
Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype


Abstract
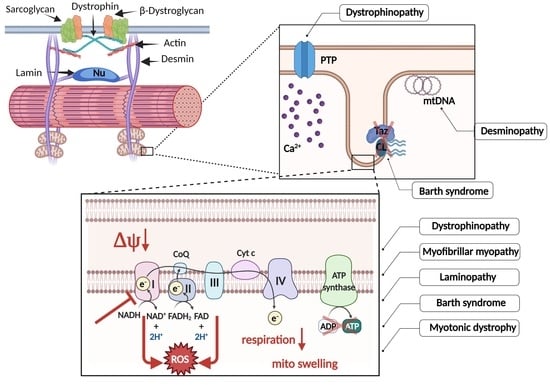
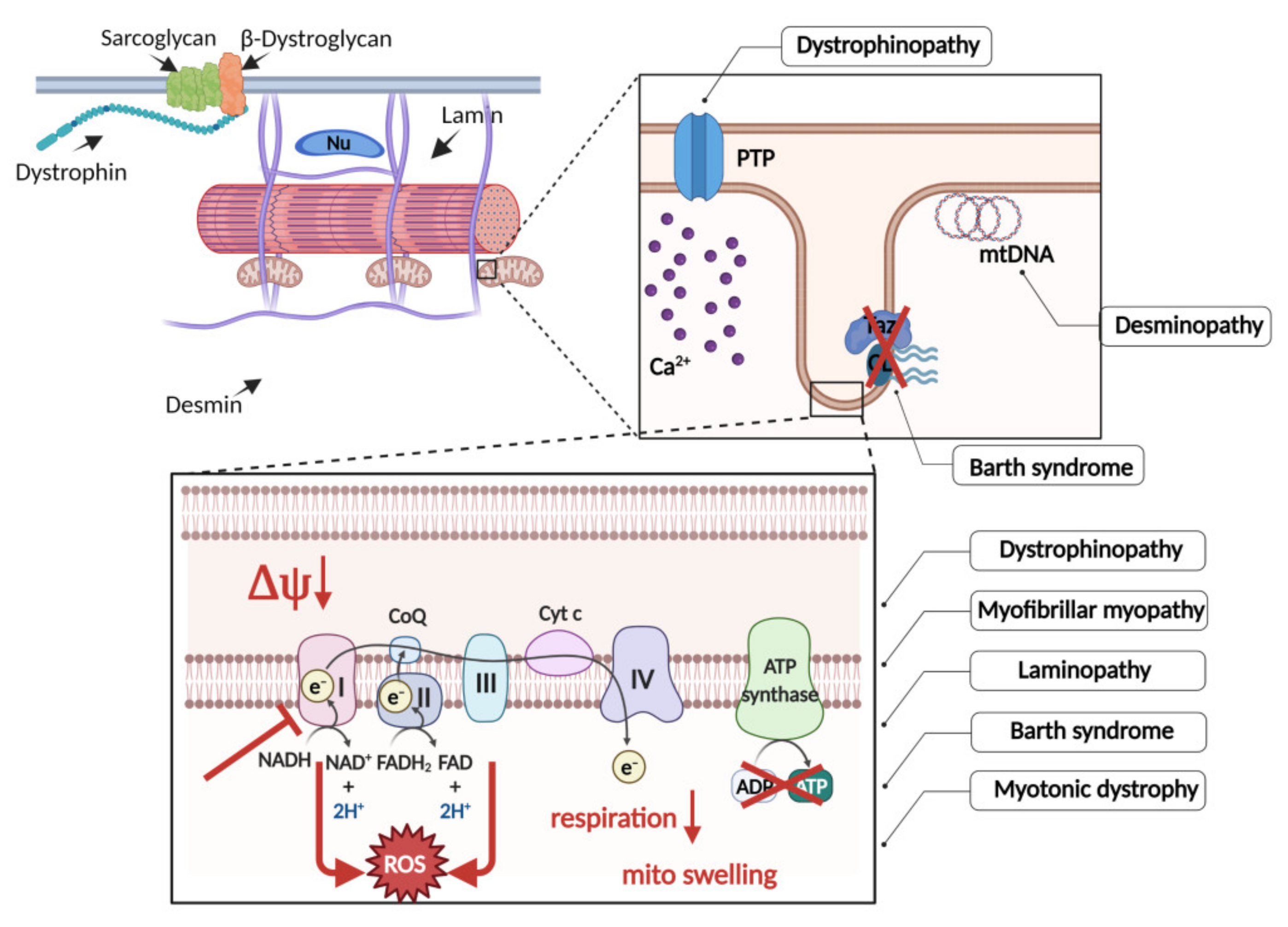
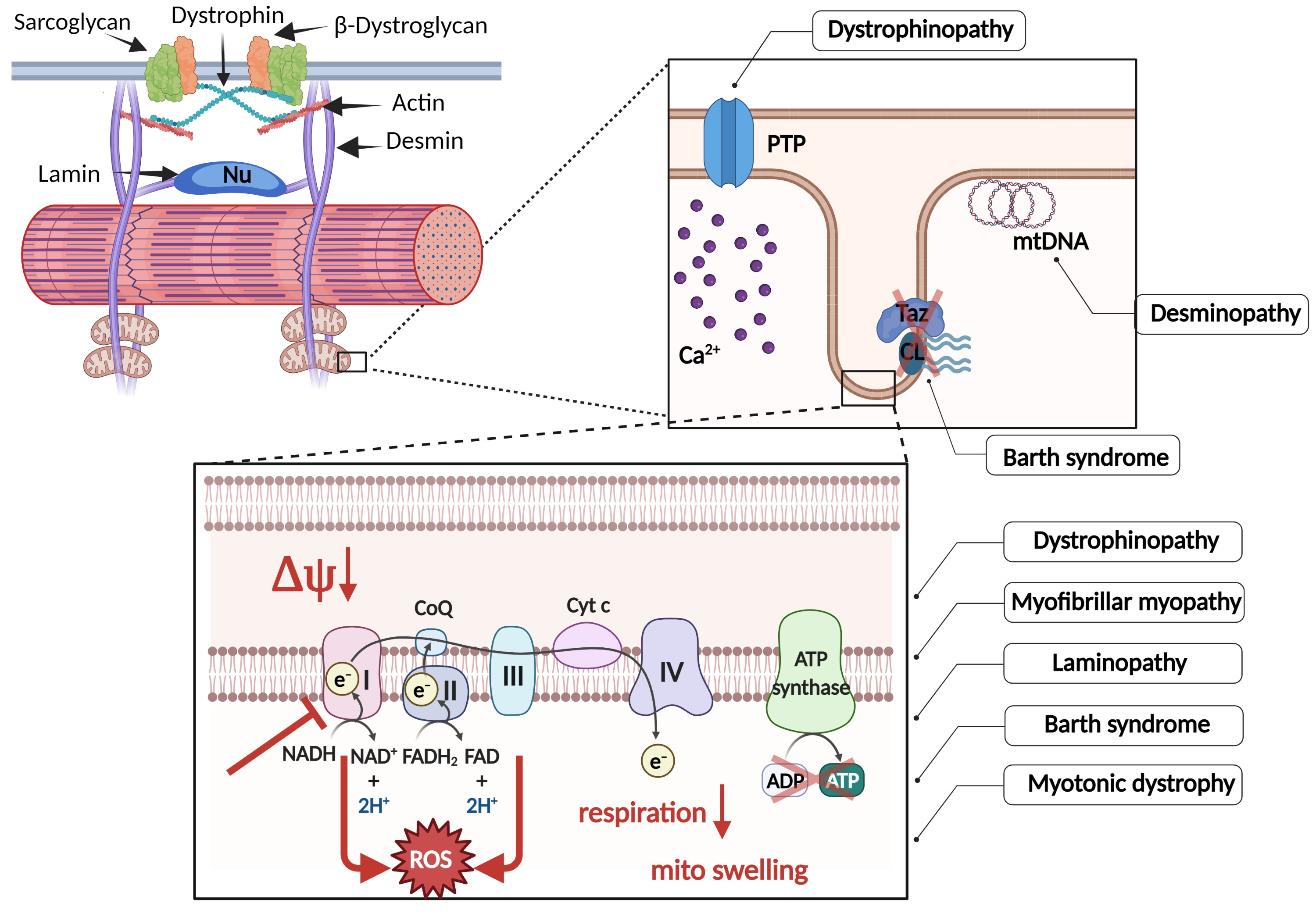{kind=link}
{kind=link}
1. Introduction
2. Role of Mitochondria in Muscle
3. Mitochondrial Dysfunction in Specific Neuromuscular Disorders
3.1. Dystrophinopathies
3.2. Desminopathies and Myofibrillar Myopathies
3.3. Lamin A and Laminopathies
3.4. Myotonic Dystrophy
3.5. Barth Syndrome
3.6. The Other Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laing, N.; Kraus, S.M.; Shaboodien, G.; Ntusi, N.A.B. An overview of the genetic basis of cardiovascular disease. S. Afr. Med. J. 2019, 109, 364–370. [Google Scholar] [CrossRef]
- Kostareva, A.; Sejersen, T.; Sjoberg, G. Genetic spectrum of cardiomyopathies with neuromuscular phenotype. Front. Biosci. 2013, 5, 325–340. [Google Scholar] [CrossRef]
- Limongelli, G.; D’Alessandro, R.; Maddaloni, V.; Rea, A.; Sarkozy, A.; McKenna, W.J. Skeletal muscle involvement in cardiomyopathies. J. Cardiovasc. Med. 2013, 14, 837–861. [Google Scholar] [CrossRef]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Sergushichev, A.; Dmitrieva, R.; Khudiakov, A.; Jorholt, J.; Smolina, N.; Sukhareva, K.; Fomicheva, Y.; et al. Truncating variant in myof gene is associated with limb-girdle type muscular dystrophy and cardiomyopathy. Front. Genet. 2019, 10, 608. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, H.W.; Powers, S.K. Mitochondrial dysfunction is a common denominator linking skeletal muscle wasting due to disease, aging, and prolonged inactivity. Antioxidants 2021, 10, 588. [Google Scholar] [CrossRef] [PubMed]
- De Mario, A.; Gherardi, G.; Rizzuto, R.; Mammucari, C. Skeletal muscle mitochondria in health and disease. Cell Calcium 2021, 94, 102357. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R.; Hoppeler, H. Exercise-induced maximal metabolic rate scales with muscle aerobic capacity. J. Exp. Biol. 2005, 208, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Bulthuis, E.P.; Adjobo-Hermans, M.J.; Willems, P.H.; Koopman, W.J. Mitochondrial Morphofunction in Mammalian Cells. Antioxid. Redox Signal. 2019, 30, 2066–2109. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, H.; Muncke, G. Idiopathic cardiomyopathy and skeletal muscle abnormality. Am. Heart J. 1975, 90, 767–773. [Google Scholar] [CrossRef]
- Smith, E.R.; Heffernan, L.P.; Sangalang, V.E.; Vaughan, L.M.; Flemington, C.S. Voluntary Muscle Involvement in Hypertrophic Cardiomyopathy. Ann. Intern. Med. 1976, 85, 566–572. [Google Scholar] [CrossRef]
- Caforio, A.L.; Rossi, B.; Risaliti, R.; Siciliano, G.; Marchetti, A.; Angelini, C.; Crea, F.; Mariani, M.; Muratorio, A. Type 1 fiber abnormalities in skeletal muscle of patients with hypertrophic and dilated cardiomyopathy: Evidence of subclinical myogenic myopathy. J. Am. Coll. Cardiol. 1989, 14, 1464–1473. [Google Scholar] [CrossRef]
- Karandreas, N.; Stathis, P.; Anastasakis, A.; Rigopoulos, A.; Piperos, P.; Theopistou, A.; Stefanadis, C.; Toutouzas, P. Electromyographic evidence of subclinical myopathy in hypertrophic cardiomyopathy. Muscle Nerve 2000, 23, 1856–1861. [Google Scholar] [CrossRef]
- Shafig, S.A.; Sande, M.; Carruthers, R.; Killip, T.; Milhorat, A.; Shafiq, S. Skeletal muscle in idiopathic cardiomyopathy. J. Neurol. Sci. 1972, 15, 303–320. [Google Scholar] [CrossRef]
- Barth, P.G.; Wanders, R.J.; Ruitenbeek, W.; Roe, C.; Scholte, H.R.; van der Harten, H.; van Moorsel, J.; Duran, M.; Dingemans, K.P. Infantile fibre type disproportion, myofibrillar lysis and cardiomyopathy: A disorder in three unrelated Dutch families. Neuromuscul. Disord. 1998, 8, 296–304. [Google Scholar] [CrossRef]
