
New Concept and Apparatus for Cytocentrifugation and Cell Processing for Microscopy Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- (1)
- Samples have to be typically removed from the special cassette before the staining step(s), the personnel working with the biological material can come into direct contact with the unfixed material.
- (2)
- Relatively large slides are used and the slides are commonly stained by immersion in the staining solution. Therefore, a large amount of staining solutions is necessary. Alternatively, special devices or a special approach has to be used, which commonly results in the prolongation of the procedure, making it more expensive.
- (3)
- A special cytocentrifuge or cytorotor is usually needed.
- (4)
- Relatively high expenses are necessary for the purchase of the cassettes used for cytocentrifugation. Consequently, it often leads to the repeated usage of these cassettes.
2. Results and Discussion
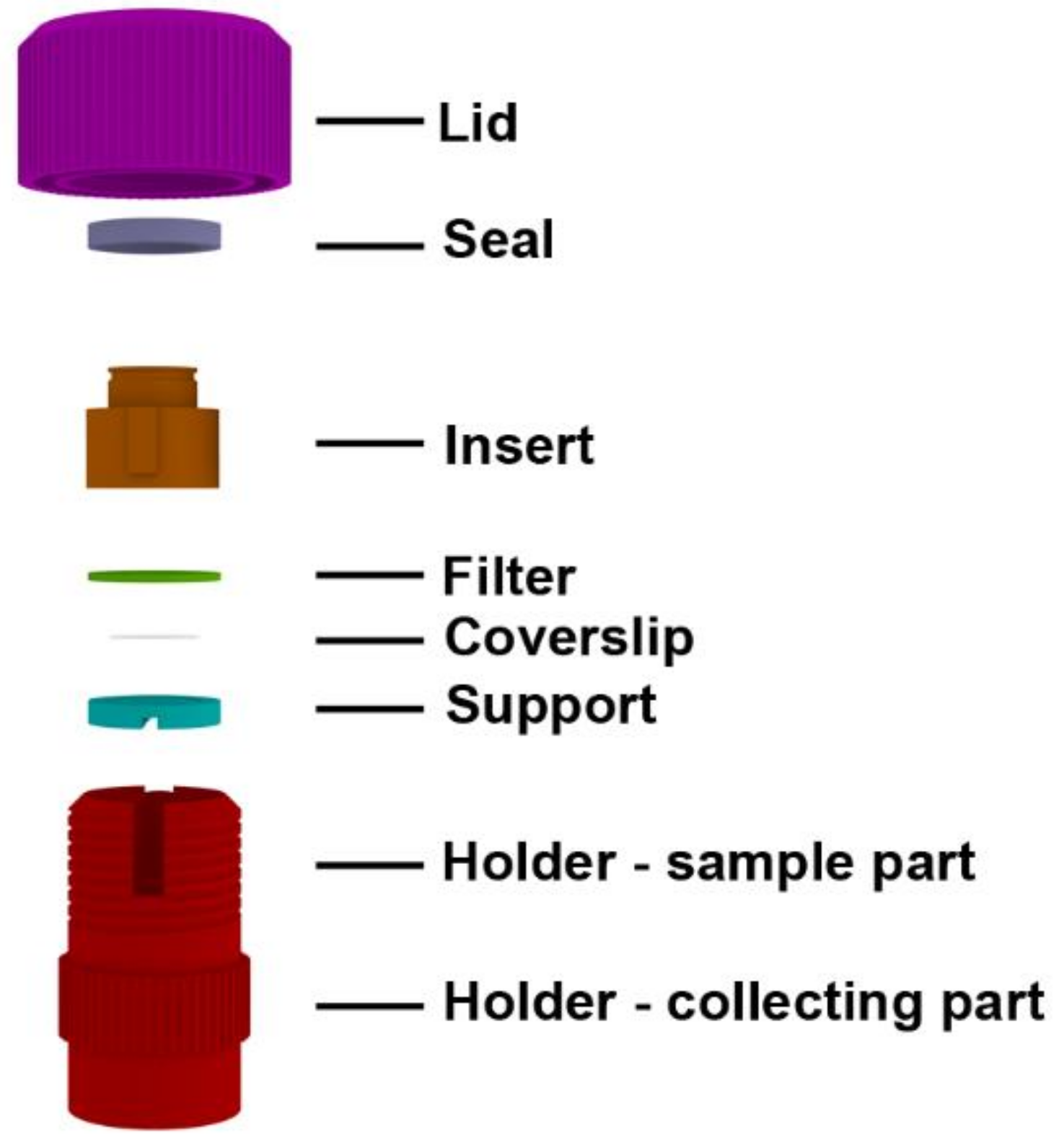
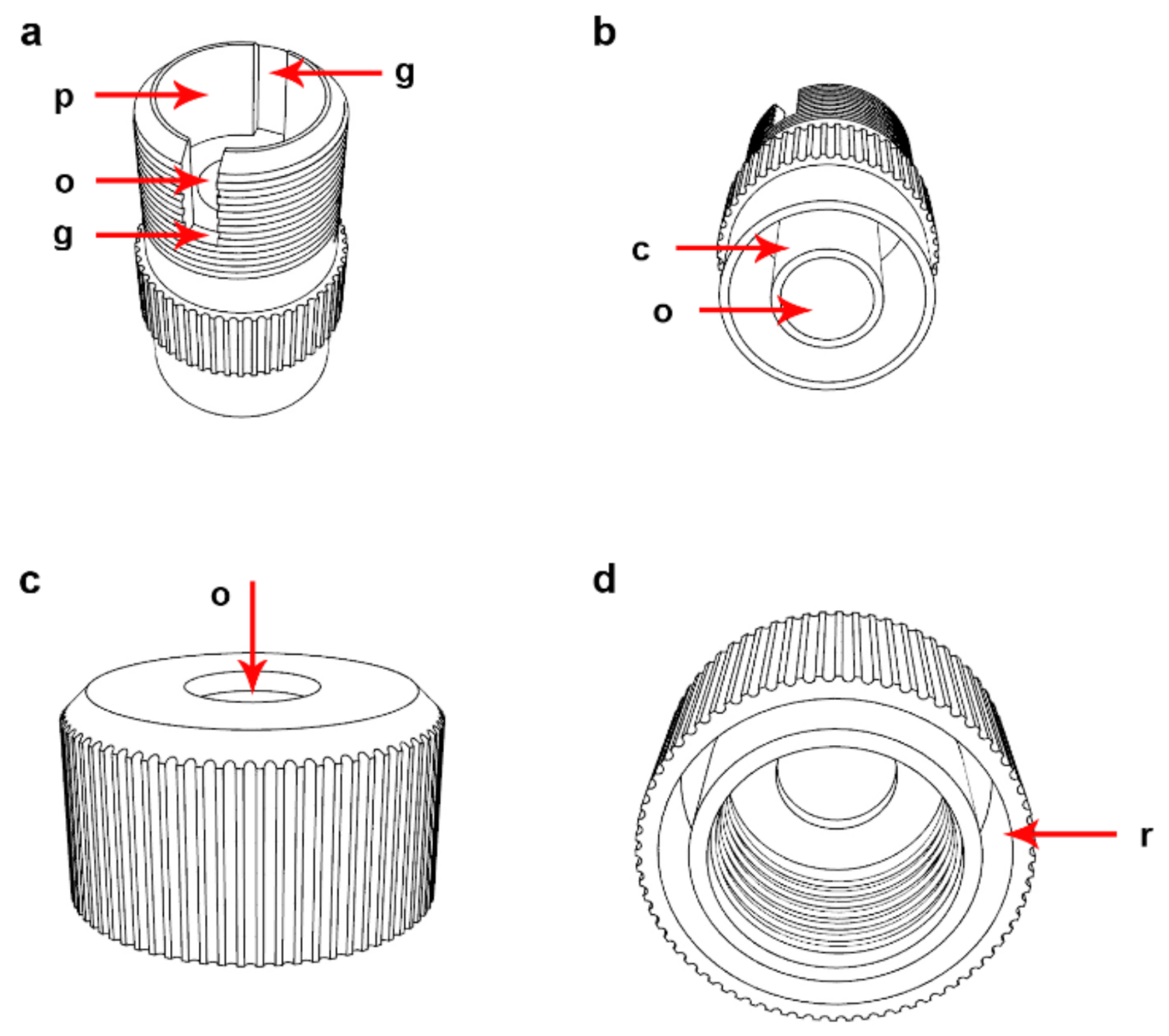
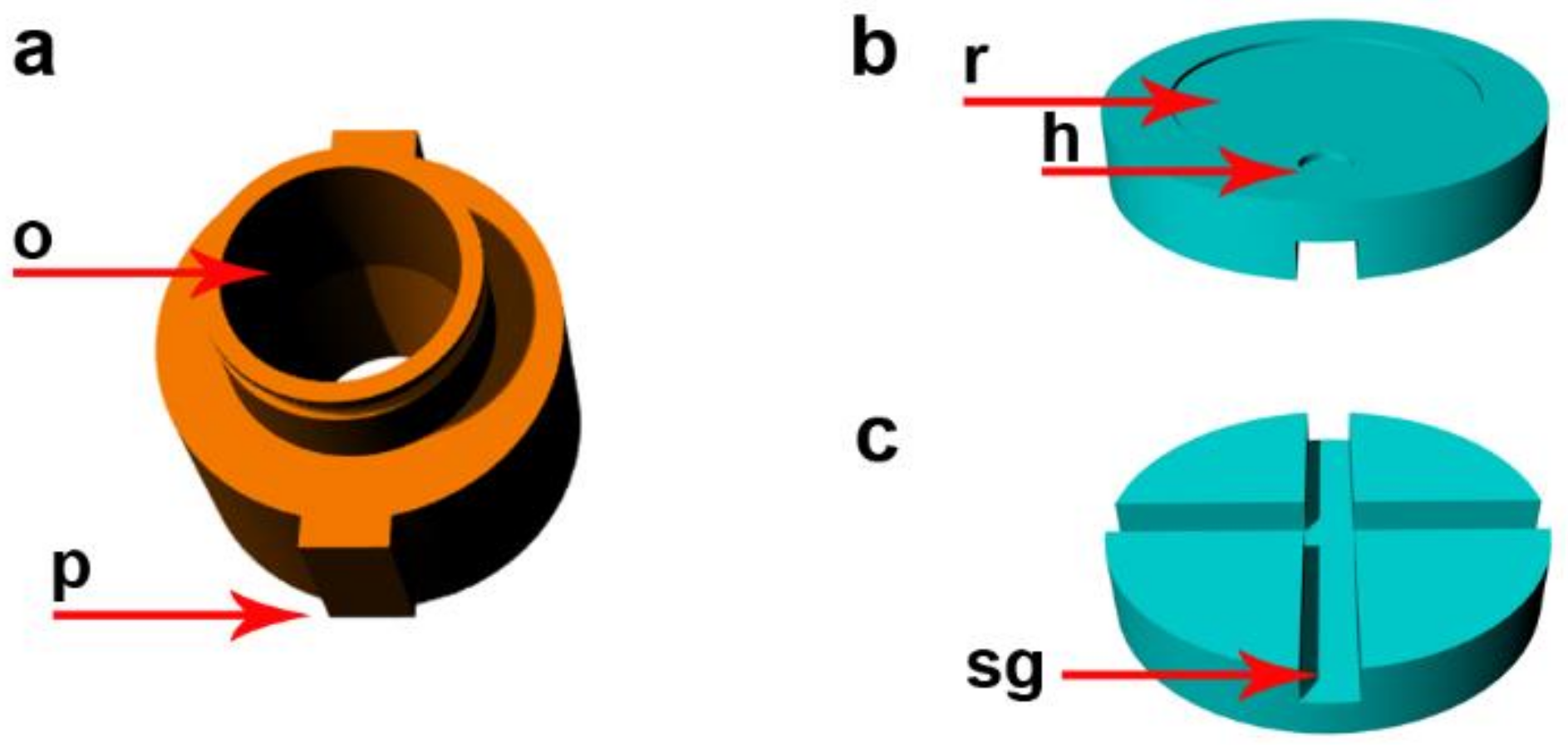
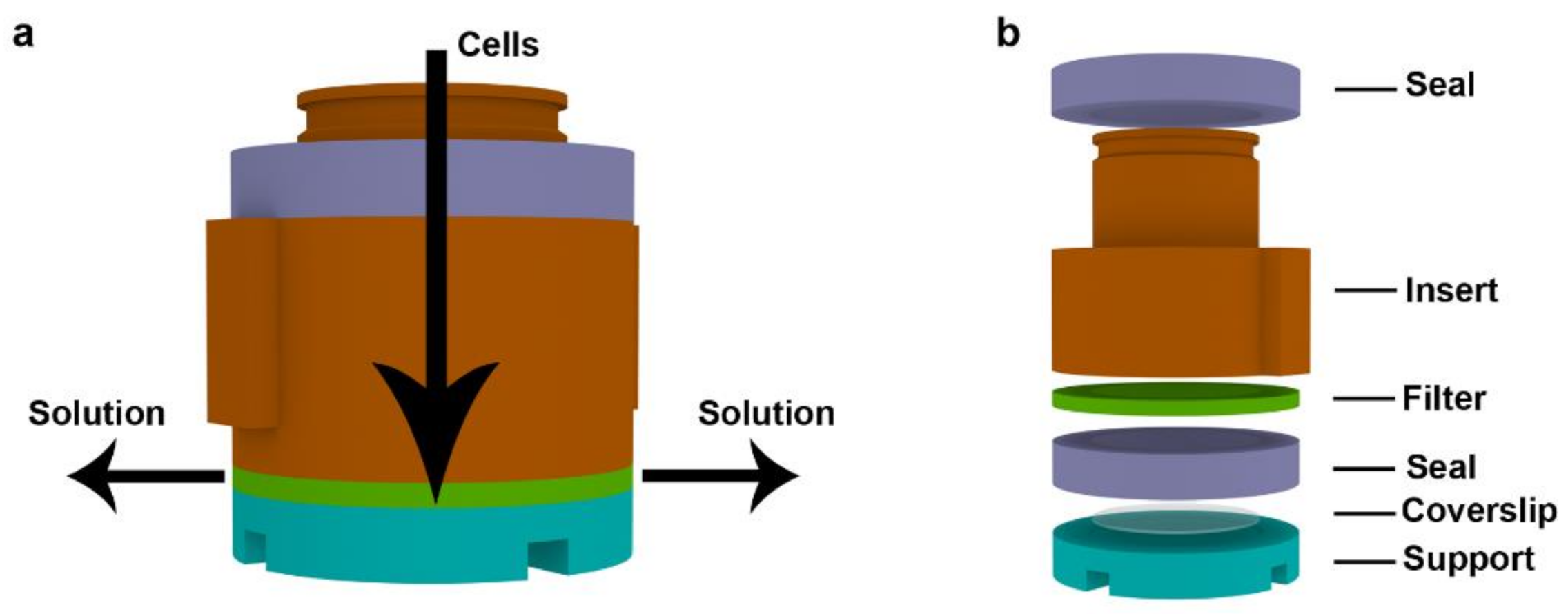
2.1. Description of the Device
2.2. Examples of the CytoTrap Use
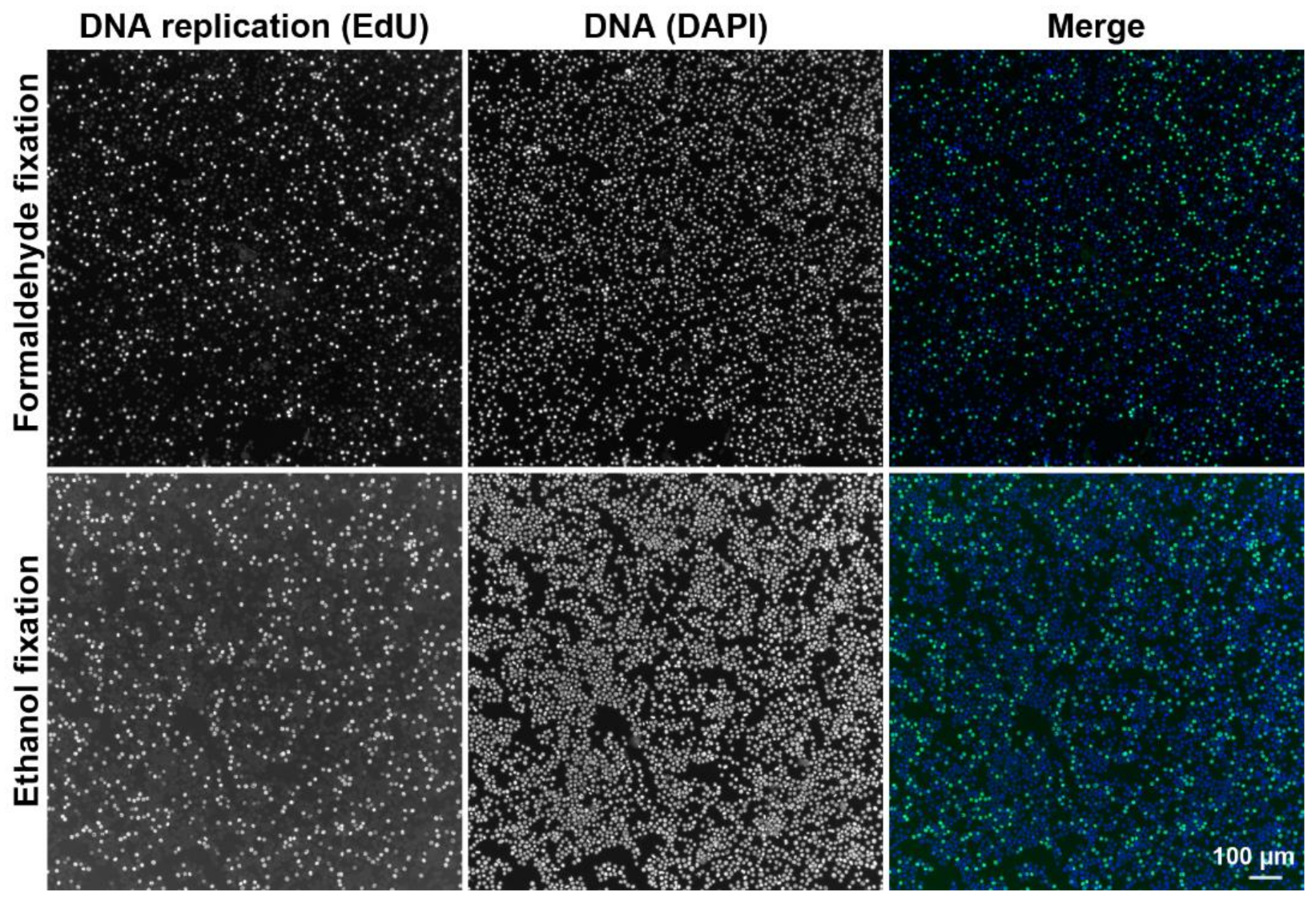
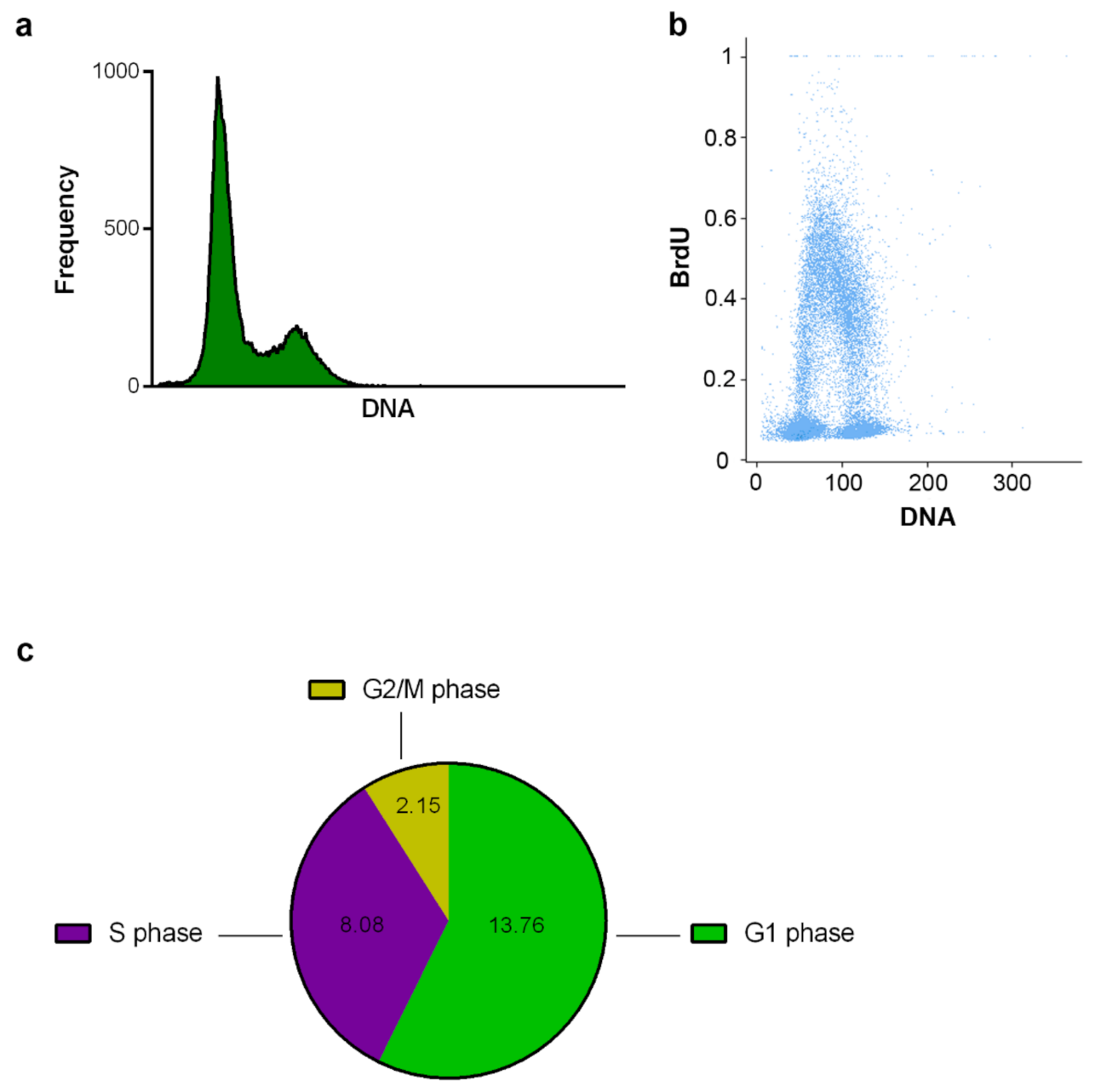
2.2.1. DNA Replication and Cell Cycle Analysis Using the CytoTrap
