tibério, I.F.; et al. Prophylactic and therapeutic treatment with the flavonone sakuranetin ameliorates LPS-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L217–L230. [Google Scholar] [CrossRef]
- Lima Rde, F.; Prado, V.F.; Prado, M.A.; Kushmerick, C. Quantal release of acetylcholine in mice with reduced levels of the vesicular acetylcholine transporter. J. Neurochem. 2010, 113, 943–951. [Google Scholar] [CrossRef]
- Prado, M.A.; Reis, R.A.; Prado, V.F.; de Mello, M.C.; Gomez, M.V.; de Mello, F.G. Regulation of acetylcholine synthesis and storage. Neurochem. Int. 2002, 41, 291–299. [Google Scholar] [CrossRef]
- Prado, V.F.; Martins-Silva, C.; de Castro, B.M.; Lima, R.F.; Barros, D.M.; Amaral, E.; Ramsey, A.J.; Sotnikova, T.D.; Ramirez, M.R.; Kim, H.G.; et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 2006, 51, 601–612. [Google Scholar] [CrossRef]
- Pinheiro, N.M.; Miranda, C.; Santana, F.R.; Bittencourt-Mernak, M.; Arantes-Costa, F.M.; Olivo, C.; Perini, A.; Festa, S.; Caperuto, L.C.; Tibério, I.; et al. Effects of VAChT reduction and α7nAChR stimulation by PNU-282987 in lung inflammation in a model of chronic allergic airway inflammation. Eur. J. Pharmacol. 2020, 882, 173239. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A.L.; Cortes-Burgos, L.A.; Cook, K.K.; Dinh, D.M.; Groppi, V.E.; Hajos, M.; Higdon, N.R.; Hoffmann, W.E.; Hurst, R.S.; Myers, J.K.; et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J. Med. Chem. 2005, 48, 905–908. [Google Scholar] [CrossRef]
- Hajós, M.; Hurst, R.S.; Hoffmann, W.E.; Krause, M.; Wall, T.M.; Higdon, N.R.; Groppi, V.E. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo [2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J. Pharmacol. Exp. Ther. 2005, 312, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Dobelis, P.; Madl, J.E.; Pfister, J.A.; Manners, G.D.; Walrond, J.P. Effects of Delphinium alkaloids on neuromuscular transmission. J. Pharmacol. Exp. Ther. 1999, 291, 538–546. [Google Scholar] [PubMed]
- Escubedo, E.; Chipana, C.; Pérez-Sánchez, M.; Camarasa, J.; Pubill, D. Methyllycaconitine prevents methamphetamine-induced effects in mouse striatum: Involvement of alpha7 nicotinic receptors. J. Pharmacol. Exp. Ther. 2005, 315, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Cockcroft, V.B.; Lunt, G.G.; Smillie, F.S.; Wonnacott, S. Methyllycaconitine: A selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett. 1990, 270, 45–48. [Google Scholar] [CrossRef]
- Papke, R.L.; Heinemann, S.F. Partial agonist properties of cytisine on neuronal nicotinic receptors containing the beta 2 subunit. Mol. Pharmacol. 1994, 45, 142–149. [Google Scholar] [PubMed]
- Happe, H.K.; Peters, J.L.; Bergman, D.A.; Murrin, L.C. Localization of nicotinic cholinergic receptors in rat brain: Autoradiographic studies with [3H]cytisine. Neuroscience 1994, 62, 929–944. [Google Scholar] [CrossRef]
- Gilbert, R.F.; Hanley, M.R.; Iversen, L.L. [3H]-Quinuclidinyl benzilate binding to muscarinic receptors in rat brain: Comparison of results from intact brain slices and homogenates. Br. J. Pharmacol. 1979, 65, 451–456. [Google Scholar] [CrossRef]
- Jayakar, S.S.; Ang, G.; Chiara, D.C.; Hamouda, A.K. Photoaffinity Labeling of Pentameric Ligand-Gated Ion Channels: A Proteomic Approach to Identify Allosteric Modulator Binding Sites. Methods Mol. Biol. 2017, 1598, 157–197. [Google Scholar] [PubMed]
- Rimele, T.J.; Rogers, W.A.; Gaginella, T.S. Characterization of muscarinic cholinergic receptors in the lower esophageal sphincter of the cat: Binding of [3H]quinuclidinyl benzilate. Gastroenterology 1979, 77, 1225–1234. [Google Scholar] [CrossRef]