- Duboc, D.; Jehenson, P.; Tamby, J.F.; Payen, J.F.; Syrota, A.; Guerin, F. Abnormalities of the skeletal muscle in hypertrophic cardiomyopathy. Spectroscopy using phosphorus-31 nuclear magnetic resonance. Arch. Mal. Coeur Vaiss. 1991, 84, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.H.; Kemp, G.J.; Taylor, D.J.; Conway, M.; Rajagopalan, B.; O’Donoghue, A.; Styles, P.; McKenna, W.J.; Radda, G.K. Abnormal skeletal muscle bioenergetics in familial hypertrophic cardiomyopathy. Heart 1997, 78, 177–181. [Google Scholar] [CrossRef][Green Version]
- Arbustini, E.; Fasani, R.; Morbini, P.; Diegoli, M.; Grasso, M.; Dal Bello, B.; Marangoni, E.; Banfi, P.; Banchieri, N.; Bellini, O.; et al. Coexistence of mitochondrial DNA and β myosin heavy chain mutations in hypertrophic cardiomyopathy with late congestive heart failure. Heart 1998, 80, 548–558. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hübner, G.; Grantzow, R. Mitochondrial cardiomyopathy with involvement of skeletal muscles. Virchows Arch. 1982, 399, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fananapazir, L.; Dalakas, M.C.; Cyran, F.; Cohn, G.; Epstein, N.D. Missense mutations in the β-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 1993, 90, 3993–3997. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001, 24, 1575–1594. [Google Scholar] [CrossRef]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The Dystrophin Glycoprotein Complex: Signaling Strength and Integrity for the Sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2015, 96, 253–305. [Google Scholar] [CrossRef]
- Hunsaker, R.H.; Fulkerson, P.K.; Barry, F.J.; Lewis, R.P.; Leier, C.V.; Unverferth, D.V. Cardiac function in Duchenne’s muscular dystrophy. Results of 10-year follow-up study and noninvasive tests. Am. J. Med. 1982, 73, 235–238. [Google Scholar] [CrossRef]
- Verhaert, D.; Richards, K.; Rafael-Fortney, J.A.; Raman, S.V. Cardiac involvement in patients with muscular dystrophies magnetic resonance imaging phenotype and genotypic considerations. Circ. Cardiovasc. Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, G.; Siller, W.G.; Wight, P.A.L.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, M.; Darras, B.T.; Ryan, M.M.; Med, M. Inherited Myopathies and Muscular Dystrophies. Semin. Neurol. 2008, 28, 250–259. [Google Scholar] [CrossRef]
- McNally, E.M.; Pytel, P. Muscle Diseases: The Muscular Dystrophies. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Duncan, C.J.; Greenaway, H.C.; Publicover, S.J.; Rudge, M.F.; Smith, J.L. Experimental production of “septa” and apparent subdivision of muscle mitochondria. J. Bioenerg. Biomembr. 1980, 12, 13–33. [Google Scholar] [CrossRef]
- Bodensteiner, J.B.; Engel, A.G. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology 1978, 28, 439–446. [Google Scholar] [CrossRef]
- Spencer, M.J.; Croall, D.E.; Tidball, J.G. Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 1995, 270, 10909–10914. [Google Scholar] [CrossRef]
- Spencer, M.J.; Tidball, J.G. Calpain concentration is elevated although net calcium-dependent proteolysis is suppressed in dystrophin-deficient muscle. Exp. Cell Res. 1992, 203, 107–114. [Google Scholar] [CrossRef]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef]
- Pauly, M.; Angebault-Prouteau, C.; Dridi, H.; Notarnicola, C.; Scheuermann, V.; Lacampagne, A.; Matecki, S.; Fauconnier, J. ER stress disturbs SR/ER-mitochondria Ca2+ transfer: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Moreira, D.; Neto, H.S.; Marques, M.J. P2Y2 purinergic receptors are highly expressed in cardiac and diaphragm muscles of mdx mice, and their expression is decreased by suramin. Muscle Nerve 2017, 55, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregón, A.; Lansman, J.B. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J. Physiol. 1994, 481, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Dura, M.; Murphy, P.W.; Deng, S.X.; Landry, D.W.; Nieman, D.; Lehnart, S.E.; Samaru, M.; LaCampagne, A.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. [Google Scholar] [CrossRef]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Wrogemann, K.; Pena, S.D.J. Mitochondrial calcium overload: A general mechanism for cell-necrosis in muscle diseases. Lancet 1976, 307, 672–674. [Google Scholar] [CrossRef]