2.2.2. Giemsa−Romanowski Staining Using the CytoTrap
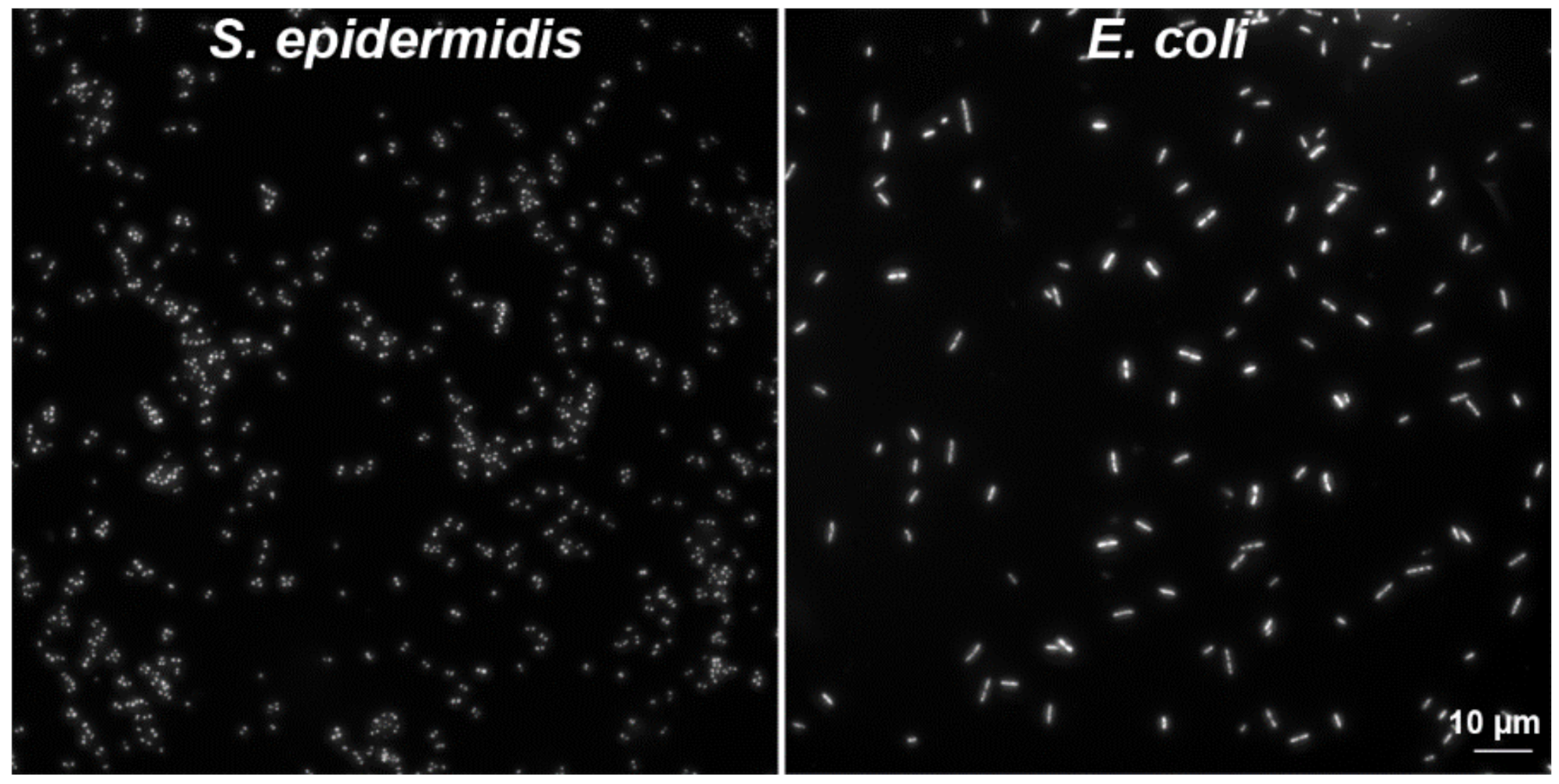
2.2.3. Another Possible Usage of the CytoTrap
3. Materials and Methods
3.1. Cell Cultures and Bacteria Cells
3.2. Preparation of Cytospin Samples
3.2.1. Preparation of Fixed Cell Samples
3.2.2. Preparation of Non-Fixed Cells by the Dry Centrifugation
3.2.3. Preparation of Ethanol-Fixed Samples by Dry Centrifugation
3.2.4. Preparation of Formaldehyde-Fixed Samples Using Wet Centrifugation