- Tsukahara, T.; Usui, H.; Taniguchi, T.; Shimohama, S.; Fujiwara, M.; Handa, H. Characterization of muscarinic cholinergic receptors in human and dog cerebral arteries. Stroke 1986, 17, 300–305. [Google Scholar] [CrossRef]
- Kox, M.; Vaneker, M.; van der Hoeven, J.G.; Scheffer, G.J.; Hoedemaekers, C.W.; Pickkers, P. Effects of vagus nerve stimulation and vagotomy on systemic and pulmonary inflammation in a two-hit model in rats. PLoS ONE 2012, 7, e34431. [Google Scholar] [CrossRef]
- De Castro, B.M.; De Jaeger, X.; Martins-Silva, C.; Lima, R.D.; Amaral, E.; Menezes, C.; Lima, P.; Neves, C.M.; Pires, R.G.; Gould, T.W.; et al. The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol. Cell. Biol. 2009, 29, 5238–5250. [Google Scholar] [CrossRef] [PubMed]
- Lips, K.S.; Luhrmann, A.; Tschernig, T.; Stoeger, T.; Alessandrini, F.; Grau, V.; Haberberger, R.V.; Koepsell, H.; Pabst, R.; Kummer, W. Down-regulation of the non-neuronal acetylcholine synthesis and release machinery in acute allergic airway inflammation of rat and mouse. Life Sci. 2007, 80, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Bittinger, F.; Kamin, W.; Zepp, F.; Meyer, E.; Schad, A.; Kirkpatrick, C.J. Dysfunction of the non-neuronal cholinergic system in the airways and blood cells of patients with cystic fibrosis. Life Sci. 2007, 80, 2253–2258. [Google Scholar] [CrossRef]
- Santana, F.P.R.; Pinheiro, N.M.; Bittencourt-Mernak, M.I.; Perini, A.; Yoshizaki, K.; Macchione, M.; Saldiva, P.H.N.; Martins, M.A.; Tibério, I.; Prado, M.A.M.; et al. Vesicular acetylcholine transport deficiency potentiates some inflammatory responses induced by diesel exhaust particles. Ecotoxicol. Environ. Saf. 2019, 167, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Santana, F.P.R.; Ricardo-da-Silva, F.Y.; Fantozzi, E.T.; Pinheiro, N.M.; Tibério, I.; Moreira, L.F.P.; Prado, M.A.M.; Prado, V.F.; Tavares-de-Lima, W.; Prado, C.M.; et al. Lung Edema and Mortality Induced by Intestinal Ischemia and Reperfusion Is Regulated by VAChT Levels in Female Mice. Inflammation 2021, 1–12. [Google Scholar]
- Sun, P.; Li, L.; Zhao, C.; Pan, M.; Qian, Z.; Su, X. Deficiency of α7 nicotinic acetylcholine receptor attenuates bleomycin-induced lung fibrosis in mice. Mol. Med. 2017, 23, 34–39. [Google Scholar] [CrossRef]
- Kistemaker, L.E.; Bos, I.S.; Hylkema, M.N.; Nawijn, M.C.; Hiemstra, P.S.; Wess, J.; Meurs, H.; Kerstjens, H.A.; Gosens, R. Muscarinic receptor subtype-specific effects on cigarette smoke-induced inflammation in mice. Eur. Respir. J. 2013, 42, 1677–1688. [Google Scholar] [CrossRef]
- Verbout, N.G.; Lorton, J.K.; Jacoby, D.B.; Fryer, A.D. Atropine pretreatment enhances airway hyperreactivity in antigen-challenged guinea pigs through an eosinophil-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1126–L1135. [Google Scholar] [CrossRef]
- Gori, S.; Alcain, J.; Vanzulli, S.; Moreno Ayala, M.A.; Candolfi, M.; Jancic, C.; Geffner, J.; Vermeulen, M.; Salamone, G. Acetylcholine-treated murine dendritic cells promote inflammatory lung injury. PLoS ONE 2019, 14, e0212911. [Google Scholar] [CrossRef] [PubMed]
- Español, A.J.; Maddaleno, M.O.; Lombardi, M.G.; Cella, M.; Martínez Pulido, P.; Sales, M.E. Treatment with LPS plus INF-γ induces the expression and function of muscarinic acetylcholine receptors, modulating NIH3T3 cell proliferation: Participation of NOS and COX. Br. J. Pharmacol. 2014, 171, 5154–5167. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.; Liu, L.; Geng, B. Muscarinic M1 and M2 receptor subtypes play opposite roles in LPS-induced septic shock. Pharmacol. Rep. 2019, 71, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Qs, L.; Ap, L.; Qw, H.; Qh, Y.; Yl, W.; Zz, Z. Roles of M(3) receptor in the effect of penehyclidine hydrochloride upregulated beta-arrestin-1 expression in LPS-stimulated HPMVEC. J. Recept. Signal. Transduct. Res. 2019, 39, 39–44. [Google Scholar] [CrossRef]