- Zulian, A.; Schiavone, M.; Giorgio, V.; Bernardi, P. Forty years later: Mitochondria as therapeutic targets in muscle diseases. Pharmacol. Res. 2016, 113, 563–573. [Google Scholar] [CrossRef]
- Heydemann, A. Skeletal muscle metabolism in duchenne and becker muscular dystrophy—Implications for therapies. Nutrients 2018, 10, 796. [Google Scholar] [CrossRef]
- Bonsett, C.A.; Rudman, A. “Oil globules” in Duchenne muscular dystrophy-History, demonstration, and metabolic significance. Med. Hypotheses 1994, 43, 327–338. [Google Scholar] [CrossRef]
- Godin, R.; Daussin, F.; Matecki, S.; Li, T.; Petrof, B.J.; Burelle, Y. Peroxisome proliferator-activated receptor γ coactivator 1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. Authors. J. Physiol. C 2012, 590, 5487–5502. [Google Scholar] [CrossRef]
- Jahnke, V.E.; van der Meulen, J.H.; Johnston, H.K.; Ghimbovschi, S.; Partridge, T.; Hoffman, E.P.; Nagaraju, K. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet. Muscle 2012, 2, 16. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Gen. 2013, 22, 153–167. [Google Scholar] [CrossRef]
- Cullen, M.J.; Jaros, E. Ultrastructure of the skeletal muscle in the X chromosome-linked dystrophic (mdx) mouse-Comparison with Duchenne muscular dystrophy. Acta Neuropathol. 1988, 77, 69–81. [Google Scholar] [CrossRef]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Even, P.C.; Decrouy, A.; Chinet, A. Defective regulation of energy metabolism in mdx-mouse skeletal muscles. Biochem. J. 1994, 304, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Gannoun-Zaki, L.; Fournier-Bidoz, S.; le Cama, G.; Chambon, C.; Millasseaub, P.; Lkger, J.J.; Dechesnea, C.A. Down-regulation of mitochondrial mRNAs in the mdx mouse model for Duchenne muscular dystrophy. FEBS Lett. 1995, 375, 268–272. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; Von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef]
- Cole, M.A.; Rafael, J.A.; Taylor, D.J.; Lodi, R.; Davies, K.E.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Gaglianone, R.B.; Santos, A.T.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Costa, M.L.; Quirico-Santos, T.; da Silva, W.S.; Mermelstein, C. Reduced mitochondrial respiration and increased calcium deposits in the EDL muscle, but not in soleus, from 12-week-old dystrophic mdx mice. Sci. Rep. 2019, 9, 1986. [Google Scholar] [CrossRef]
- Passaquin, A.C.; Renard, M.; Kay, L.; Challet, C.; Mokhtarian, A.; Wallimann, T.; Ruegg, U.T. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord. 2002, 12, 174–182. [Google Scholar] [CrossRef]
- Liang, R.C.R. Studies on mitochondria from dystrophic skeletal muscle of mice. Biochem. Med. Metab. Biol. 1986, 36, 172–178. [Google Scholar] [CrossRef]
- Schuh, R.A.; Jackson, K.C.; Khairallah, R.J.; Ward, C.W.; Spangenburg, E.E. Measuring mitochondrial respiration in intact single muscle fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Zhang, H.; Ropelle, E.R.; Sorrentino, V.; Mázala, D.A.G.; Mouchiroud, L.; Marshall, P.L.; Campbell, M.D.; Ali, A.S.; Knowels, G.M.; et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global parylation. Sci. Transl. Med. 2016, 8, 361ra139. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, M.; Choi, J.J.; Lee, N.H.; Jeong, H.; Anderson, S.E.; Han, W.M.; Aliya, B.; Peykova, T.Z.; Verma, S.; García, A.J.; et al. Transplantation of Muscle Stem Cell Mitochondria Rejuvenates the Bioenergetic Function of Dystrophic Muscle. bioRxiv 2020. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef]