3.3. Preparation of Bacteria Samples by Wet Centrifugation
3.4. Preparation of DNA Halo Samples
3.5. Isolated Mitochondria
3.6. Sample Preparation by the Conventional Cytocentrifuge
3.7. Staining Protocols
3.7.1. DAPI Staining
3.7.2. Giemsa−Romanowski Staining
3.7.3. Labeling of DNA Replication by EdU
3.7.4. Labeling of DNA Replication by BrdU
3.8. Fluorescence Microscopy
3.9. Data Evaluation
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bots, G.T.A.; Went, L.N.; Schaberg, A. Een toestel voor cytologisch onderzoek van de liquor cerebrospinalis. Ned. T. Geneeskd 1963, 107, 445–446. [Google Scholar]
- Dore, C.F.; Balfour, B.M. A device for preparing cell spreads. Immunology 1965, 9, 403–405. [Google Scholar]
- Watson, P. A slide centrifuge: An apparatus for concentrating cells in suspension onto a microscope slide. J. Lab. Clin. Med. 1966, 68, 494–501. [Google Scholar] [PubMed]
- Stokes, B.O. Principles of cytocentrifugation. Lab. Med. 2004, 35, 434–437. [Google Scholar] [CrossRef]
- Bucci, D. Preparation of Cytological Slides from Mononuclear Cell Fraction of Bone Marrow Aspirate or Peripheral Blood; Biorepositories and Biospecimen Research Branch, National Cancer Institute: Bethesda, MD, USA, 2014. Available online: https://brd.nci.nih.gov/brd/sop/download-pdf/1088 (accessed on 21 May 2018).