- Lara, A.; Damasceno, D.D.; Pires, R.; Gros, R.; Gomes, E.R.; Gavioli, M.; Lima, R.F.; Guimarães, D.; Lima, P.; Bueno, C.R., Jr.; et al. Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol. Cell. Biol. 2010, 30, 1746–1756. [Google Scholar] [CrossRef]
- Van Westerloo, D.J.; Giebelen, I.A.; Florquin, S.; Bruno, M.J.; Larosa, G.J.; Ulloa, L.; Tracey, K.J.; van der Poll, T. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006, 130, 1822–1830. [Google Scholar] [CrossRef]
- Van Westerloo, D.J.; Giebelen, I.A.; Florquin, S.; Daalhuisen, J.; Bruno, M.J.; de Vos, A.F.; Tracey, K.J.; van der Poll, T. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J. Infect. Dis. 2005, 191, 2138–2148. [Google Scholar] [CrossRef]
- Schelegle, E.S.; Walby, W.F. Vagal afferents contribute to exacerbated airway responses following ozone and allergen challenge. Respir. Physiol. Neurobiol. 2012, 181, 277–285. [Google Scholar] [CrossRef][Green Version]
- Hofer, S.; Eisenbach, C.; Lukic, I.K.; Schneider, L.; Bode, K.; Brueckmann, M.; Mautner, S.; Wente, M.N.; Encke, J.; Werner, J.; et al. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit. Care Med. 2008, 36, 404–408. [Google Scholar] [CrossRef]
- Jeremias, I.C.; Victorino, V.J.; Barbeiro, H.V.; Kubo, S.A.; Prado, C.M.; Lima, T.M.; Soriano, F.G. The Role of Acetylcholine in the Inflammatory Response in Animals Surviving Sepsis Induced by Cecal Ligation and Puncture. Mol. Neurobiol. 2016, 53, 6635–6643. [Google Scholar] [CrossRef]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 2013, 27, 5072–5082. [Google Scholar] [CrossRef] [PubMed]
- Lind, R.J.; Hardick, D.J.; Blagbrough, I.S.; Potter, B.V.; Wolstenholme, A.J.; Davies, A.R.; Clough, M.S.; Earley, F.G.; Reynolds, S.E.; Wonnacott, S. [3H]-Methyllycaconitine: A high affinity radioligand that labels invertebrate nicotinic acetylcholine receptors. Insect Biochem. Mol. Biol. 2001, 31, 533–542. [Google Scholar] [CrossRef]
- Zhang, J.; Steinbach, J.H. Cytisine binds with similar affinity to nicotinic alpha4beta2 receptors on the cell surface and in homogenates. Brain Res. 2003, 959, 98–102. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinheiro, N.M.; Banzato, R.; Tibério, I.; Prado, M.A.M.; Prado, V.F.; Hamouda, A.K.; Prado, C.M. Acute Lung Injury in Cholinergic-Deficient Mice Supports Anti-Inflammatory Role of α7 Nicotinic Acetylcholine Receptor. Int. J. Mol. Sci. 2021, 22, 7552. https://doi.org/10.3390/ijms22147552
Pinheiro NM, Banzato R, Tibério I, Prado MAM, Prado VF, Hamouda AK, Prado CM. Acute Lung Injury in Cholinergic-Deficient Mice Supports Anti-Inflammatory Role of α7 Nicotinic Acetylcholine Receptor. International Journal of Molecular Sciences. 2021; 22(14):7552. https://doi.org/10.3390/ijms22147552
Chicago/Turabian StylePinheiro, Nathalia M., Rosana Banzato, Iolanda Tibério, Marco A. M. Prado, Vânia F. Prado, Ayman K. Hamouda, and Carla M. Prado. 2021. "Acute Lung Injury in Cholinergic-Deficient Mice Supports Anti-Inflammatory Role of α7 Nicotinic Acetylcholine Receptor" International Journal of Molecular Sciences 22, no. 14: 7552. https://doi.org/10.3390/ijms22147552
APA StylePinheiro, N. M., Banzato, R., Tibério, I., Prado, M. A. M., Prado, V. F., Hamouda, A. K., & Prado, C. M. (2021). Acute Lung Injury in Cholinergic-Deficient Mice Supports Anti-Inflammatory Role of α7 Nicotinic Acetylcholine Receptor. International Journal of Molecular Sciences, 22(14), 7552. https://doi.org/10.3390/ijms22147552