- Azzolin, L.; Basso, E.; Argenton, F.; Bernardi, P. Mitochondrial Ca2+ transport and permeability transition in zebrafish (Danio rerio). Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1775–1779. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Roy, S.; Šileikyte, J.; Schiavone, M.; Neuenswander, B.; Argenton, F.; Aubé, J.; Hedrick, M.P.; Chung, T.D.Y.; Forte, M.A.; Bernardi, P.; et al. Discovery, Synthesis, and Optimization of Diarylisoxazole-3-carboxamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2015, 10, 1655–1671. [Google Scholar] [CrossRef]
- Šileikytė, J.; Devereaux, J.; de Jong, J.; Schiavone, M.; Jones, K.; Nilsen, A.; Bernardi, P.; Forte, M.; Cohen, M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. ChemMedChem 2019, 14, 1771–1782. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Šileikytė, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165. [Google Scholar] [CrossRef]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef]
- Hnia, K.; Ramspacher, C.; Vermot, J.; Laporte, J. Desmin in muscle and associated diseases: Beyond the structural function. Cell Tissue Res. 2015, 360, 591–608. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Taylor, M.R.G.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef]
- Schwarz, N.; Leube, R. Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function. Cells 2016, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mericskay, M.; Agbulut, O.; Butler-Browne, G.; Carlsson, L.; Thornell, L.E.; Babinet, C.; Paulin, D. Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J. Cell Biol. 1997, 139, 129–144. [Google Scholar] [CrossRef]
- Kay, L.; Li, Z.; Mericskay, M.; Olivares, J.; Tranqui, L.; Fontaine, E.; Tiivel, T.; Sikk, P.; Kaambre, T.; Samuel, J.L.; et al. Study of regulation of mitochondrial respiration in vivo. An analysis of influence of ADP diffusion and possible role of cytoskeleton. Biochim. Biophys. Acta Bioenerg. 1997, 1322, 41–59. [Google Scholar] [CrossRef]
- Fidziańska, A.; Kotowicz, J.; Sadowska, M.; Goudeau, B.; Walczak, E.; Vicart, P.; Hausmanowa-Petrusewicz, I. A novel desmin R355P mutation causes cardiac and skeletal myopathy. Neuromuscul. Disord. 2005, 15, 525–531. [Google Scholar] [CrossRef]
- Vernengo, L.; Chourbagi, O.; Panuncio, A.; Lilienbaum, A.; Batonnet-Pichon, S.; Bruston, F.; Rodrigues-Lima, F.; Mesa, R.; Pizzarossa, C.; Demay, L.; et al. Desmin myopathy with severe cardiomyopathy in a Uruguayan family due to a codon deletion in a new location within the desmin 1A rod domain. Neuromuscul. Disord. 2010, 20, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Goudeau, B.; Simon, M.C.; Fischer, D.; Eggermann, T.; Clemen, C.S.; Li, Z.; Reimann, J.; Xue, Z.; Rudnik-Schöneborn, S.; et al. On noxious desmin: Functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum. Mol. Genet. 2003, 12, 657–669. [Google Scholar] [CrossRef]
- McCormick, E.M.; Kenyon, L.; Falk, M.J. Desmin common mutation is associated with multi-systemic disease manifestations and depletion of mitochondria and mitochondrial DNA. Front. Genet. 2015, 6, 199. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Wittig, I.; Peeva, V.; Eggers, B.; Heidler, J.; Chevessier, F.; Kley, R.A.; Barkovits, K.; Strecker, V.; Berwanger, C.; et al. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol. 2016, 132, 453–473. [Google Scholar] [CrossRef]
- Vincent, A.E.; Grady, J.P.; Rocha, M.C.; Alston, C.L.; Rygiel, K.A.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Mitochondrial dysfunction in myofibrillar myopathy. Neuromuscul. Disord. 2016, 26, 691–701. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, J.; Chen, S.; Zhu, M.; Hong, D. Mitochondrial proteomics reveal potential targets involved in mitochondrial abnormalities of desminopathy. Clin. Neuropathol. 2017, 36, 15–22. [Google Scholar] [CrossRef]
- Kubánek, M.; Schimerová, T.; Piherová, L.; Brodehl, A.; Krebsová, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansíková, H.; Zeman, J.; Paleček, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Sjöberg, G.; Bruton, J.; Zhang, S.J.; Balogh, J.; Gudkova, A.; Hedberg, B.; Edström, L.; Westerblad, H.; Sejersen, T. Mice expressing L345P mutant desmin exhibit morphological and functional changes of skeletal and cardiac mitochondria. J. Muscle Res. Cell Motil. 2008, 29, 25–36. [Google Scholar] [CrossRef]