- Sen, R.; Hasija, S.; Kalra, R.; Garg, S.; Singh, A.; Megha. Morphometric analysis and immunocytochemical staining on cytospin preparation in effusion cytology: A study. J. Cytol. Histol. 2015, 6, 314. [Google Scholar]
- Oertel, J.; Oertel, B.; Dorken, B. Detection of small numbers of cells characteristic for haematological disorders in peripheral blood (the deep diff). Clin. Lab. Haematol. 2002, 24, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.G.; Halford, J.A. Evaluation of thin-layer methods in urine cytology. Cytopathology 2001, 12, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Chapinrobertson, K.; Dahlberg, S.E.; Edberg, S.C. Clinical and laboratory analyses of cytospin-prepared gram stains for recovery and diagnosis of bacteria from sterile body-fluids. J. Clin. Microbiol. 1992, 30, 377–380. [Google Scholar] [CrossRef] [PubMed]
- da Cunha Santos, G.; Saieg, M.A. Preanalytic specimen triage: Smears, cell blocks, cytospin preparations, transport media, and cytobanking. Cancer Cytopathol. 2017, 125, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Marcos, R.; Santos, M.; Marrinhas, C.; Correia-Gomes, C.; Caniatti, M. Cytocentrifuge preparation in veterinary cytology: A quick, simple, and affordable manual method to concentrate low cellularity fluids. Vet. Clin. Path. 2016, 45, 725–731. [Google Scholar] [CrossRef]
- Hu, X.; Laguerre, V.; Packert, D.; Nakasone, A.; Moscinski, L. A simple and efficient method for preparing cell slides and staining without using cytocentrifuge and cytoclips. Int. J. Cell Biol. 2015, 2015, 813216. [Google Scholar] [CrossRef]
- Krishnamurthy, V.; Satish, S.; Doreswamy, S.M.; Vimalambike, M.G. Comparison of cell preparations between commercially available filter cards of the cytospin with custom made filter cards. J. Clin. Diagn. Res. 2016, 10, EC18–EC20. [Google Scholar] [CrossRef]
- Stadler, C.; Skogs, M.; Brismar, H.; Uhlen, M.; Lundberg, E. A single fixation protocol for proteome-wide immunofluorescence localization studies. J. Proteom. 2010, 73, 1067–1078. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Yao, Q. Copper-based biomaterials for bone and cartilage tissue engineering. J. Orthop. Translat. 2021, 29, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Byerley, J.J.; Teo, W.K. Oxidation of formaldehyde by copper(I1) in aqueous solution. Can. J. Chem. 1969, 47, 3355–3360. [Google Scholar] [CrossRef]
- Ligasova, A.; Vydrzalova, M.; Burianova, R.; Bruckova, L.; Vecerova, R.; Janostakova, A.; Koberna, K. A new sensitive method for the detection of mycoplasmas using fluorescence microscopy. Cells 2019, 8, 1510. [Google Scholar] [CrossRef]
- Maurya, D.K.; Halo, J. An imagej program for semiautomatic quantification of DNA damage at single-cell level. Int. J. Toxicol. 2014, 33, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Mendez, J. Cohesin organizes chromatin loops at DNA replication factories. Gene Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef]
- Ligasova, A.; Liboska, R.; Rosenberg, I.; Koberna, K. The fingerprint of anti-bromodeoxyuridine antibodies and its use for the assessment of their affinity to 5-bromo-2’-deoxyuridine in cellular DNA under various conditions. PLoS ONE 2015, 10, e0132393. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Liboska, R.; Friedecky, D.; Micova, K.; Adam, T.; Ozdian, T.; Rosenberg, I.; Koberna, K. Dr Jekyll and Mr Hyde: A strange case of 5-ethynyl-2’-deoxyuridine and 5-ethynyl-2’-deoxycytidine. Open Biol. 2016, 6, 150172. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Strunin, D.; Friedecky, D.; Adam, T.; Koberna, K. A fatal combination: A thymidylate synthase inhibitor with DNA damaging activity. PLoS ONE 2015, 10, e0117459. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Konecny, P.; Frydrych, I.; Koberna, K. Cell cycle profiling by image and flow cytometry: The optimised protocol for the detection of replicational activity using 5-Bromo-2’-deoxyuridine, low concentration of hydrochloric acid and exonuclease III. PLoS ONE 2017, 12, e0175880. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Strunin, D.; Koberna, K. A new method of the visualization of the double-stranded mitochondrial and nuclear DNA. PLoS ONE 2013, 8, e66864. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. ilastik: Interactive machine learning for (bio) image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ligasová, A.; Koberna, K. New Concept and Apparatus for Cytocentrifugation and Cell Processing for Microscopy Analysis. Int. J. Mol. Sci. 2021, 22, 7098. https://doi.org/10.3390/ijms22137098
Ligasová A, Koberna K. New Concept and Apparatus for Cytocentrifugation and Cell Processing for Microscopy Analysis. International Journal of Molecular Sciences. 2021; 22(13):7098. https://doi.org/10.3390/ijms22137098
Chicago/Turabian StyleLigasová, Anna, and Karel Koberna. 2021. "New Concept and Apparatus for Cytocentrifugation and Cell Processing for Microscopy Analysis" International Journal of Molecular Sciences 22, no. 13: 7098. https://doi.org/10.3390/ijms22137098
APA StyleLigasová, A., & Koberna, K. (2021). New Concept and Apparatus for Cytocentrifugation and Cell Processing for Microscopy Analysis. International Journal of Molecular Sciences, 22(13), 7098. https://doi.org/10.3390/ijms22137098