- Smolina, N.; Khudiakov, A.; Knyazeva, A.; Zlotina, A.; Sukhareva, K.; Kondratov, K.; Gogvadze, V.; Zhivotovsky, B.; Sejersen, T.; Kostareva, A. Desmin mutations result in mitochondrial dysfunction regardless of their aggregation properties. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165745. [Google Scholar] [CrossRef]
- Smolina, N.; Bruton, J.; Sjoberg, G.; Kostareva, A.; Sejersen, T. Aggregate-prone desmin mutations impair mitochondrial calcium uptake in primary myotubes. Cell Calcium 2014, 56, 269–275. [Google Scholar] [CrossRef]
- Claeys, K.G.; Fardeau, M.; Schröder, R.; Suominen, T.; Tolksdorf, K.; Behin, A.; Dubourg, O.; Eymard, B.; Maisonobe, T.; Stojkovic, T.; et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul. Disord. 2008, 18, 656–666. [Google Scholar] [CrossRef]
- Dittmer, T.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynska, A.; Gonzalo, S.; Shanahan, C.; Askjaer, P. The nuclear lamina in health and disease. Nucleus 2016, 7, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Magagnotti, C.; Bachi, A.; Zerbini, G.; Fattore, E.; Fermo, I.; Riba, M.; Previtali, S.C.; Ferrari, M.; Andolfo, A.; Benedetti, S. Protein profiling reveals energy metabolism and cytoskeletal protein alterations in LMNA mutation carriers. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 970–979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caron, M.; Auclair, M.; Donadille, B.; Béréziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Sieprath, T.; Corne, T.D.J.; Nooteboom, M.; Grootaert, C.; Rajkovic, A.; Buysschaert, B.; Robijns, J.; Broers, J.L.V.; Ramaekers, F.C.S.; Koopman, W.J.H.; et al. Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus 2015, 6, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Carboni, N.; Bernasconi, P. Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 2016, 5, 33. [Google Scholar] [CrossRef]
- Boschmann, M.; Engeli, S.; Moro, C.; Luedtke, A.; Adams, F.; Gorzelniak, K.; Rahn, G.; Mähler, A.; Dobberstein, K.; Krüger, A.; et al. LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance. J. Clin. Endocrinol. Metab. 2010, 95, 1634–1643. [Google Scholar] [CrossRef]
- Ignatieva, E.V.; Ivanova, O.A.; Komarova, M.Y.; Khromova, N.V.; Polev, D.E.; Kostareva, A.A.; Sergushichev, A.; Dmitrieva, R.I. LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile. Genes 2020, 11, 1057. [Google Scholar] [CrossRef]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed]
- André, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in skeletal muscle myogenesis, growth, and regeneration in myotonic dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Lam, L.T.; Pham, Y.C.N.; Man, N.T.; Morris, G.E. Characterization of a monoclonal antibody panel shows that the myotonic dystrophy protein kinase, DMPK, is expressed almost exclusively in muscle and heart. Hum. Mol. Genet. 2000, 9, 2167–2173. [Google Scholar] [CrossRef]
- Cho, D.H.; Tapscott, S.J. Myotonic dystrophy: Emerging mechanisms for DM1 and DM2. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 195–204. [Google Scholar] [CrossRef]
- Harmon, E.B.; Harmon, M.L.; Larsen, T.D.; Yang, J.; Glasford, J.W.; Perryman, M.B. Myotonic dystrophy protein kinase is critical for nuclear envelope integrity. J. Biol. Chem. 2011, 286, 40296–40306. [Google Scholar] [CrossRef]
- Van Herpen, R.E.M.A.; Oude, R.J.A.O.; Wijers, M.; Bennink, M.B.; van de Loo, F.A.J.; Fransen, J.; Wieringa, B.; Wansink, D.G. Divergent Mitochondrial and Endoplasmic Reticulum Association of DMPK Splice Isoforms Depends on Unique Sequence Arrangements in Tail Anchors. Mol. Cell. Biol. 2005, 25, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Smith, D.B.J.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Shimokawa, M.; Yamamoto, M.; Kameda, N.; Mizusawa, H.; Baba, T.; Terada, N.; Fujii, Y.; Ohno, S.; Ishiura, S.; et al. Decreased expression of myotonic dystrophy protein kinase and disorganization of sarcoplasmic reticulum in skeletal muscle of myotonic dystrophy. J. Neurol. Sci. 1999, 162, 38–50. [Google Scholar] [CrossRef]
- Yamada, H.; Nakagawa, M.; Higuchi, I.; Horikiri, T.; Osame, M. Detection of DNA fragmentation of myonuclei in myotonic dystrophy by double staining with anti-emerin antibody and by nick end-labeling. J. Neurol. Sci. 2000, 173, 97–102. [Google Scholar] [CrossRef]
- Loro, E.; Rinaldi, F.; Malena, A.; Masiero, E.; Novelli, G.; Angelini, C.; Romeo, V.; Sandri, M.; Botta, A.; Vergani, L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 2010, 17, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, D.; Lombardi, V.; Mancuso, M.; Martelli, F.; Sighieri, C.; Rocchi, A.; Tovani, S.; Siciliano, G.; Murri, L. Potential involvement of ubiquinone in myotonic dystrophy pathophysiology: New diagnostic approaches for new rationale therapeutics. Neurol. Sci. 2000, 21, S979–S980. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Mancuso, M.; Tedeschi, D.; Manca, M.L.; Renna, M.R.; Lombardi, V.; Rocchi, A.; Martelli, F.; Murri, L. Coenzyme Q10, exercise lactate and CTG trinucleotide expansion in myotonic dystrophy. Brain Res. Bull. 2001, 56, 405–410. [Google Scholar] [CrossRef]
- Gramegna, L.L.; Giannoccaro, M.P.; Manners, D.N.; Testa, C.; Zanigni, S.; Evangelisti, S.; Bianchini, C.; Oppi, F.; Poda, R.; Avoni, P.; et al. Mitochondrial dysfunction in myotonic dystrophy type 1. Neuromuscul. Disord. 2018, 28, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Pantic, B.; Trevisan, E.; Citta, A.; Rigobello, M.P.; Marin, O.; Bernardi, P.; Salvatori, S.; Rasola, A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef] [PubMed]
- Ophuis, R.J.A.O.; Wijers, M.; Bennink, M.B.; van de Loo, F.A.J.; Fransen, J.A.M.; Wieringa, B.; Wansink, D.G. A tail-anchored myotonic dystrophy protein kinase isoform induces perinuclear clustering of mitochondria, autophagy and apoptosis. PLoS ONE 2009, 4, e8024. [Google Scholar] [CrossRef]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.M.; Van’T Veer-Korthof, E.T.; van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- D’Adamo, P.; Gedeon, L.; Janssen, A.; Bolhuis, S.; Barth, P.A.; Wilson, P.G.; Haan, M.; Patton, E.; Green, M.A.; Zammarchi, A.J.; et al. The x-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am. J. Hum. Genet. 1997, 61, 862–867. [Google Scholar] [CrossRef]
- Neuwald, A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997, 7, R462–R466. [Google Scholar] [CrossRef]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the respiratory chain together: Cardiolipin is required for super complex formation in the inner mitochondrial membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial Respiratory Chain Super complexes Are Destabilized in Barth Syndrome Patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Tsoras, M.; Göring, H.H.H.; Hug, P.; Chuck, G.; Tombragel, E.; McGraw, C.; Schlotman, J.; Ralston, M.A.; Hug, G. Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Investig. 2002, 82, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Xu, Y.; Stokes, D.L.; Schlame, M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Investig. 2007, 87, 40–48. [Google Scholar] [CrossRef]
- El-Hafidi, M.; Correa, F.; Zazueta, C. Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165744. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Barth, P.G.; Valianpour, F.; Bowen, V.M.; Lam, J.; Duran, M.; Vaz, F.M.; Wanders, R.J.A. X-Linked Cardioskeletal Myopathy and Neutropenia (Barth Syndrome): An Update. Am. J. Med. Genet. 2004, 126, 349–354. [Google Scholar] [CrossRef]
- Brandner, K.; Mick, D.U.; Frazier, A.E.; Taylor, R.D.; Meisinger, C.; Rehling, P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: Implications for Barth syndrome. Mol. Biol. Cell 2005, 16, 5202–5214. [Google Scholar] [CrossRef]
- Chen, S.; He, Q.; Greenberg, M.L. Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 2008, 68, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V.; Greenberg, M.L. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 2002, 364, 317–322. [Google Scholar] [CrossRef]
- Shen, Z.; Li, Y.; Gasparski, A.N.; Abeliovich, H.; Greenberg, M.L. Cardiolipin regulates mitophagy through the protein kinase C pathway. J. Biol. Chem. 2017, 292, 2916–2923. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Condell, M.; Plesken, H.; Edelman-Novemsky, I.; Ma, J.; Ren, M.; Schlame, M. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 11584–11588. [Google Scholar] [CrossRef]
- Khuchua, Z.; Yue, Z.; Batts, L.; Strauss, A.W. A zebrafish model of human barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 2006, 99, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Vaz, F.; Houtkooper, R.H.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 2011, 286, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Soustek, M.S.; Falk, D.J.; Mah, C.S.; Toth, M.J.; Schlame, M.; Lewin, A.S.; Byrne, B.J. Characterization of a transgenic short hairpin RNA-induced murine model of tafazzin deficiency. Hum. Gene Ther. 2011, 22, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 2016, 8, 139–154. [Google Scholar] [CrossRef]
- Le, C.H.; Benage, L.; Specht, K.S.; Puma, L.C.L.; Mulligan, C.M.; Heuberger, A.; Prenni, J.E.; Claypool, S.M.; Chatfield, K.C.; Sparagna, G.C.; et al. Tafazzin deficiency impairs CoA-dependent oxidative metabolism in cardiac mitochondria. J. Biol. Chem. 2020, 295, 12485–12497. [Google Scholar] [CrossRef]
- Greenwell, A.A.; Gopal, K.; Altamimi, T.; Saed, C.T.; Wang, F.; Tabatabaei-Dakhili, S.A.; Ho, K.L.; Zhang, L.; Eaton, F.; Kruger, J.L.; et al. Barth Syndrome-Related Cardiomyopathy is Associated with a Reduction in Myocardial Glucose Oxidation. Am. J. Physiol. Circ. Physiol. 2021, 320, H2255–H2269. [Google Scholar] [CrossRef]
- Goncalves, R.L.S.; Schlame, M.; Bartelt, A.; Brand, M.D.; Hotamışlıgil, G.S. Cardiolipin deficiency in Barth syndrome is not associated with increased superoxide/H2O2 production in heart and skeletal muscle mitochondria. FEBS Lett. 2021, 595, 415–432. [Google Scholar] [CrossRef]
- Ghosh, S.; Ball, W.B.; Madaris, T.R.; Srikantan, S.; Madesh, M.; Mootha, V.K.; Gohil, V.M. An essential role for cardiolipin in the stability and function of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2020, 117, 16383–16390. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, S.; Guo, X.; Li, Y.; Ogurlu, R.; Lu, F.; Prondzynski, M.; de la Serna Buzon, S.; Ma, Q.; Zhang, D.; et al. Increased Reactive Oxygen Species-Mediated Ca2+/Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome. Circulation 2021, 143, 1894–1911. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV gene therapy prevents and reverses heart failure in a murine knockout model of barth syndrome. Circ. Res. 2020, 126, 1024–1039. [Google Scholar] [CrossRef]
- Southern, W.M.; Nichenko, A.S.; Qualls, A.E.; Portman, K.; Gidon, A.; Beedle, A.M.; Call, J.A. Mitochondrial dysfunction in skeletal muscle of fukutin-deficient mice is resistant to exercise- and 5-aminoimidazole-4-carboxamide ribonucleotide-induced rescue. Exp. Physiol. 2020, 105, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Pambianco, S.; Giovarelli, M.; Perrotta, C.; Zecchini, S.; Cervia, D.; di Renzo, I.; Moscheni, C.; Ripolone, M.; Violano, R.; Moggio, M.; et al. Reversal of Defective Mitochondrial Biogenesis in Limb-Girdle Muscular Dystrophy 2D by Independent Modulation of Histone and PGC-1α Acetylation. Cell Rep. 2016, 17, 3010–3023. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Oliveira, C.C.; Steinz, M.; Schneiderat, P.; Mulder, H.; Durbeej, M. Bioenergetic Impairment in Congenital Muscular Dystrophy Type 1A and Leigh Syndrome Muscle Cells. Sci. Rep. 2017, 7, 45272. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.F.; Huang, Y.Y.; Lu, W.S.; Huang, Y.H.; Xian, J.; Wei, H.Q.; Huang, Q. The Caveolin-3 P104L mutation of LGMD-1C leads to disordered glucose metabolism in muscle cells. Biochem. Biophys. Res. Commun. 2017, 486, 218–223. [Google Scholar] [CrossRef]
- Gonzalez Coraspe, J.A.; Weis, J.; Anderson, M.E.; Munchberg, U.; Lorenz, K.; Buchkremer, S.; Carr, S.; Zahedi, R.P.; Brauers, E.; Michels, H.; et al. Biochemical and pathological changes result from mutatedcaveolin-3 in muscle. Skelet. Muscle 2018, 8, 28. [Google Scholar] [CrossRef]
- Shah, D.S.; Nisr, R.B.; Stretton, C.; Krasteva-Christ, G.; Hundal, H.S. Caveolin-3 deficiency associated with the dystrophy P104L mutation impairs skeletal muscle mitochondrial form and function. J. Cachexia Sarcopenia Muscle 2020, 11, 838–858. [Google Scholar] [CrossRef] [PubMed]
- Gayathri, N.; Alefia, R.; Nalini, A.; Yasha, T.C.; Anita, M.; Santosh, V.; Shankar, S.K. Dysferlinopathy: Spectrum of pathological changes in skeletal muscle tissue. Indian J. Pathol. Microbiol. 2011, 54, 350–354. [Google Scholar] [CrossRef]
- Liu, F.; Lou, J.; Zhao, D.; Li, W.; Zhao, Y.; Sun, X.; Yan, C. Dysferlinopathy: Mitochondrial abnormalities in human skeletal muscle. Int. J. Neurosci. 2016, 126, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul. Disord. 2016, 26, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Edgett, B.A.; Huber, J.S.; Polidovitch, N.; Schlattner, U.; Backx, P.H.; Simpson, J.A.; Perry, C.G.R. Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. J. Physiol. 2020, 598, 1377–1392. [Google Scholar] [CrossRef]
- Shao, Z.; Koh, W.; Ni, Y.; Li, W.; Agatisa-Boyle, B.; Merkurjev, D.; Tang, W.H.W. RNA Sequence Analyses throughout the Course of Mouse Cardiac Laminopathy Identify Differentially Expressed Genes for Cell Cycle Control and Mitochondrial Function. Sci. Rep. 2020, 10, 6632. [Google Scholar] [CrossRef]
- Clemen, C.S.; Stöckigt, F.; Strucksberg, K.H.; Chevessier, F.; Winter, L.; Schütz, J.; Bauer, R.; Thorweihe, J.M.; Wenzel, D.; Schlötzer-Schrehardt, U.; et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015, 129, 297–315. [Google Scholar] [CrossRef]
- Fountoulakis, M.; Soumaka, E.; Rapti, K.; Mavroidis, M.; Tsangaris, G.; Maris, A.; Weisleder, N.; Capetanaki, Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. J. Mol. Cell Cardiol. 2005, 38, 461–474. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX Is an Essential Component of Mitochondrial Na+/Ca2+ Exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; de Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef]
- Kazmierczak, K.; Liang, J.; Yuan, C.-C.; Yadav, S.; Sitbon, Y.H.; Walz, K.; Ma, W.; Irving, T.C.; Cheah, J.X.; Gomes, A.V.; et al. Slow-twitch skeletal muscle defects accompany cardiac dysfunction in transgenic mice with a mutation in the myosin regulatory light chain. FASEB J. 2019, 33, 3152–3166. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ignatieva, E.; Smolina, N.; Kostareva, A.; Dmitrieva, R. Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype. Int. J. Mol. Sci. 2021, 22, 7349. https://doi.org/10.3390/ijms22147349
Ignatieva E, Smolina N, Kostareva A, Dmitrieva R. Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype. International Journal of Molecular Sciences. 2021; 22(14):7349. https://doi.org/10.3390/ijms22147349
Chicago/Turabian StyleIgnatieva, Elena, Natalia Smolina, Anna Kostareva, and Renata Dmitrieva. 2021. "Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype" International Journal of Molecular Sciences 22, no. 14: 7349. https://doi.org/10.3390/ijms22147349
APA StyleIgnatieva, E., Smolina, N., Kostareva, A., & Dmitrieva, R. (2021). Skeletal Muscle Mitochondria Dysfunction in Genetic Neuromuscular Disorders with Cardiac Phenotype. International Journal of Molecular Sciences, 22(14), 7349. https://doi.org/10.3390/ijms22147349